
Abstract
Pathogenesis of neurodegenerative diseases involves dysfunction of mitochondria, one of the most important cell organelles in the brain, with its most prominent roles in producing energy and regulating cellular metabolism. Here we investigated the effect of transferring active intact mitochondria as a potential therapy for Alzheimer’s disease (AD), in order to correct as many mitochondrial functions as possible, rather than a mono-drug related therapy. For this purpose, AD-mice (amyloid-β intracerebroventricularly injected) were treated intravenously (IV) with fresh human isolated mitochondria. One to two weeks later, a significantly better cognitive performance was noticed in the mitochondria treated AD-mice relative to vehicle treated AD-mice, approaching the performance of non-AD mice. We also detected a significant decrease in neuronal loss and reduced gliosis in the hippocampus of treated mice relative to untreated AD-mice. An amelioration of the mitochondrial dysfunction in brain was noticed by the increase of citrate-synthase and cytochrome c oxidase activities relative to untreated AD-mice, reaching activity levels of non-AD-mice. Increased mitochondrial activity was also detected in the liver of mitochondria treated mice. No treatment-related toxicity was noted. Thus, IV mitochondrial transfer may possibly offer a novel therapeutic approach for AD.
INTRODUCTION
Alzheimer’s disease (AD) is a multifactorial neurodegenerative disease affecting many cellular pathways, including protein aggregation, mitochondrial dysfunction, oxidative stress, and neuroinflammation, leading ultimately to neuronal death. Functional mitochondria are critical for the normal function of all cells, and they are considered the ‘powerhouses of cells’, providing energy via ATP generation [1]. Due to their limited glycolytic capacity, neurons are highly dependent on mitochondrial function for energy production [2]. We have recently shown that impairing mitochondrial function induced AD/tauopathy brain pathology (Lahiani-Cohen et al., unpublished data).
Most anti-AD therapies have focused so far on anti-protein (particularly amyloid) aggregation strategies, but most studies have failed in the clinic. Targeting the mitochondrial associated impairments may provide a preferential strategy, due to the involvement of mitochondria in many processes implicated in neurodegeneration: oxidative stress (via reactive oxygen species (ROS) generation) [3], apoptosis [4], iron metabolism [5], energy imbalance [6], and interaction with amyloid-β (Aβ) and phosphorylated tau protein [7–10]. However, since it affects many pathways, targeting mitochondrial dysfunction is not a trivial goal. First, learned from the limited success of antioxidant therapies in AD [11], and also from the dose-dependent toxicity of the mitochondria-targeted ubiquinone in neural stem cells [12]. Moreover, targeting one of the mitochondrial deficits, e.g., by enhancing energy production, may be ineffective, since the deficit may be due to the mitochondrial dynamics or iron metabolism. Other complexities involved in targeting mitochondria are the paradoxical effects of the mitochondrial function, particularly since mitochondria are the major producers of ROS (modulators of cellular function but also of cell death) while at the same time targets of ROS toxicity [13]; and also the opposite interaction of amyloid with the voltage-dependent anion channel-1 (enhancing the conductance while also blocking the channel pore) [7]. These make the mitochondrial therapy difficult to precisely regulate. To overcome these limitations, we suggest as an AD therapy to transfer active mitochondria as intact cell organelles, thereby allowing a broad range of activities with accurate and physiological regulation. Mitochondrial transfer, a process by which isolated mitochondria can be incorporated in vitro into cells by simple incubation, has been reported in 1982 by Clark (“mitochondrial transformation”) [14]. The functionality of the transformed mitochondria inside the recipient cells was demonstrated by increased ATP production, oxygen consumption, and proliferation in cell [14–18]. Moreover, injections, mostly local but also systemic, of isolated mitochondria in experimental animal models were reported: mitigating ischemia injury in the liver [19, 20], heart [21–23], and brain [24], symptoms of acute respiratory distress syndrome [25] and spinal injury [26], amelioration of schizophrenia symptoms [27] and Parkinson’s disease related motor deficits in rodents [28, 29], using autogenic but also xenogenic sources (reviewed in [30, 31]).
We recently reported mechanistic aspects of transfer of intact viable mitochondria into cells, indicating that isolated human mitochondria enter many cell types, and are beneficial in mitochondria-defective cells. Heparan sulfated polysaccharide molecules, as well as the outer membrane and its proteins integrity are crucial for this process. Transmission electron microscopy analysis revealed that the transferred mitochondria interact directly with cells, in macropinocytosis or macropinocytosis-like mechanisms, in line with our pharmacological inhibition tests [32].
So far, using isolated mitochondria to treat cognitive diseases has not been reported. We report here that AD-mice treated IV with human isolated mitochondria showed a significant amelioration of cognitive deficits, neuronal loss, and gliosis, accompanied by an increase in mitochondrial activity in the brain, and in the liver.
METHODS
Mitochondria isolation
HeLa DsRed2-mito cells [HeLa cells previously transfected by us with the plasmid DsRed2-Mito (encodes fusion of Discosoma sp. red fluorescent protein and the mitochondrial targeting sequence, MTS, from subunit-VIII of human cytochrome c oxidase, which targets the protein to the mitochondria)] were collected by trypsinization, suspended in PBS, and centrifuged (5 min, 250 g) twice. Mitochondrial isolation procedures were performed at 4°C or on ice and in the dark (to prevent fluorescence decay). The centrifuged cells were re-suspended in mitochondrial isolation buffer (320 mM sucrose, 5 mM Tris-HCl, pH 7.4, 2 mM EGTA), and homogenized with a Dounce homogenizer. Nuclei and cell debris were removed by two centrifugations (5 min, 3000 g) and the supernatant was collected. The supernatant was then centrifuged (10 min, 12,000 g) and the mitochondrial pellet was re-suspended in mitochondrial isolation buffer. Mitochondrial concentration was determined by Bradford-assay. All the experiments were performed with freshly isolated mitochondria. We have shown that this isolation protocol keeps the mitochondria intact and viable, inducing beneficial effects in mitochondria-defective-cells [32].
Induction of AD-model (Aβ-ICV)
Ten-week-old male C57BL/6 mice were used (in experiment #1 we used C57BL/6 mice originated from Jackson’s Lab; in experiments #2 and #3 we used mice purchased from Envigo. Israel; C57BL/6JRccHsd). Mice were IP anaesthetized with ketamine 3 mg/kg and cepetore 0.03 mg/kg. Stereotaxic coordinates used for the intracerebroventricular (ICV) injection in mm from the bregma: –0.2 mm anteroposterior,±1 mm mediolateral, dorsoventral –2.3 mm. Small holes were drilled, and an injector (27 G, 35 mm) was guided successively into the lateral ventricles. Amyloid-beta (Aβ1–42 amyloid peptide (Sigma Aldrich A9810) (82 pmol/μl, dissolved in sterile PBS) (AD-mice) or PBS only [in the sham-mice (non-AD-mice)] were injected into the lateral ventricles, using a 10 μl Hamilton microsyringe mounted on an automated pump (Razel-Scientific-Instruments), at the rate of 1 μl/min, receiving a total of 5 μl. Injectors were let in place for 5 min to avoid flow back of solution (modified from [33]). Animal experimentation was approved by the Institutional Ethics Committee.
