
Abstract
Background:
Mitochondrial dysfunction is an early feature of Alzheimer’s disease (AD) and miR-195 is involved in mitochondrial disorder through targeting MFN-2 protein in hippocampal neurons of AD.
Objective:
To clarify if administration of miR-195 inhibitor could enhance the memory deficits through improving hippocampal neuron mitochondrial dysfunction in SAMP8 mice.
Methods:
The expression of miR-195 was detected by RT-qPCR in primary hippocampal neurons and HT-22 cells treated with Aβ1–42. Morris water maze (MWM) was used to assess the learning and memory function in SAMP8 mice administrated with antagomir-195. Transmission electron microscopy was employed to determine the morphological changes of synapses and mitochondria of hippocampus in SAMP8 mice. Mitochondrial respiration was measured using a high-resolution oxygraph.
Results:
The expression of miR-195 were upregulated in the primary hippocampal neurons and HT-22 cells induced by Aβ1–42. Inhibition of miR-195 ameliorated the mitochondrial dysfunction in HT-22 cells induced by Aβ1–42, including mitochondrial morphologic damages, mitochondrial membrane potential, respiration function, and ATP production. Administration of antagomir-195 by the third ventricle injection markedly ameliorated the cognitive function, postsynaptic density thickness, length of synaptic active area, mitochondrial aspect ratio, and area in hippocampus of SAMP8 mice. Finally, antagomir-195 was able to promote an increase in the activity of respiratory chain complex CI and II in SAMP8 mice.
Conclusion:
This study demonstrated that miR-195 inhibitor ameliorated the cognitive impairment of AD mice by improving mitochondrial structure damages and dysfunction in the hippocampal neurons, which provide an experimental basis for further exploring the treatment strategy of AD.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disease that is projected to grow from about 46.8 million people in 2015 to 74.7 million in 2030 worldwide [1]. The long prodromal phase of AD is characterized by cognitive decline and behavioral disturbances. At the pathological level, AD is featured by two well-known microscopical phenomena: amyloid-β (Aβ) plaques and neurofibrillary tangles [2, 3]. There is currently no effective way to cure AD or prevent the progression of AD.
Studies have shown that oxidative stress plays important roles in the pathogenesis of AD [4]. The regulation and expression of microRNAs (miRNAs) have become an emerging field to determine the mechanisms of regulating oxidative stress [5]. miRNAs are a class of short (21∼22 nucleotides), single-stranded and endogenous non-coding RNAs, acting at a post-transcriptional level and fine-tuning the expression of its target protein-encoding genes. Recently, miRNAs, being inherently stable and easy to manage, have been reported as key regulators of the complicated network in the development of AD [6]. Moreover, the importance of miRNAs and their emerging roles in AD are becoming increasingly evident. Cell-based, as well as clinical sample-based, studies involving miRNA have shed much light on understanding the underlying mechanism of AD.
Mitochondrial fusion protein 2 (mitofusin-2, MFN-2) is localized in the mitochondrial outer membrane and the endoplasmic reticulum-mitochondria junction in cells [7]. Studies have shown that the expression of MFN-2 is downregulated in the hippocampus of AD patients, and the amplitude of MFN-2 is 1.1 to 4.7 times in the frontal cortex of different Braak stages [8]. Leal et al confirmed that modulation of MFN-2 expression could affect γ-secretase activity and Aβ generation in AD model [9]. Decreased MFN-2 protein is found in the brain of AD patients and models of AD, which suggests that the impaired mitochondrial fusion is involved in the pathological process of AD [10, 11]. Through miRNA microarray analysis, we have identified a series of miRNAs induced by oxidative stress were dysregulation in AD [12]. Particularly, the expression of miR-195 in hippocampus was significantly upregulated in SAMP8 mice compared with SAMR1 mice, and miR-195 directly regulated MFN-2 protein expression in hippocampus in vitro and in vivo, suggesting that MFN-2 played a critical role in the mitochondrial disorder during the progression of AD and miR-195 might be a potential new therapeutic target for treatment of AD [15]. However, if administration of miR-195 inhibitor could enhance the memory function in AD remains unclear.
Therefore, based on our previous research [12], we hypothesized that inhibition of miR-195 expression may attenuate mitochondrial structure and function damage in hippocampal neurons and improve the cognitive impairment in AD. In this study, the expression patterns of miR-195 were determined in primary hippocampal neurons and HT-22 cells induced by Aβ1–42. Then, the changes in the structure and function of mitochondria in hippocampal neurons by inhibiting miR-195 were investigated in vitro and in vivo. Finally, the intervention effects of miR-195 inhibitor on the cognitive impairment were observed and an improvement in the mitochondrial respiration dysfunction was detected in SAMP8 mice. This study will provide an experimental basis for further exploring the treatment strategy of AD.
MATERIALS AND METHODS
Animals and cells
Six-month-old SAMR1 and SAMP8 male mice in clean grade, weighing 28–30 g, were purchased from The Animal Center of Beijing University Medical Department (Beijing, China). They were kept in normal circadian rhythm with temperature 22±2°C and relative humidity 40–60%. All experimental procedures were approved by the Animal Care and Use Committee of the First Hospital of Hebei Medical University.