Mitochondrial transfer treatment
Two days after the induction of the AD-model (Day 0 in Fig. 1), the mice received a single IV injection of the isolated mitochondria [200 μg/mouse, containing 50–55 mU (milliunits = nmol/min) CS and 46–51 mU COX equivalents, respectively] or of vehicle only (mitochondrial buffer) into the tail vein. Non-AD-mice were IV injected with mitochondrial buffer only (n = 5/group, experiment #1). This experiment was followed by a separate and independent similar experiment (yet with some differences in the behavioral studies) [non-AD (n = 4), AD (n = 5), AD-treated (n = 8); experiment #2]. In experiments #1–2, behavioral studies were performed, followed by scarification on day 15 following mitochondrial injection. In experiment #3, animals were sacrificed on day 5 following mitochondrial injection (n = 4/group) (Fig. 1).
Study design.
In order to test whether the IV tail vein injected mitochondria arrive at the brain and/or at the liver, an organ which has a cross talk with the brain, AD-mice, non-AD mice, and naïve (non ICV operated) mice were IV injected with mitochondria or buffer.
To test the safety of the mitochondria transfer, 10-week-old C57BL male mice (n = 3) received an IV injection of the isolated mitochondria (200 μg/mouse). A clinical follow-up of 8 h was performed. In addition, 10-week-old mice (n = 4) received 2 IV injections of the mitochondria (200 μg/mouse) or vehicle only per week for 4 weeks. A clinical follow-up for the general health was conducted. Subsequently, the animals were sacrificed, and the internal organs were macroscopically tested.
To test whether mitochondrial transfer may cause an inflammatory/immune response, C57BL male mice (n = 7–9/group) received one IV injection of mitochondria (200 μg/mouse or vehicle only) and were tested for the presence of the inflammatory marker tumor necrosis factor alpha (TNF-α) (ELISA KIT, R&D Systems, Inc, Minneapolis, MN) in serum 10 days following treatment (a time point which was reported for testing allo-response to IP mitochondrial injection [34]). To test whether the inflammatory marker is present in the serum of the treated mice at an earlier stage, we IV injected mitochondria to additional C57BL male mice (n = 8/group), and collected serum on day 4 following injection of mitochondria (or vehicle only).
Behavioral studies
At 5–13 days following the IV mitochondrial treatment, cognitive tests were performed (study design presented in Fig. 1).
Fear conditioning
Fear conditioning aims to evaluate fear memory: contextual (hippocampus mediated) and cued (amygdale mediated). The test device (Panlab) consists of two compartments and has a grid floor capable of generating low electrical current for a given time period. The animal was put inside the device and given a 2 s, 0.5 mA shock coupled to a tone for two repeats. After 24 h, the freezing time of the animal after exposure to either the acoustic tone (cued fear conditioning) or the visual context (context fear conditioning) was measured (Packwin) out of a total 5-min exposure [35].
Y-maze
This test evaluates short-term memory. The Y-maze is a three-arm maze with all arms at equal angles, 30 cm in length and 5 cm in width with walls 12 cm high. Mice were initially placed in the middle, and the sequence of arm entries were recorded manually for each mouse over an 8-min period. The triads with all three arms represented (i.e., ABC, CAB, or BCA but not ABB) – were considered ‘correct triads’ [36].
Radial arm water maze (RAWM)
This maze evaluates working memory, spatial learning, and cognitive ability [37, 38]. The apparatus consists of 6 arms, 30 cm in length, converging in a central 40x40 cm pool, filled with water to cover a plexiglass platform. The platform was located at the end of one arm. During each trial, the mouse was released from the end of a pseudo randomly selected arm (counterbalanced across trials) and was required to navigate to a submerged platform at the end of a goal arm in 60 s. For each animal, the platform location remained consistent throughout the experiment. Mice were run for 30 trials in total (15 on each day) and the average of every three trails was determined as a ‘block’ (for example, block 5 is the average of trails 13–15 of day 1). On day 1, the animal was trained to locate the platform (13 trials alternating between hidden and visible platforms and the 3 last trails with hidden platforms). On day 2, all 15 trials were with the platform hidden. The last 3 trials of each day, i.e., block 5 & 10, were considered “test trials”. In addition, block 6 was calculated for long term memory analysis, as the mice had a 24 h break before this block [39]. Entry into the wrong arm was designated as an error. Number of errors for each animal was tracked and scored. Animals were automatically tracked using EthoVision XT (Noldus).
Open-field habituation
This test evaluates long term non-associative, non-aversive spatial learning [40] by measuring the decrease in the exploratory activity of the animal in a test session carried out 24 h after the first exploration session (delta of 2nd session –1st session). Animals were exposed to a novel environment by placing them in a 40 cm × 50 cm × 60 cm open field box. The distance walked was measured for a 5-min period (EthoVision). Twenty-four hours later animals were re-exposed to the same environment and the distance they walked was recorded. Bigger delta between days (shorter distance on the test session compared to the first session) represented intact learning.
Histopathological studies
After finalizing the behavioral tests (about 2 weeks after mitochondrial injection) mice were anesthetized with a lethal dose of pentobarbital and perfused via the ascending aorta with ice-cold PBS, followed by cold 4% paraformaldehyde in PBS. The brains were deep frozen in dry ice and serial 10 μM sections were made. Histochemical analysis of the gross arrangement of the tissue was performed [41], using the H&E staining (briefly, 5 min hematoxylin, tap water, 1 min eosin Y, tap water, and hydromount). Immunofluorescent staining to determine the degree of neuronal loss was performed using the antibody against the neuronal marker (anti-NeuN, Millipore Biotest clone A60; 1 : 500), the microglia rabbit anti-Iba1 (Wako-Pure-Chemical Industries, Ltd., VA, USA; 1 : 250) and astrocytes Rabbit anti-GFAP (Dako, Denmark; 1 : 250). Goat anti-mouse Cy3 (Jackson Immuno-Research, West Grove, PA, USA), goat anti-rabbit Alexa Fluor 488 and goat anti-mouse Alexa Fluor 488 (Invitrogen, Carmarillo, CA, USA) were used as secondary Abs, respectively. Briefly, tissue sections were fixed in 4% formaldehyde (10 min, room temperature), washed in Optimax (BioGenex Laboratories, San Ramon, CA, USA) and blocked in Cas Block (Invitrogen, Carmarillo, CA, USA) (10 min, room temperature). The tissues were then labeled with the appropriate primary antibody (overnight at 4°C or 1 h at room temperature) and detected with the appropriate secondary antibody (30 min, room temperature). The nuclei were stained with DAPI. The sections were imaged by confocal microscopy (X20, X40, X60, Olympus BX63). For NeuN analysis, cells were counted manually in a blinder manner (similarly to the way used by others [42]) in the DG and CA1 zones in the hippocampus (4 sections per animal). For IbaI and GFAP analysis, signal intensity was integrated to measure fluorescence signal density strength. Quantification was analyzed by Image Pro Research software.
To test the distribution of injected DsRed2-mitochondria, AD-mice and non-AD-mice were sacrificed 2 h after IV mitochondria or buffer injection. In addition, 50 min following IV treatment with mitochondria or buffer, AD-mice and naïve mice (n = 2/group) were sacrificed. Brains and livers were collected, fixed in cold 4% paraformaldehyde, deep-frozen in dry ice and then cut into serial 10 μM sections. Histological analysis was performed using the anti-RFP (Red fluorescent protein) antibody (MBL Co Ltd). Briefly, tissue sections were fixed in 4% formaldehyde (10 min, room temperature), washed in PBS, permeabilized in 3% Triton/PBS, quenched with 3% H2O2/DDW and blocked in Cas Block with 3% Triton (Invitrogen, Carmarillo, CA, USA) (10 min, room temperature). The tissues were then labeled with anti-RFP (MBL 1 : 100) overnight at 4°C or 1 h at room temperature and detected by anti-HRP (1 : 1000, 1 h, room temperature) and DAB Qanto kit (Thermo scientific). The nuclei were stained with hematoxylin.
Imaging analysis
Brains and livers were put to an imaging analysis to detect the DsRed signal using the In Vitro Imaging System (IVIS Lumina LT Series III, Perkin Elmer). Excitation was at 535 nm, and Emission- DsRed filter: 575–650 nm.
Enzymatic assays
Enzymatic assays were performed in homogenates of pooled hippocampal and cortex samples of mice sacrificed on day 15 following mitochondrial transfer, and of hippocampal and cortex as well as liver samples of mice sacrificed on day 5 following mitochondrial transfer. All samples were prepared in mitochondrial isolation buffer (see above). Duplicates of pooled samples were performed. Citrate synthase (CS) activity was measured in the presence of acetyl-CoA and oxaloacetate by monitoring the liberation of CoASH coupled to 5’,5’-dithiobis (2-nitrobenzoic) acid at 412 nm. Cytochrome c oxidase (COX) activity was measured by monitoring the oxidation of reduced cytochrome c at 550 nm [43]. Results were expressed as nmol/min/mg protein (milliunits = nmol/min). Measurements were performed in a double beam spectrophotometer UVKON XS (Secomam France).
For testing the functionality of the mitocondria preparation in mouse serum, we used a similar protocol to that used by Shi et al. [29]. Serum aliquots (400 μL) collected from healthy mice were incubated with mitochondria ( 200 μg) at 37°C for 0, 15, 30 min, and 1 h. Then samples were centrifuged at 3000 g for 10 min at 4°C. The mitochondria were collected for measuring the mitochondrial enzymatic activity.
Statistics
Behavioral and histological data were analyzed using the one way ANOVA, further analyzed using post-hoc analysis, or using the planned contrast, and, when mentioned the unpaired t-test. The comparison of qualitative variables between two groups was carried out using the Chi-square test. Statistical analysis was performed using IBM SPSS Statistics V.23.
RESULTS
Mitochondrial transfer ameliorated cognitive deficits in the AD-mice
Experiment #1
AD-mice treated with one single IV injection of mitochondria two days after induction of the AD-model were tested for their cognitive performance in comparison to that of the untreated (buffer only) AD-mice and non-AD-mice. A trend of better performance of the mitochondria-treated AD-mice relative to the untreated AD-mice, reaching the performance of the non-AD-mice in the fear conditioning test (higher percentage of freezing), was noticed already 5–6 days after the IV mitochondria injection: while a significant difference between AD-mice and the non-AD-mice was noticed (t-test, p = 0.02), mitochondria-treated AD-mice showed a similar behavior to that of non-AD-mice, with a trend toward better performance relative to untreated AD-mice (p = 0.09) (Fig. 2A). In tone conditioning, which is dependent on amygdala rather than the hippocampus, no difference was found between the groups, as expected in this model (data not shown). Improvement of cognitive performance was further evident during the next days, as follows: testing the spatial working memory in the Y-maze apparatus 8 days after mitochondrial IV injection revealed that mitochondria-treated AD-mice performed significantly better (lower correct triplets ratio) than untreated-AD-mice, reaching the performance of non-AD-mice (one-way ANOVA between the tested groups [f(2,13)=5.01, p = 0.02]; in Bonferroni post-hoc analysis: mitochondria-treated versus untreated AD-mice (p = 0.02)) (Fig. 2B). Also using the RAWM, to assess the spatial learning and memory deficits 12–13 days following the IV mitochondria injection, revealed that the mitochondria-treated AD-mice performed significantly better (less errors) than the untreated AD-mice, reaching the performance of the non-AD-mice, as presented in block 10, which is the last block of day 2 and the final block of the test (one-way ANOVA [f (2, 12)=4.82, p = 0.02]; Tukey post-hoc analysis showed a significant difference between mitochondria-treated versus untreated AD-mice (p = 0.03), with more errors by untreated AD-mice than non-AD mice (trend, p = 0.06)) (Fig. 2CI). Similar results were noticed in block 6, the first block of day 2, which may represent long-term memory (one-way ANOVA between the tested groups [f (2, 12)=5.7, p = 0.01]; Tukey Post-Hoc analysis showed that mitochondria-treated mice had less errors than untreated AD-mice (p = 0.02), and untreated AD-mice had more errors than non-AD mice (p = 0.03)) (Fig. 2CII). The same was noted in block 5, the last block of day 1 (one-way ANOVA [f (2, 12) 8.86, p = 0.004]; Tukey post-hoc analysis showed that mitochondria-treated had less errors than untreated AD-mice (p = 0.003)) (data not shown).

Mitochondria-treated AD-mice performed better in cognitive tests than untreated mice, reaching the performance of non-AD-mice (experiment #1). A) Contextual fear conditioning test (5 days following mitochondrial injection). Significantly worse performance (less freezing behavior) of AD-mice than non-AD-mice (t-test, p = 0.02) was observed, with mitochondria treated AD-mice showing similar behavior to non-AD-mice (high freezing behavior), and a trend toward better performance compared to untreated AD-mice (p = 0.09). B) Y-maze test (8 days following mitochondrial injection). One-way ANOVA test revealed a significant difference among the tested groups [f(2,13)=5.01, p = 0.02]. AD-mice treated with mitochondria performed better (higher ratio) than untreated AD-mice (p = 0.02), approaching the performance of non-AD-mice, with no statistical difference between treated AD-mice and non-AD-mice. AD-mice performed worse than non-AD mice (t-test, p = 0.03). C). RAWM test (12–13 days following mitochondrial injection). I) At block 10 (last block of day 1): one-way ANOVA yielded a significant difference between the groups [f (2, 12)=4.82, p = 0.02]. Tukey Post-Hoc revealed that AD-mice treated with mitochondria performed better (less errors) than untreated AD-mice (p = 0.03). There was no difference between non-AD-and treated AD-mice, while AD-mice performed worse than non-AD mice [more errors, trend p = 0.06]. II) At block 6 (first block of day 2): one-way ANOVA between the tested groups [f (2, 12)=5.7, p = 0.01]; Tukey Post-Hoc analysis: mitochondria-treated mice had less errors than untreated AD-mice (p = 0.02), and untreated AD-mice had more errors than non-AD mice (p = 0.03).
Experiment #2
This experiment used the same AD-model and IV mitochondria injection as experiment #1. Yet, to further investigate efficacy at the early stage (5 days after treatment), which has shown a trend of benefit in fear conditioning in experiment #1, we tested in experiment #2 whether the improvement in performance in Y-maze (which showed efficacy on day 8 in experiment #1) will be evident already on day 5. We also added another test for analyzing the efficacy of the mitochondrial treatment, the open-field habituation test, (used to evaluate long term non-associative, spatial and non-aversive learning) on days 12–13, for further confirmation of the beneficial effect that was detected in experiment #1 at this stage in the RAWM.