The primary hippocampal neurons were isolated from the Kunming male mice in postnatal day 1 (The Animal Center of Hebei Medical University, Shijiazhuang, China) as described previously[13]. Cells were collected for experiments after culturing for 7–10 days. HT-22 hippocampal neuron cells were purchased from Life Technologies (Waltham, MA, USA). Cells were grown in DMEM supplemented with 10% FBS.
Immunofluorescence
The hippocampal neurons were identified by im-munofluorescence staining of hippocampal neuron-specific marker microtubule-associated protein 2 (MAP-2, Abcam,USA), as previously described[14]. The primary hippocampal neurons were photograp-hed under a fluorescence microscope (Nikon, Tokyo, Japan).
Cell viability and reactive oxygen species (ROS) measurement
HT-22 cells were treated with different concentrations of Aβ1–42 (0, 25, 50, 100, 200, or 400μmol/L) for 24 h. Cell viability was measured using Cell Counting Kit-8 (CCK-8) determination kit Assay (Solarbio, Beijing, China) following the manufacturer’s instructions. The cells were measured for absorbance using a microplate reader (Promega, Madison, MI, USA) (λ= 450 nm) and the relative cell viability values were calculated as % of control cultures which were averaged to define the 100%. HT-22 cells were allocated into four groups: Control group (1μmol/L of DMSO) and Aβ1–42 group (100, 200 and 300μmol/L). Neurons were incubated with dichlorodihydrofluoresce in diacetate (DCFH-DA) to detect ROS, followed by flow cytometry analysis (FACSCalibur, DB, USA).
Development of AD cell model and drug treatment
An AD cell model was established by inducing neuron damage using Aβ1–42 (ChinaPeptides, Shanghai, China), as previously described [14]. Primary neurons and HT-22 cells were divided into two groups: Control group (1μmol/L of DMSO) and Aβ1–42 group (100μmol/L of Aβ1–42).
To observe neuronal mitochondria morphology, mitochondrial membrane potential, and the concentration of ATP, HT-22 cells were allocated into four groups: Control group (1μmol/L of DMSO), Aβ1–42 group (100μmol/L of Aβ1–42), inhibitor-NC +Aβ1–42group (cells were incubated with 100 nmol/L of inhibitor-NC and 100μmol/L of Aβ1–42 for 24 h) and miR-195 inhibitor+Aβ1–42 group (cells were incubated with 100 nmol/L of miR-195 inhibitor and 100μmol/L of Aβ1–42 for 24 h).
Quantitative reverse transcription polymerase chain reaction (qRT-PCR) assay
Total RNA from HT-22 cells were isolated by using Easteptm Super Total RNA Extraction kit (Promega, Madison, WI, USA). Real-time qPCR assay was performed with All-in-oneTM miRNA qRT-PCR detection kit (GeneCopoeia, Rockville, MD, USA) using the ABI 7500 sequence detection system (Applied Biosystems, Foster City, CA, USA). The data were normalized to the housekeeping gene GAPDH.
Mitochondria morphology
HT-22 cells were labeled with MitoTracker Red CMXRos (300 nmol/L) and Hoechst33258 (Sigma, St. Louis, MO, USA) for 30 min at 37°C, then washed cells with assay buffer. Images were captured by a confocal laser scanning microscopy (CLSM) (MODEL CK40-F200, LYMPUS, Tokyo, Japan).
Mitochondrial membrane potential detection
Mitochondrial membrane potential (ΔΨm) in HT-22 cells was determined using the JC-10 mitochon-drial transmembrane potential assay kit (Solarbio, Beijing, China). The cells were then examined using CLSM (MODEL CK40-F200, LYMPUS, Tokyo, Japan). JC-10 monomer (green, excitation 515 nm, emission 529 nm) and JC-10 aggregate (red, excitation 585 nm, emission 590 nm) were measured.
Determination of intracellular adenosine triphosphate (ATP) levels
ATP levels in neurons were measured using an ATP Bioluminescence Assay Kit (S0026, Beyotime Institute of Biotechnology, Shanghai, China) according to the manufacturer’s instructions. Each sample was measured using luminometer (Multiskan Ascent, Thermo Fisher, Waltham, MA, USA). The concentrations of ATP in the samples were calculated according to the standard curve.
Measurement of mitochondrial respiration
The measurements of mitochondrial respiration in HT-22 cells and mouse hippocampal tissues were performed with an Oroboros Oxygraph-2K (Oroboros Instruments, Innsbruck, Austria) [15]. Protocols were based on O2k manual titrations for the substrate-uncoupler-inhibitor titration (SUIT) protocols for mitochondrial preparations. The amplified signal from the oxygen was recorded on a computer at sampling intervals of 2 s using Datlab software (Oroboros Instruments, Innsbruck, AT). ComplexI Respiration was quantified by addition of Adenosine5’diphosphate, potassium salt (ADP, 2.5 mmol/L). Succinate (10 mmol/L) addition then stimulated electron transfer system maximum respiration (ETS max). Complex II Respiration was measured by addition of rotenone (Rot, 0.5μmol/L).
Brain stereotaxic injection
The 6-month-old SAMR1 and SAMP8 male mice were anesthetized with 10% chloral hydrate (0.004 ml/g) by intraperitoneal injection, fixed on the brain stereotaxic instrument, the top of the head was depilated, the skin was disinfected, and the middle of the head was incision. Before the exposure, according to the stereotactic map of the mouse brain, 0.5 mm from the front and 1.1 mm on both sides of the midline, the micro-injector was inserted vertically from the brain surface by 2.2 mm. Slowly injected 1μl of antagomir-195 (500μmol/L, Guangzhou RiboBio, Guangzhou, China) and control miRNA into the third ventricle. The injection time was 1μl/min on each side, and the needle was left for 5 min. All procedures were performed under aseptic conditions, and the skin incision was antibacterial with penicillin to suture the wound.
Thirty-six SAMP8 mice were randomly assigned to one of the following three groups (n = 12 mice per group): SAMP8 mice, SAMP8 mice plus antagomir NC group (NC group), SAMP8 mice plus antagomir-195 group (Antagomir-195 group). Twelve SAMR1 mice were used as the blank control group.
Morris water maze (MWM) test
Three weeks post injection, the MWM test was executed to evaluate the behaviors of mice in every group. Two days before the experiment, the mice were housed in a water maze laboratory to familiarize themselves with the environment. The day before the test, the mice were placed in the maze and allowed them to swim freely for 1 min to adapt to stress factors. The place navigation test was used to assess the learning capability of the mice in the water maze. Mice were asked to swim ad libitum for 1 min in a water maze to search for an underwater platform and the time it took to reach the platform was recorded. When the mice failed to reach the platform within 1 min, they were manually guided to the platform and allowed to stand on the platform for 30 s. This trial had been completed per day for five consecutive days. The spatial probe test was used for assessing the memory retention capability (day 7). After the underwater platform was removed, the mice were asked to swim ad libitum for 60 s to record the total residence time in the target quadrant.
Sample collection and hematoxylin-eosin (HE) staining
Subsequently, the mice were anaesthetized with 10% chloral hydrate through intraperitoneal injection. Supine fixation with the heart exposed. Subsequently, brain was quickly taken away by craniotomy. Next, the hippocampus was separated instantly, and the hippocampus tissues were obtained and rapidly placed in liquid nitrogen for detection.
After being dewaxed and hydrated, the histopathological status of hippocampal tissues in mice was evaluated by histopathological examination and photography. ATP levels were measured using an ATP Bioluminescence Assay Kit (S0026, Beyotime Institute of Biotechnology, Shanghai, China).
Immunohistochemistry
The hippocampal tissue sections were immunohistochemically stained for MFN2 using rabbit primary anti-MFN2 antibody (1 : 100; abcam USA) and secondary goat anti-rabbit IgG (DBA, Milan, Italy). These methods have been described previously [4].
Western blot assay
Hippocampal tissue protein contents of mice were determined by the BCA method (Solarbio, Beijing, China). The extracted proteins were electrophoresed on 10% polyacrylamide gel. The proteins were then transferred onto polyvinylidene fluoride membranes. The membranes were blocked with 5% nonfat dry milk solution in 0.1% TBS/Tween-20 for 1 h at room temperature. The membranes were then incubated overnight at 4°C with different primary antibodies: β-actin (1 : 5000; Proteintech Group, Wuhan, China), MFN2 (1 : 4000, Proteintech group, Wuhan, China), followed by supplement with the corresponding secondary antibody. The density of bands was measured using ImageJ analysis software (Wright Cell Imaging Facility, Toronto, ON, Canada).
Transmission electron microscopy (TEM)
The hippocampal tissues were fixed by 4% glutaraldehyde and 1% osmium tetroxide. Next, the tissues were dehydrated in ethanol, embedded in Epon 812. The ultramicrostructure of hippocampal neurons were observed under a TEM (Hitachi, Tokyo, Japan). Photos of 3 random sections were obtained, yielding at least 100 mitochondria per mouse for morphometric analysis using ImageJ. The stereological method for analysis of synapses and mitochondria was described previously [16, 17].
Statistical analysis
The data were analyzed using SPSS version 21.0 and the values are shown as the mean±SD. Significant differences between values were determined using a one-way analysis of variance followed by least significant difference post hoc tests. The threshold for the statistical significance was p < 0.05.
RESULTS
The expression of miR-195 was upregulated in the primary hippocampal neurons and HT-22 cells induced by Aβ1–42
Neuronal marker MAP-2 staining was performed to identify the purity of the hippocampal neurons from neonatal rats. The positive reaction showed red fluorescence, which was distributed in the cell bodies and dendrites of neurons. The results showed that the cultured primary hippocampal neurons had a purity of 96.4% (Fig. 1a) which can be used for subsequent experiments.