Using the Y-maze, we noted that, while AD-mice performed significantly worse than non-AD-mice, AD-mice treated with mitochondria performed better than untreated AD-mice, approaching the performance of non-AD-mice (one-way ANOVA: [f(2,14)=7.55, p < 0.001)]; Tukey post hoc analysis: AD-mice versus non-AD-mice (p < 0.001), mitochondria-treated versus untreated AD-mice (p = 0.003); with no statistical difference between treated AD-mice and non-AD-mice) (Fig. 3A).

Mitochondria-treated AD-mice showed better cognitive performance in the cognitive tests compared to untreated mice, approaching the performance of non-AD-mice (experiment #2). A) Y-maze test (8 days following mitochondrial injection). One-way ANOVA showed a significant difference between the groups [f(2,14)=7.55, p < 0.001]. Tukey Post-Hoc analysis showed a significantly worse performance of AD-mice relative to the non-AD-group (p < 0.001), and a significantly better performance of treated AD-mice relative to untreated AD-mice (p = 0.03). B) Open field habituation test. Mitochondria-treated AD-mice showed a better cognitive performance (bigger delta of distance between days) than untreated AD-mice (t-test, p = 0.03), reaching the performance of non-AD-mice. AD-mice performed worse than non-AD-mice (t-test, p = 0.05), There was no difference between non-AD- and treated AD-mice.
Open-field habituation revealed that 12–13 days after IV mitochondria injection, mitochondria-treated AD-mice showed a better cognitive performance (bigger delta of distance between days) than untreated AD-mice (t-test, p = 0.03), reaching the performance of non-AD-mice. AD-mice performed worse than non-AD-mice (t-test, p = 0.05) (Fig. 3B).
These results show that following one single IV injection of isolated human mitochondria, cognitive deficits were significantly ameliorated in AD-mice, starting already 5 days after treatment initiation and lasting at least until day 12–13.
Mitochondrial transfer reduced neuronal disarrangement, neuronal loss and gliosis in the hippocampus of AD-mice
Using H&E staining, we noted in AD-mice smaller pyramidal cells in the CA3 zone, with a distorted arrangement (as characteristic to this model) compared to the compactly arranged and organized cells in non-AD-mice [4 of 5 mice (80%), and only 1 of 5 mice (20%), showed normal appearance in the non-AD-mice and AD-mice, respectively, χ2 test, p = 0.0007]. AD-mice that were treated with the mitochondria showed a higher percentage of mice with normal appearance [3 out of 5 mice (60%)], relative to untreated AD-mice (trend, p = 0.06), approaching the percentage in non-AD mice (Fig. 4).

Mitochondria-treated AD-mice showed reduced neuronal disarrangement compared to untreated mice, approaching that of normal-mice. Using the H&E staining, a significantly lower percentage of AD-mice, relative to non-AD-mice, showed a compactly arranged and organized appearance of the pyramidal cells in the CA3 zone of the hippocampus, while most AD-animals showed rather small, with a distorted arrangement cells (χ2 test, p = 0.0007). The cells of mitochondria-treated AD-mice showed a higher percentage of mice with arranged and organized cells relative to untreated AD-mice (trend, p = 0.06), and no statistical difference with the non-AD-group.
NeuN staining for neurons in the hippocampus revealed that, while there was a significantly lower count (higher neuronal loss) in AD-mice than in non-AD-mice, the mitochondria-treated AD-mice had a significantly higher neuronal count than untreated AD-mice, reaching that of non-AD-mice, as follows: in the dentate gyrus (DG): 65.11±5.15, 94.22±7.39 and 101.9±5.84 cells in the AD-mice, non-AD-mice, and mitochondria treated AD-mice, respectively (one way ANOVA among the tested groups [f(2,24)=9.81, p = 0.001]; Tukey post hoc analysis: AD-mice versus non-AD-mice (p = 0.008), mitochondria-treated versus untreated (vehicle only) AD-mice (p = 0.001), with no statistical difference between the treated AD-mice and the non-AD-mice) (Fig. 5A). Similar differences were detected also in the CA1 : 33.67±2.47, 47.01±3.67 and 45.1±1.67 cells in the AD-mice, non-AD-mice, and mitochondria-treated AD-mice, respectively (one way ANOVA between the tested groups [f(2,24)=6.26, p = 0.006]; Bonferroni post hoc analysis: AD-mice versus non-AD-mice (both receiving vehicle only, p = 0.008), mitochondria-treated versus untreated (vehicle only) AD-mice (p = 0.03); with no statistical difference between treated AD-mice and non-AD-mice) (Fig. 5B).
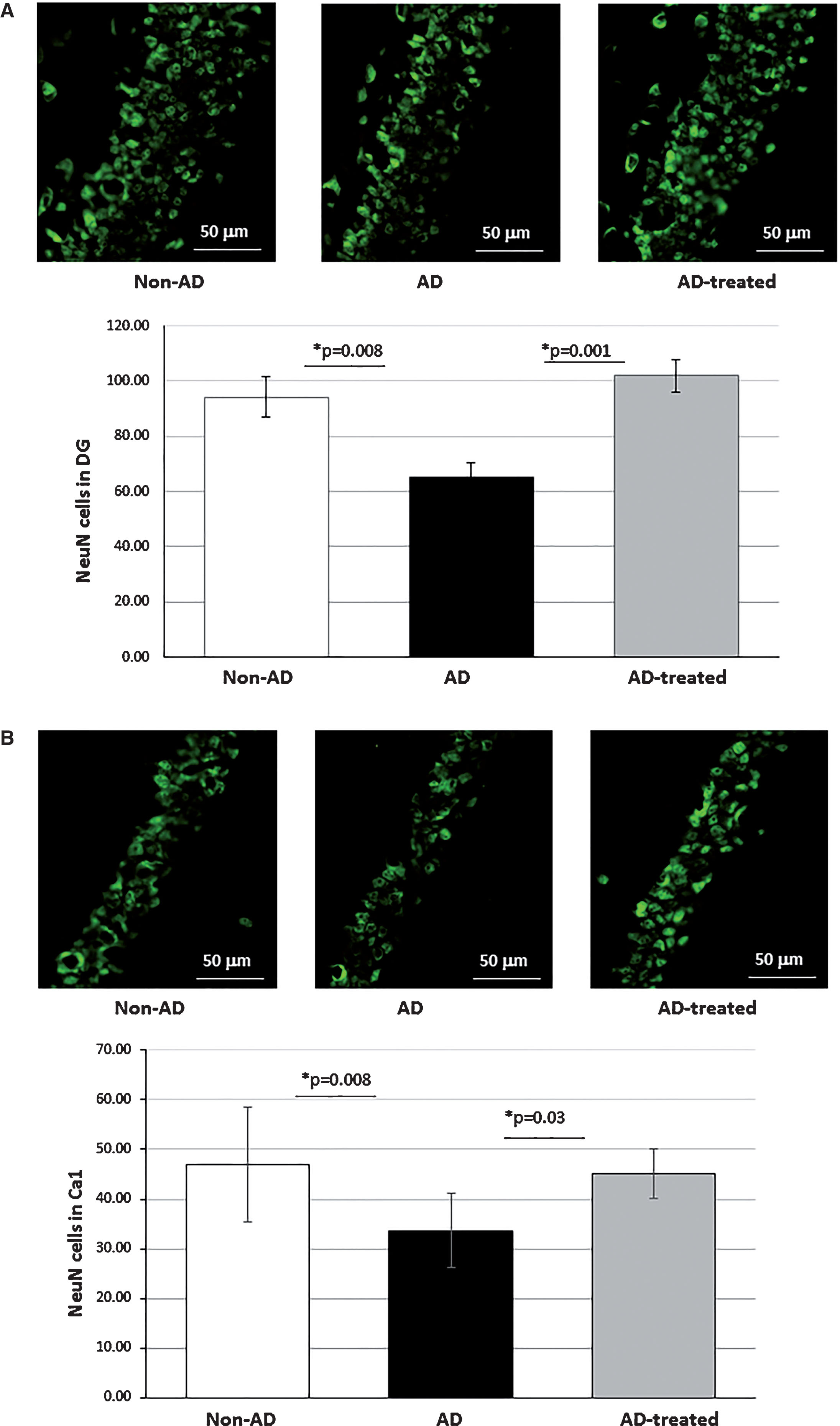
Mitochondria-treated AD-mice showed reduced neuronal loss in the hippocampus compared to untreated mice, approaching the neuronal count of non-AD-mice. A) NeuN staining for neurons in the DG: one way ANOVA among the tested groups ([f(2,24)=9.81, p = 0.001]; Tukey post hoc analysis: AD-mice versus non-AD-mice (p = 0.008), mitochondria-treated versus untreated (vehicle only) AD-mice (p = 0.001); no statistical difference between treated AD-mice and non-AD-mice. B) NeuN staining in the CA1: one way ANOVA between the tested groups [f(2,24)=6.26, p = 0.006]; Bonferroni post hoc analysis: AD-mice versus non-AD-mice (both receiving vehicle only, p = 0.008), mitochondria-treated versus untreated (vehicle only) AD-mice (p = 0.03); with no statistical difference between treated AD-mice and non-AD-mice.
A decrease in gliosis following mitochondria injection was noted using the Iba1 staining for microglia and the GFAP staining for astrocytes. In the DG, a significant lower staining with IbaI in the treated AD-mice relative to the untreated AD-mice was noticed, as follows: 13.8±1.54 and 21.7±2.43 cells, respectively, while non-AD-mice showed 17.56±1.87 cells (one way ANOVA among the tested groups ([f(2,25)=3.9, p = 0.03]; Tukey post hoc analysis revealed that mitochondria-treated AD-mice showed a lower microglial burden than untreated AD-mice (p = 0.02); with no statistical difference between the treated AD-mice and the non-AD-mice) (Fig. 6A). GFAP astrocyte staining in the CA1 showed that, while significantly higher in AD-mice than in non-AD-mice, AD-mice treated with mitochondria showed lower astrocytic density than untreated AD-mice, reaching the level of non-AD-mice, as follows: 88,146.89±7882.58, 64,471.56±8353.63, and 61,324.5±6626.02 GFAP area density in the AD-mice, non-AD-mice, and mitochondria-treated AD-mice, respectively (planned contrast analysis [f(2,26)=3.62, p = 0.04)]: AD-mice versus non-AD-mice (p = 0.03), mitochondria-treated versus untreated AD-mice (p = 0.02)) (Fig. 6B). Also in the DG, mitochondria-treated AD-mice had lower area density of GFAP than untreated AD-mice: 85,370.5±5,602.88 versus 115,937.8±10,229 area density, respectively. Non-AD-mice showed 108,843 cells (planned contrast revealed a trend toward a difference among the tested groups ([f(2,24)=3.05, p = 0.06]; with mitochondria-treated versus untreated AD-mice had a lower astrocytic density (p = 0.02)).
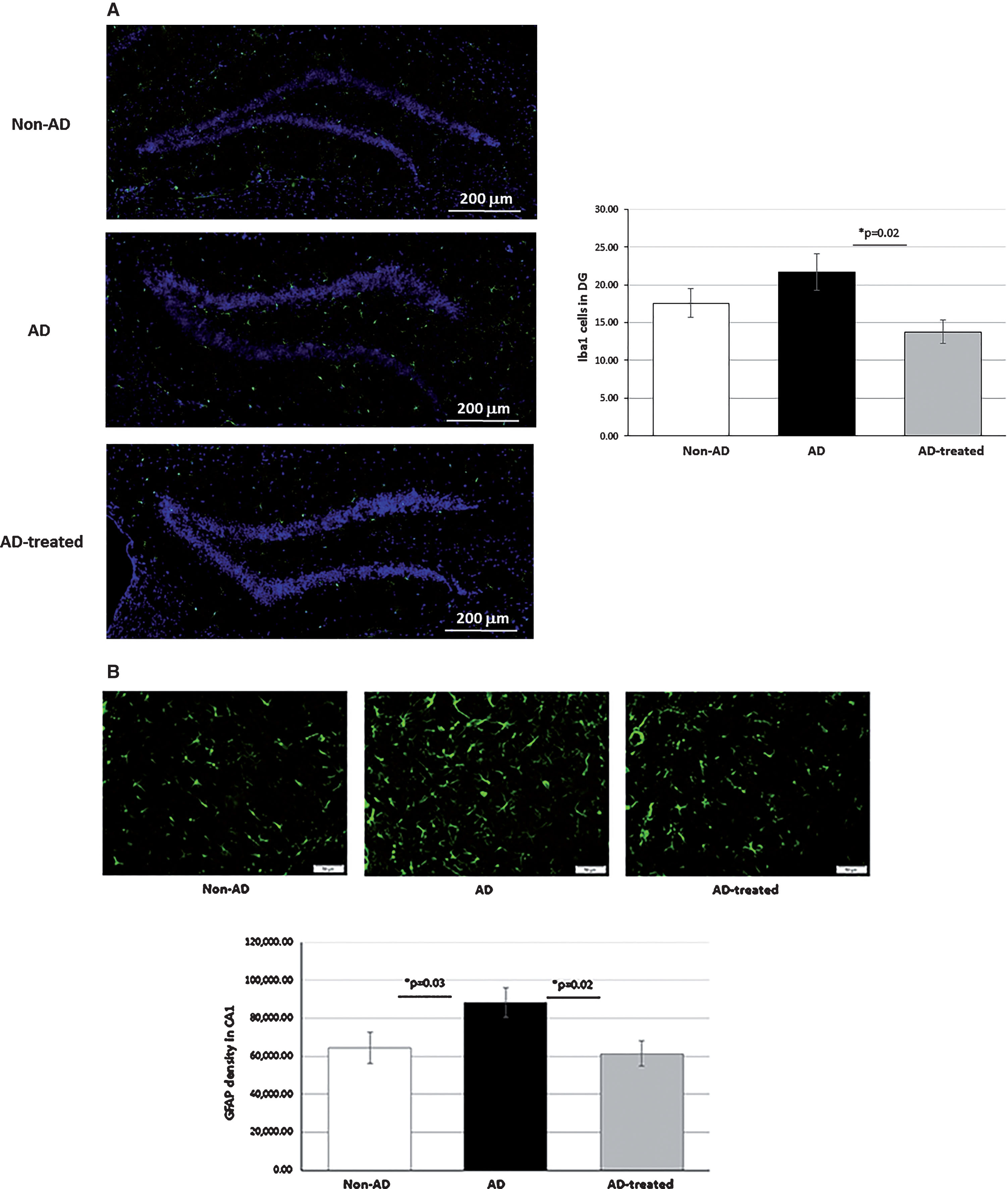
Mitochondria-treated AD-mice showed reduced gliosis in the hippocampus compared to untreated mice, approaching that of non-AD-mice. A) Iba1 staining for microglia in the DG: one-way ANOVA test revealed a significant difference among the tested groups ([f(2,25)=3.9, p = 0.03]. Tukey post hoc analysis revealed that AD-mice treated with mitochondria showed less microgliosis than untreated AD-mice (p = 0.02). There was no statistical difference between treated AD-mice and non-AD mice. B) GFAP staining for astrocytes in the CA1: one-way ANOVA test revealed a significant difference among the tested groups ([f(2,26)=3.62, p = 0.04]; planned contrast analysis: AD-mice showed more astrogliosis than non-AD-mice (p = 0.03), mitochondria-treated showed less astrogliosis than untreated AD-mice (p = 0.02), with no statistical difference between treated AD-mice and non-AD-mice.