Expression levels of miR-195 were increased in oxidative stressed hippocampal neurons induced by Aβ1–42. a) Identification of mouse primary hippocampal neurons by immunofluorescence staining. Scale bar = 20μm. b) Cell viability of HT-22 cells treated with different concentrations of Aβ1–42 (0, 25, 50, 100, 200, 400μmol/L). c,d) The levels of ROS generation in HT-22 cells induced by Aβ1–42. HT-22 cells were treated with different concentrations of Aβ1–42 (0, 100, 200, 300μmol/L) for 24 h, and ROS generation was detected using flow cytometry. e) Quantitative real-time RT-PCR analysis of miR-195 levels in mouse primary hippocampal neurons and HT-22 cells treated with of Aβ1–42 (100μmol/L). All data were obtained from three independent experiments performed in duplicates. The values are the mean±SD.
The cytotoxicity of Aβ1–42 for neurons was detected by CCK8 assay as shown in Fig. 1b, treatment with different concentrations of Aβ1–42 significantly decreased cell viability. And when the concentration of Aβ1–42 was 100μmol/L, the cell viability dropped to about 60% compared to control (p < 0.001). Based on these results, the condition of treatment cells with 100μmol/L Aβ1–42 for 24 h was used in subsequent experiment. It has been reported that Aβ1–42 reduced respiratory chain complex activities, disrupted mitochondrial electron transport chain, enhanced ROS levels, and caused mitochondrial damage [18]. Next, we investigated the effect of Aβ1–42 on oxidative stress in HT-22 cells. We found that Aβ1–42 induced an increase in ROS production in HT-22 cells by flow cytometry (FCM) assay (Fig. 1c). To validate the oxidative stress-induced increase in miR-195, real-time PCR results showed that, after Aβ1–42 stimulation, the expression of miR-195 were significantly higher than control in primary hippocampal neurons and HT-22 cells (p = 0.001, p = 0.003) (Fig. 1e). These results suggested that Aβ1–42 induced the oxidative stress mediated the increase of miR-195 expression in primary hippocampal neurons and HT-22 cells.
MiR-195 inhibitor improved mitochondrial dysfunction in HT-22 cells induced by Aβ1–42
Recent evidence indicates that mitochondria in AD brain cells exhibit abnormal morphology, such as swollen, fragmented with decreased size [19]. Therefore, we assessed whether miR-195 inhibitor improves mitochondrial damage in HT-22 cells induced by Aβ1–42. Healthy mitochondria in untreated cells appeared oval or round distributing in a linear or tubular network, whereas damaged mitochondria in Aβ1–42-treated cells exhibited swollen shape, which appeared as a fine granular structure. Notably, pretreatment cells with miR-195 inhibitor (100μmol/L) for 24 h before the addition of Aβ1–42 improved mitochondrial damage compared to inhibitor-NC group. But mitochondrial morphology of cells incubated with inhibitor-NC following stimulation with Aβ1–42 was basically the same as that of Aβ1–42-treated cells, without significant alterations (Fig. 2a).
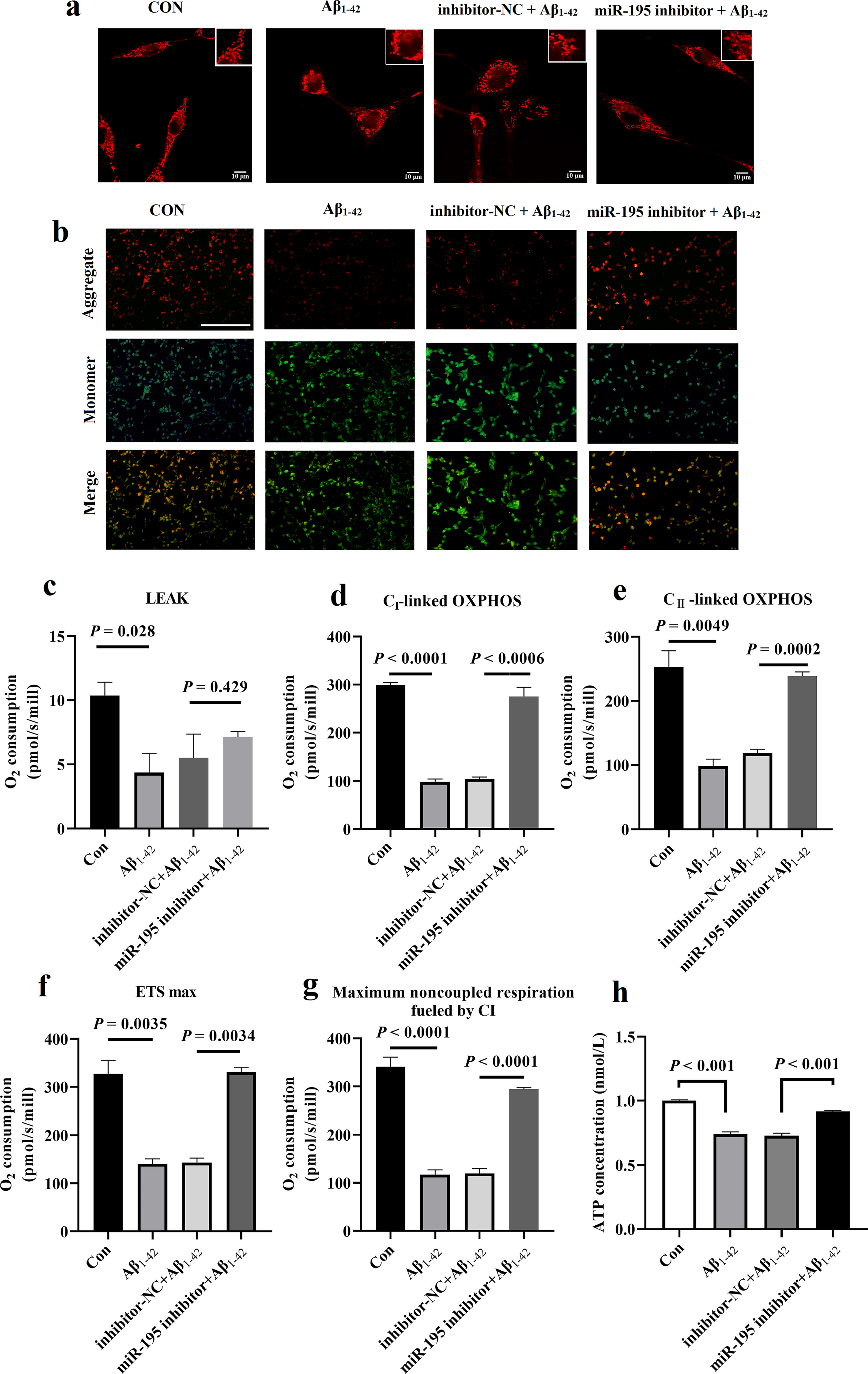