Mitochondria transfer increased mitochondrial activity in the brains of AD-mice
To test the mitochondrial functionality, we measured the activities of two mitochondrial enzymes involved in energy production: CS, an ubiquitous mitochondrial matrix Krebs cycle enzyme, and COX, the 4th complex of the electron transfer chain. As presented in Table 1, testing the mitochondrial functionality on day 15 following mitochondrial transfer (after completing the behavioral studies) revealed a partial decrease in activity of CS and COX in the hippocampus of AD-mice relative to non-AD-mice [75.5% COX activity (a decrease of 24.6%) and 89.2% CS activity (a decrease of 10.81%) of non-AD-mice], indicative of mitochondria dysfunction in this AD-model. Analysis of these parameters in the mitochondria-treated AD-mice revealed a correction of mitochondrial dysfunction in AD-mice (an increase of 61.1% in COX activity and 41.5% in CS activity, relative to untreated AD-mice), reaching the levels, or slightly above, of those of non-AD-mice (an increase to 121.6% and 125.76% in COX- and CS-activity, respectively, relative to non-AD-mice). Similar trends were detected in the cortex, particularly an increase (of 29.5%), in COX activity in mitochondria-treated AD-mice relative to untreated AD-mice, showing a 121.6% activity relative to non-AD-mice. When testing the mitochondrial enzymatic activity in the brain of mice at a stage closer to the mitochondrial transfer, on day 5 following the transfer (experiment #3), some similar trend for effect on COX activity was detected, yet smaller and not reaching a statistical significance: 451.80±29.70 mU (considered as 100 %) in non-AD mice, 426.66±29.70 mU in untreated AD-mice [94.44 %, (decrease of 5.56% relative to non-AD mice)], and 452.18±35.21 mU in mitochondria treated AD-mice (increase of 5.98% relative to untreated AD-mice, reaching the activity of non-AD mice). Some small increase (6.96%) in CS activity in mitochondria treated AD-mice relative to untreated AD-mice was also noticed: 493.58±43.99 mU versus 461.47 29.50 mU, respectively (with 464.52±6.64 mU CS activity in non-AD mice). Such differences in enzymatic activities were not noticed in the cortex among groups (data not shown). This may point to some beneficial effect, yet limited, starting already as early as 5 days following mitochondrial transfer, progressing into a more pronounced effect on day 15 following transfer.
Enzymatic activities
*Tissue pool (n = 4); ** nmol/min/mg protein.
IV injected mitochondria are detected in the liver
To test if the mitochondria injected IV into the tail vein reach the brain, and if they are detected in the liver, the peripheral organ that has a substantial cross talk with the brain, these two organs were stained with anti-RFP Ab to detect the injected Dsred2-labelled mitochondria. As shown in Figure 7A, a strong signal of DesRed staining was detected in the liver of mitochondria-injected AD-mice 2 h following injection, but not in the buffer-injected mice, indicating the arrival of the injected mitochondria to the liver and their presence there. DsRed staining was also detected in non-AD-mice 2 h after injection and in AD-mice and naïve mice 50 min after mitochondria injection (data not shown). No DsRed was detected in the brain of all the mitochondria- or buffer-injected mice.
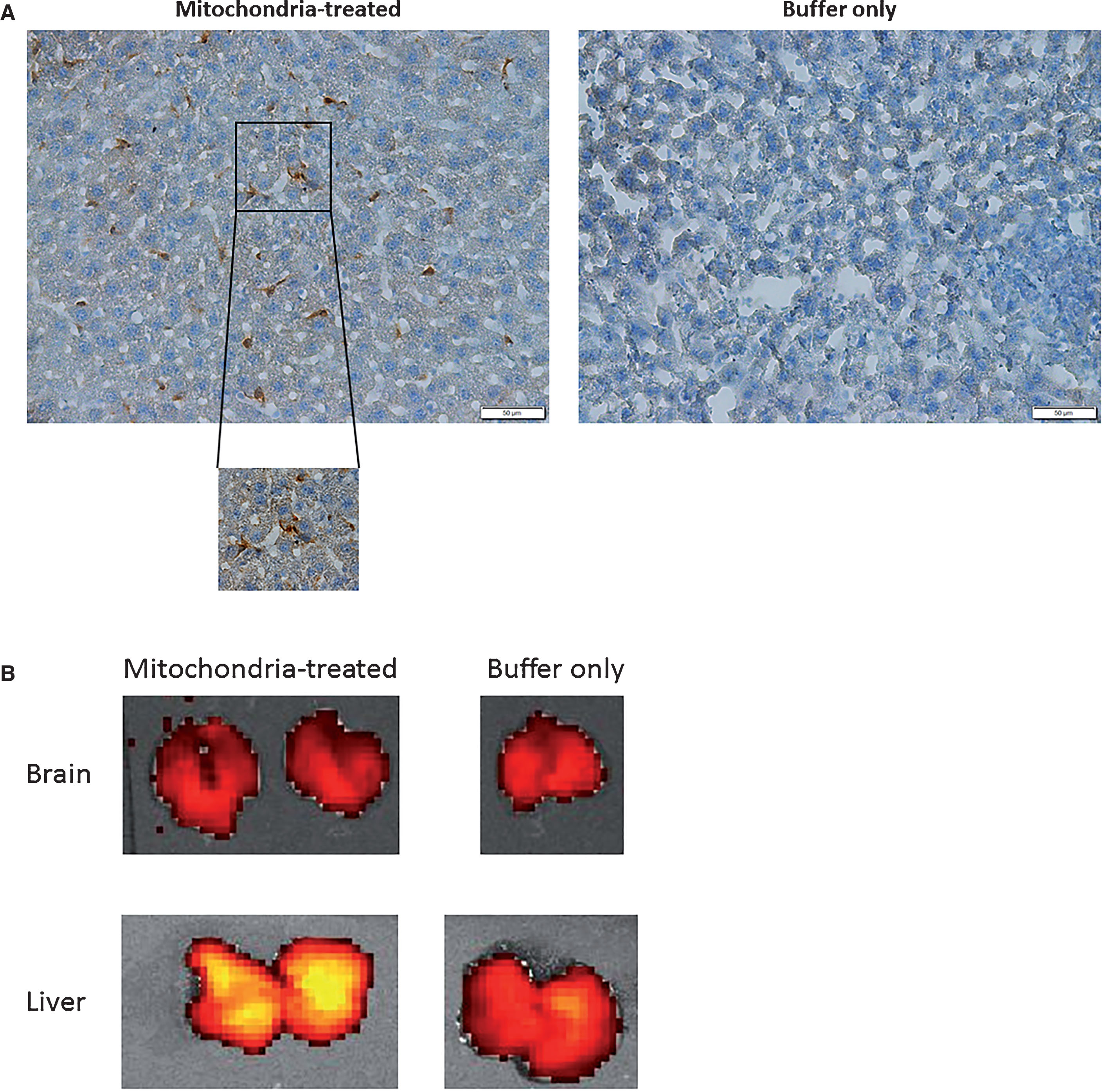
IV injected mitochondria are detected in the liver. A) Immunohistochemical analysis. DsRed2 staining of labelled mitochondria in the liver 2 h after the IV tail vein injection of mitochondria revealed that mitochondria are present in the liver of the mitochondria injected AD-mice but not in the liver of untreated (buffer-only) mice (AD). B) IVIS analysis. No signal in the brains of AD-mice 2 h following injection of mitochondria relative to buffer injected. For comparison, a strong signal of DsRed is demonstrated in the liver of AD-mouse following mitochondrial injection (no signal was detected in the liver of a buffer injected naïve mouse).
In addition, we also followed the injected Dsred2-labelled mitochondria using the IVIS for the detection of the DsRed signal 2 h following IV injection of mice. No signal in the brain of AD-mice injected with mitochondria relative to vehicle injected AD-mice was noticed, while a strong signal of DsRed was detected in the liver following mitochondrial injection (Fig. 7B). No DsRed signal was also detected in the brain of non-AD mice following IV mitochondrial injection (data not shown).
Mitochondria transfer increased mitochondrial activity in the livers of AD-mice
To test whether the beneficial effect of the mitochondrial transfer has a beneficial effect on the liver, we tested the mitochondrial enzymatic activity in mitochondria treated AD-mice, untreated AD-mice, and also in the non-AD mice, on day 5 following mitochondrial transfer. Interestingly, we detected that AD-mice have reduced COX and CS activity in the liver relative to non-AD mice [COX: 440.00±28.53 mU versus 588.57±18.34 mU, respectively (p = 0.009), decrease of 33.76%. CS: 88.97±5.18 mU versus 108.01±3.99 mU, respectively (p = 0.045), decrease of 12.15% ], suggesting a mitochondrial dysfunction in the AD-mice. Importantly, we found that the mitochondria treated AD-mice showed an increase in liver mitochondrial activity relative to the untreated AD-mice [COX: 569.52±31.29 mU versus 440.00±28.53 mU, respectively (p = 0.038), increase of 32.75%. CS: 96.62±5.14 mU versus 88.97±5.18 mU, respectively, increase of 7.92% ], approaching the mitochondrial activities of non-AD mice. This points that the IV injected mitochondria not only enter the liver but also improves the mitochondrial activity, correcting the mitochondrial enzymatic deficit. This may support the hypothesis that the beneficial effect of mitochondrial transfer may, at least in part, be mediated by the liver mitochondrial response, with possible some cross talk between the liver and brain. What may further support this possibility is the finding that while a robust increase in mitochondrial function was noticed in the liver already on day 5 following the transfer, at this stage the increase in the brain was only limited (see above), if at all, while was pronounced on day 15 (see above). This may point to a possible downstream effect of the brain to the liver response.
Administration of isolated mitochondria to the mice was safe and did not cause an immune response
The eight-hour clinical follow-up of the mice receiving one IV injection of isolated mitochondria revealed a normal behavior of the mice. In a separate experiment, in which naive mice received two weekly IV injections of mitochondria (200 μg/mouse) or vehicle only for 4 weeks, clinical follow-up for general health showed normal behavior, without differences between mitochondria-treated or vehicle only mice. Following sacrifice, no macroscopic differences in the internal organs were detected: spleen size was similar in both groups, liver and lungs were intact, and no hemorrhages were observed in the brains. These results indicate that IV administration of DsRed2-mitochondria isolated from Hela cells is safe when given as a single dose or as a repeated treatment.
When testing the level of the inflammatory marker TNF-α in the serum of injected mice on day 10 (a time point which was reported for testing allo-response to IP mitochondrial injection [35]), no increase was detected in the mitochondria injected mice relative to buffer injected mice (2.53±0.25 versus 2.98±0.44 pg/ml, respectively). To test whether the inflammatory marker is present in the serum of the treated mice at an earlier stage, we IV injected mitochondria to additional C57BL male mice and collected serum on day 4 following injection of mitochondria (or vehicle only). No increase in TNF-α was detected in the mitochondria injected mice relative to buffer injected mice (4.22±0.25 versus 5.24±0.59 pg/ml, respectively). These results show that no immune response was evident at two different time points following mitochondrial transfer (and maybe even some possible trend of decrease in the TNF-α level).
Evidence for mitochondria functionality in mouse serum
To examine the functionality of the mitochondrial preparation in mouse serum, the mitochondria were incubated with mouse serum for several time points, then the activity of the CS and COX enzymes was determined. The results showed that the mitochondria were functional in serum at least for 1 h, as follows: 50.8 mU (100%), 37.1 mU (73.0%), 39.3 mU (77.36%), 34.4 mU (67.71%) CS activity at 0, 15, 30 min, and 1 h, respectively. There was also evidence for COX activity in the mitochondria preparation following incubation in the mouse serum: 47.7 mU (100%), 30.9 mU (64.77%), 32.7 mU (68.55%), 19.8 mU (42.51%) COX activity at 0, 15, 30 min, and 1 h, respectively.
DISCUSSION
Mitochondrial dysfunction is a major component in neurodegeneration and in AD. Thus, combating this dysfunction is a central goal in the therapy of this disease. However, the multifaceted, and even opposed, modes of involvement of the mitochondria and its complex regulation make the goal of targeting mitochondrial deficits a very challenging one. Our strategy aims to overcome this limitation by using the intact mitochondria organelle, thereby allowing it to act as a whole organelle rather than targeting only one of its abnormal functions. In this way, a broad range of activities, with a physiological mode of regulation, can be achieved. For this purpose we used, for the treatment of AD-mice, fresh and active mitochondria isolated using a protocol which keeps them intact and viable, and which we have shown to induce beneficial effects in mitochondria-defective-cells [32]. Our results showed that transfer of active and fresh mitochondria isolated from human cells, delivered in a single IV injection, ameliorated cognitive deficits, neuronal disarrangement, neuronal loss and gliosis and corrected mitochondrial enzymatic activities in the treated AD-mice.
Our strategy is novel since it uses active and fresh unmodified mitochondria for the treatment of AD-mice. So far, this strategy has not been reported as a therapy for cognitive diseases. While it was reported to show efficacy against motor deficits in a pharmacologically induced PD-model [29], such a beneficial effect was evident by another group only when using IC injected, modified mitochondria (in the form Pep-1 (a cell-penetrating peptide) conjugated mitochondria) but not as unmodified mitochondria [28]. Our results, showing high efficacy of unmodified mitochondria, is advantageous since it does not alter the original components of these cell organelles, thereby keeping its original mode, and avoiding non-physiological effects. Moreover, the IV delivery used by us is obviously advantageous, compared to the IC mode of delivery.
The high efficacy and, importantly, the high safety profile reported here, using non-allogeneic human mitochondria to treat mice—may be explained by the homology between mouse and human mitochondrial DNA [44], which is particularly high in the fusion proteins [45]. We may anticipate that using allogeneic human unmodified mitochondria in the clinic to treat AD patients may be even more effective, due to a full homology. This may be further supported by the results of Chang et al. [28], who showed that allogeneic peptide-mediated mitochondrial delivery in the PD-rat model had a more robust effect than xenogeneic delivery.