Inhibition of miR-195 ameliorated the mitochondrial dysfunction of HT-22 cells induced by Aβ1–42. a) Representative images of mitochondrial morphology in HT-22 cells by MitoRed staining. Scale bar = 10μm. b) Mitochondrial membrane potential (ΔΨm) was monitored with JC-10 staining in HT-22 cells. The red fluorescence indicated that theΔΨm of HT-22 was normal, but the green fluorescence suggested the decrease or loss of ΔΨmin HT-22 cells. Scale bar = 20μm. c-g) The mitochondrial respiratory functions were measured by a high resolution respirometry (Oxygraph-2k) including LEAK (c), OXPHOS CI (d), OXPHOS CII (e), ETS max (f), and Maximum noncoupled respiration fueled by CI (g). h) The intracellular ATP levels in HT-22 cells were detected by ATP Bioluminescence Assay Kit. The HT-22 cells were incubated with DMEM medium containing DMSO (1μmol/L), or Aβ1–42 (100μmol/L), or inhibitor-NC (100 nmol/L)+Aβ1–42 (100μmol/L) for 24 h, or miR-195 inhibitor (100 nmol/L)+Aβ1–42 (100μmol/L) for 24 h. The values are the mean±SD (n = 3).
As shown in Fig. 2b, the neurons in control group showed bright red fluorescence but no green fluorescence, indicating that the neuron mitochondrial membrane potential (ΔΨm) of the control group was normal. In the Aβ1–42-stimulated cells, red fluorescence intensity decreased significantly, and green fluorescence intensity increased significantly, indicating that Aβ1–42 induced a significant decrease in ΔΨm. Compared with the Aβ1–42 group, ΔΨm of cells incubated with inhibitor-NC following stimulation with Aβ1–42 has no significant changes. It is worth mentioning that red fluorescence increased significantly, and the green fluorescence intensity decreased significantly in the pre-treated cells with miR-195 inhibitor, indicating that miR-195 inhibitor significantly improved the decrease of ΔΨm in neurons induced by Aβ1–42.
Assessment of mitochondrial respiration represents a functional evaluation of mitochondrial homeostatic state [20]. To study the effect of miR-195 inhibitor on the alterations of the main mitochondrial metabolic functions, we performed high-resolution respirometry experiments in HT-22 cells. We found that miR-195 inhibitor showed a tendency toward improved the oxygen consumption of complex I & II-linked respiration (Fig. 2d, e). Analysis of OXPHOS in isolated mitochondria from HT-22 cells revealed that miR-195 inhibitor also promotes an increase in ETS max and noncoupled respiration fueled by CI (Fig. 2f, g). But measurement of leak-linked oxygen consumption did not show a significant difference in respiration between inhibitor-NC group and miR-195 inhibitor group (Fig. 2c). We also measured the changes in intracellular ATP after the treatment of miR-195 inhibitor since miR-195 is also known to affect mitochondrial function. Our results showed that Aβ1–42-treated HT-22 cells had lower intracellular ATP levels compared to control cells, but miR-195 inhibitor could rescue the decrease of intracellular ATP levels induced by Aβ1–42 (Fig. 2h). Together, these results indicated that miR-195 inhibitor improves mitochondrial damage and dysfunction in HT-22 cells induced by Aβ1–42.
Inhibition of miR-195 enhanced spatial learning and memory function, and improved pathological damage to hippocampal neurons in SAMP8 mice
To investigate whether miR-195 inhibitor im-proves the cognitive deficits in SAMP8 mice, memory functions of 6-month-old mice were examined by the MWM test after administration of antagomir-195 by brain stereotaxic injection (Fig. 3a). The results of MWM test showed that intraventricular injection of antagomir-195 markedly increased the number of platform crossing (Fig. 3b) and the time spent in the target quadrant (Fig. 3c) in SAMP8 mice compared with the NC group. These results demonstrated that miR-195 inhibitor could improve the cognitive function of SAMP8 mice.
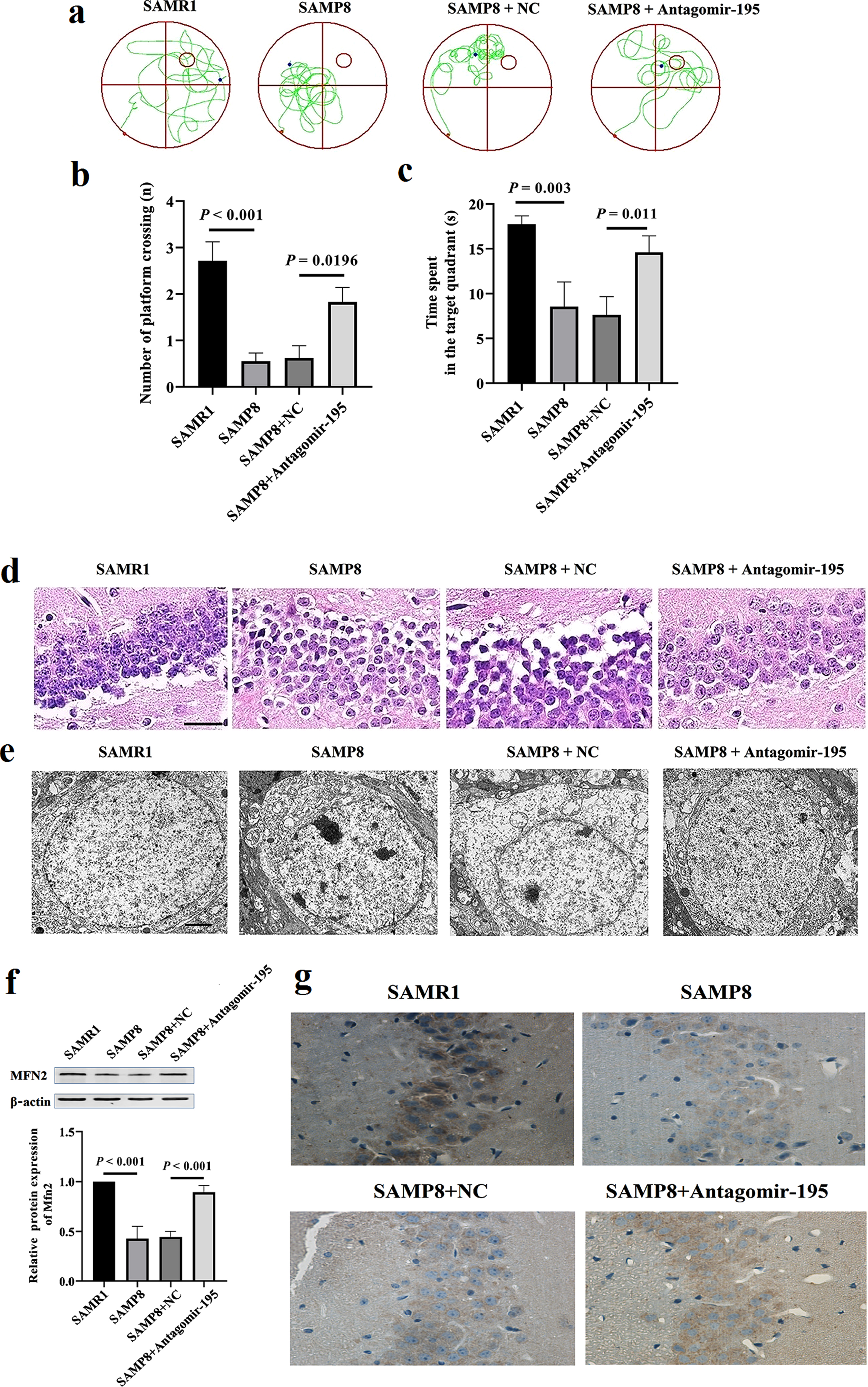
Administration of miR-195 inhibitor enhanced the memory function and improved the hippocampal neuron nucleus structure in SAMP8 mice. a) The representative swimming path for SAMP8 or SAMR1 mice to find the platform in Morris Water Maze Test after administration of miR-195 inhibitors by brain stereotaxic injection. b) Number of platform crossing and (c) time spent in target quadrant results from Morris water maze test in 6-month SAMP8 or SAMR1 mice. d) The HE staining of hippocampus tissues in SAMP8 or SAMR1 mice (n = 6). Scale bar = 50μm. e) The hippocampal neuron nucleus structure in SAMP8 or SAMR1 mice by TEM. Scale bar = 2μm. f) Antagomir-195 significantly increased the level of MFN2 protein in the hippocampus of SAMP8 mice. Western blot analysis was performed to detect the hippocampal SYT1 protein in SAMP8 mice injected with antagomir-195. β-actin was used as a control for the normalization of samples. The values are the means±SD (n = 6). g) Representative photos of the signal levels of immunohistochemical staining of MFN2 protein in the hippocampus from 6-month-old SAMP8 or SAMR1 mice (×40).