Transfer of mitochondria is a naturally occurring physiological process, also termed mitochondrial exchange. Neurons can release damaged mitochondria and transfer them to astrocytes for disposal and recycling [46]. The ability to exchange mitochondria may represent a potential mode of cell-cell signaling in the CNS, and astrocytes could provide a source for functional mitochondria that enter into neurons. Transient focal cerebral ischemia in mice induced astrocytic mitochondria entry to adjacent neurons that amplified cell survival signals [47]. This naturally occurring physiological process strongly supports the concept of mitochondrial transfer as a therapy for brain diseases with mitochondrial dysfunction. Therefore, the concept of mitochondrial transfer aims not only at delivering intact organelles possessing multi original components and functions, but also at mimicking an endogenous process aimed at protecting the CNS from injury/insults. First, the concept of mitochondrial transfer for correcting mitochondrial deficits was studied in mitochondria-deficient cells [15–18, 48], and then in mitochondria-defective cells by others and by us [32, 49]. Therapy, mostly local but also systemic, was studied in animals under various conditions with a mitochondrial dysfunction component, yet not addressing the effect of cognitive diseases. We are now showing efficacy of unmodified mitochondria, for the first time in AD, by IV delivery.
Mitochondrial transfer presented here seems to ameliorate the mitochondrial dysfunction detected in AD-mice, as expressed by the increased activities of the two mitochondrial enzymes, which participate in the energy production: CS, a ubiquitous mitochondrial matrix Krebs cycle enzyme, and COX, the 4th complex of the electron transfer chain. The mitochondrial transfer therapy was administered to AD-mice two days following Aβ-ICV-injection, at a stage when cognitive impairment may already be evident [50, 51]. Therefore, we can hypothesize that this therapy did not only prevent the generation of the Aβ-induced damage but also corrected it, or interfered with its propagation. This Aβ-ICV-injected AD model represents the toxicity of the major AD culprit, the Aβ peptide, including neuronal loss and neuroinflammation. Furthermore, the model provides a tool for interfering with disease pathogenesis even before development of amyloid plaques and neurofibrillary tangles. This pharmacological acute model is used to model AD and in drug discovery. It is suggested that this model is suitable to study the role of Aβ in the neurological and behavioral changes in early stages of amyloid pathologies [52–54]. Additional studies in transgenic models for AD will allow us to see whether mitochondrial transfer is also beneficial in chronic models of AD, which present amyloid as well as of tau pathology.
Whether the beneficial effect of mitochondrial transfer is mediated directly by the entrance of the mitochondria into the brain or by a response of the brain to the effect of the injected mitochondria in the periphery is an important question. That the brain is not completely blocked to the entrance of macromolecules [55] or to the penetration of cells under inflammatory conditions, is well established [56]. However, we did not detect a DsRed2 signal of the labelled mitochondria in the brain of IV (tail vein) mitochondria injected AD-mice. Possibly, the injected mitochondria are highly diluted when passing from the tail vein to the circulation (via the heart) and only a small fraction may arrive at the brain, and be undetectable by imaging and histological staining of the DsRed label. We did detect the DsRed2 signal in the liver of the mitochondria IV injected mice, suggesting uptake of the injected mitochondria by the liver. As an organ which gets the highest blood supply, the accumulation of the IV injected mitochondria in the liver much more than in the brain (even without the challenge of penetrating the blood-brain-barrier) is actually reasonable. Our results show not only the uptake of the injected mitochondria by the liver but also an increase in the mitochondrial enzymatic activity in the AD-mice injected with the mitochondria. The possibility that the responsiveness of the liver to the transfer of healthy mitochondria (by uptake of the injected mitochondria and by the increase in mitochondrial enzymatic activity) can affect brain functions and ameliorate AD-related deficits fits with the established phenomenon of cross talk between the liver and the brain, such as via metabolic/endocrine signals. Such conditions are evident in hepatic encephalopathy, a brain dysfunction caused by liver insufficiency; it manifests as a wide spectrum of neurological or psychiatric abnormalities ranging from subclinical alterations to coma [57]. Cross talk is also evident in diabetes: AD-brain abnormalities are evident in diabetic patients and in experimental mouse models of diabetes, such as systemic injection of streptozotocin, an insulin producing beta-cell toxin [58], suggesting that metabolic signals can act on the brain distally from other organs [58, 59]. Another condition is the nonalcoholic fatty liver disease (NAFLD), which significantly influences the cognitive deficit and tissue volume reduction, with NAFLD patients having a much higher risk of cognitive impairment [60, 61]. Animal studies further support these finding [62]. The interplay between liver and brain may have a particular relevance in our Aβ-ICV AD-model, presenting the other direction of the cross talk, as we demonstrated here by the finding of a mitochondrial dysfunction (reduced enzymatic activity) evident in the AD-mice relative to the non-AD mice. That injection of the amyloid into the brain can affect the liver is further supported by the report that ICV amyloid injection induced IL-6 mRNA expression in lymph nodes and liver, suggesting that the Aβ1 - 42 induced IL-6 may influence immunological and hepatic functions [63]. IL-6-mediated impairment of mitochondrial functions in hepatic cells has been reported, showing an increase in superoxide levels and reduction of the mitochondrial membrane potential [64]. In addition, translocation of signal transducer and activator of transcription 3 (STAT3) to the mitochondria of immune cells, modifying the mitochondrial membrane, has been reported [65]. Therefore, it can be assumed that the intact injected mitochondria which have a beneficial effect on mitochondrial dysfunction in the liver, may ameliorate the cognitive and AD-brain pathology as well as the accompanied mitochondrial deficits, via metabolic /endocrine signaling to the brain. What may further support this possibility is the finding that while a robust increase in mitochondrial function was noticed in the liver already on day 5 following the transfer, at this stage the increase in the brain mitochondrial activity was only limited, if at all, and was pronounced on day 15. That the mitochondrial responsiveness in liver precedes this effect in the brain may point to a possible downstream effect of the brain to the liver response. The way such a cross talk is taking place requires further investigation. Possible candidates are growth factors, like the fibroblast growth factors which have been reported to mediate the cross talk between liver and brain during prolonged fasting [66]. Therefore, we may hypothesize that beside the possibility that some fraction of the exogenous mitochondria can act directly on the brain, the effect of the mitochondria can be also mediated by other organs, particularly the liver.
The increase in mitochondrial enzymatic activity (some even above baseline) could be due to increased mitochondrial content, with physiological and pharmacological factors, including some stress, possibly activating the mitochondrial biogenesis [67, 68]. Future research will be needed to elucidate the mechanism.
Although we did not encounter safety problems, when further developing this therapeutic approach and moving to clinical trials, both efficacy as well as safety aspects need to be further addressed.
Our results suggest that transfer of active intact mitochondria, aimed at efficiently mimicking mitochondrial function, is beneficial to treat AD-deficits, correcting cognitive deficits, brain pathology, and mitochondrial defects in an AD-mouse-model. Mitochondrial transfer can be easily applied in humans, since collecting mitochondria from an autologous source, e.g., self-skin cells, can be a feasible procedure. Thus, this novel therapeutic approach may be clinically applicable.
Footnotes
ACKNOWLEDGMENTS
This study was supported in part by the Israel Ministry of Science, Technology and Space (3010853) Knowledge Center Program. We would like to thank The Wohl Institute for Translational Medicine Hadassah Hebrew University Medical Center for the IVIS acquisition and analysis. We would like also to thank Eran Nitzan for his technical support.