H&E staining was conducted to assess the effects of antagomir-195 on the morphology damages of brains in SAMP8 mice. The results of HE staining depicted that the hippocampal neurons of mice in control group were intact, well arranged, and the nucleus centered with clear staining. In contrast, SAMP8 mouse hippocampal neurons were arranged in disorder, which were also featured by the irregular morphology of cells, the enlarged gap, and the deepened cytoplasm staining. However, these neural damages were alleviated after administration of antagomir-195 in SAMP8 mice (Fig. 3d). TEM results demonstrated that there were normal ultrastructure of hippocampal neurons, intact cell structure, and uniform distributed chromatin in SAMR1 mice. In SAMP8 mice, the results of cellultra structure showed chromatin side set and incomplete nuclear membrane as well as significantly shrinkages, swelling of mitochondria, vacolation, and endoplasmic reticulum dilation in the hippocampal neurons, while in the antagomir group, they showed better morphology of neurons and intact nuclear membrane, as well as a few shrinkages, mild pyknosis of the nucleus, uniform distribution of chromatin in the nucleus, and most normal cytoplasmic organelle (Fig. 3e). Western blot results revealed that the levels of MFN-2 protein in the hippocampus of SAMP8 were increased in antagomir group (Fig. 3f). The immunochemical staining also revealed SAMP8 mice with antagomir-195 exhibited significant higher MFN-2 levels than SAMP8 mice with NC in the CA3 subfield of hippocampus (Fig. 3g). These results demonstrated that inhibition of miR-195 enhanced spatial learning and memory function and improved pathological damage to hippocampal neurons through upregulation of MFN-2 protein in SAMP8 mice.
Administration of miR-195 inhibitor alleviated synaptic degradation and improved mitochondrial morphology in the hippocampus of SAMP8 mice
In this study, the ultrastructural changes in syna-pses of the hippocampus were observed by TEM. The pre-synaptic and postsynaptic membranes were clear and with complete outlines, and synaptic vesicles and the postsynaptic densities (PSDs) density were abundant in hippocampal nervous tissues of SAMR1 mice. In SAMP8 mice, the synaptic structure was blurred, the synaptic vesicles in the pre-synaptic membrane were reduced, and the thickness of PSD was decreased in hippocampus. Through administration of antagomir-195, the synaptic structure of the mice was clear, the synaptic vesicles in the pre-synaptic membrane increased, the distribution was denser, and the density of PSD was thicker compared with NC group (Fig. 4a). Quantitatively, the thickness of PSD thinned, and length of synaptic active area shortened in SAMP8 mice compared with SAMR1 mice (p < 0.001, Fig. 4b; p = 0.001, Fig. 4c). After administration of miR-195 inhibitors, PSD thickness and synaptic active area were normalized to levels of NC group (p = 0.014, Fig. 4b; p = 0.0006, Fig. 4c). These results indicated that inhibition of miR-195 protects the synaptic structure of the hippocampus from damage in the SAMP8 mice.
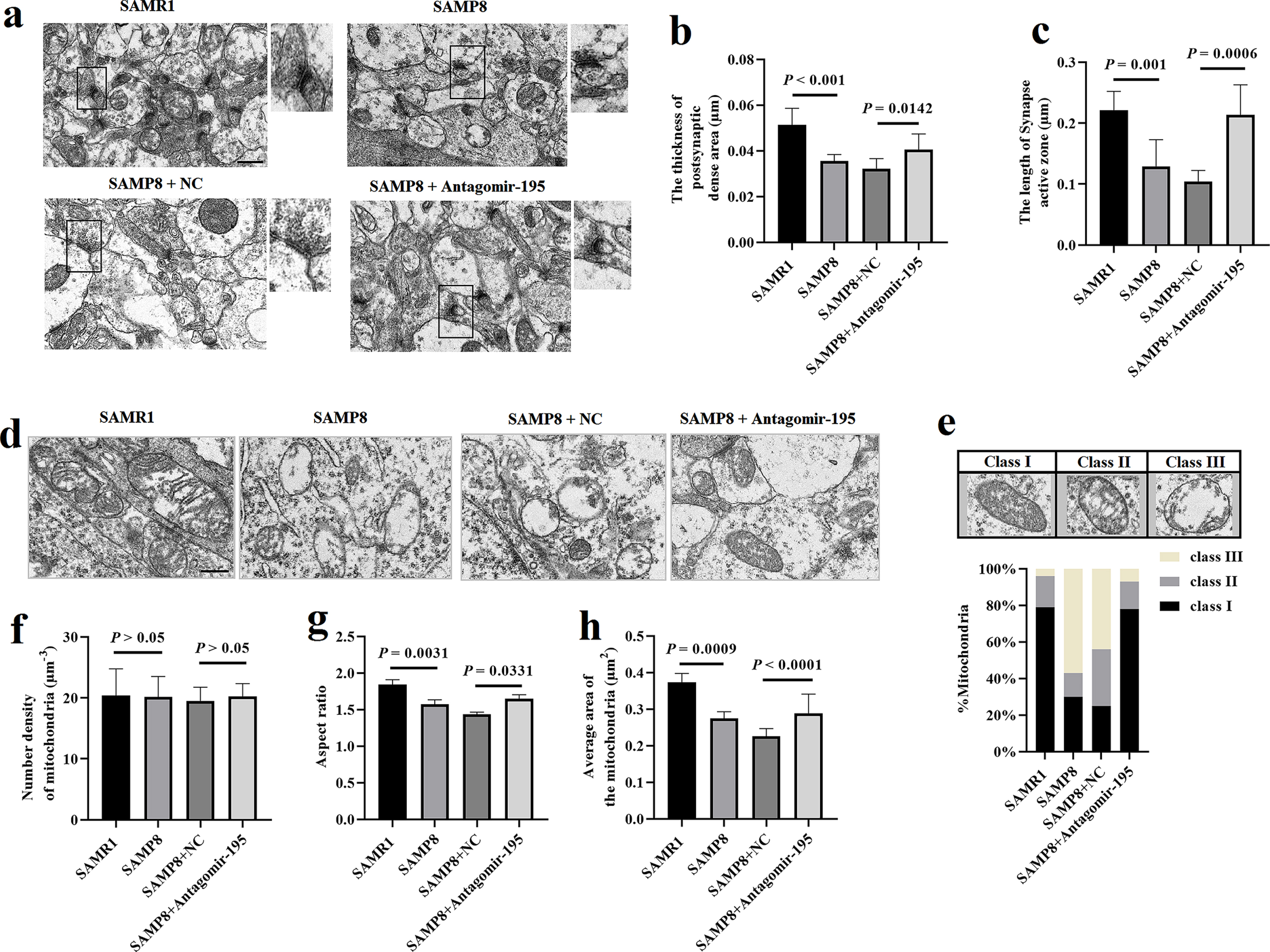
Delivery of antagomir-195 ameliorated the synaptic degradation and mitochondrial structure damage in the hippocampal neurons of SAMP8 mice. a) Morphology of synapses in the hippocampal neurons of SAMP8 or SAMR1 mice after delivery of antagomir-195 by brain stereotaxic injection under TEM (n = 6). Scale bar = 0.5μm. b,c) Comparison of ultrastructural interface parameters of the hippocampal neuron synapses in SAMP8 or SAMR1 mice. d) The ultrastructure of mitochondria by electron microscopy in the hippocampal neurons of SAMP8 or SAMR1 mice (n = 6). Scale bar = 0.5μm. e) Representative images of mitochondria classes I, II, and III (upper) and their quantitative distribution (lower) in the hippocampal neurons of SAMP8 or SAMR1 mice (n = 6). f-h) Quantitative graphs of the mitochondria number density (f), aspect ratio (g) and mitochondria area (h) in the hippocampal neurons of SAMP8 or SAMR1 mice. The values are the mean±SD (n = 6).
As shown in the TEM images (Fig. 4d), the mitochondria of the hippocampal neurons in SAMR1 mice were uniform in size, mostly ovalor long tubular, the mitochondrial membrane structure was intact, and the mitochondria were clearly visible. In SAMP8 mice, hippocampal mitochondria showed more fragmentation, mitochondrial membrane structure was damaged, mitochondria were not clear, and mitochondria showed more vacuolation. However, mitochondria in antagomir-195 group exhibited a relatively intact structure. We analyzed in depth mitochondrial alterations and classified mitochondria morphology in three categories (class I: fairly dark mitochondria, filled with regular distributed cristae; class II: mitochondria with disrupted cristae and loss of matrix density; class III: empty mitochondria) (Fig. 4e). Quantification of mitochondria subclass revealed that while NC group exhibited 25% of mitochondria class I, 31% class II, 44% class III, the antagomir-195 group displayed an enhancement of mitochondria class I (78%), a drastic reduction of mitochondria class II (15%) and III (7%) (Fig. 4i). It was shown that the antagomir-195 group displayed larger mitochondria (increased area and aspect ratio), compared to NC group (p = 0.0331, p < 0.0001, Fig. 4f-h).
Delivery of antagomir-195 improved the mitochondrial respiratory dysfunction in hippocampal neurons of SAMP8 mice
The mitochondrial respiration of the hippocampus was measured by OROBOROS Oxygraph 2K to determine whether delivery of antagomir-195 had an impact on mitochondrial respiration capacity in SAMP8 mice. Mitochondria from the hippocampus of SAMP8 mice displayed reduced proton leak, CI & CII-linked OXPHOS, ETS max and maximum noncoupled respiration by CI compared to those from SAMR1 mice (Fig. 5b-f). Measurement of leak-linked oxygen consumption in the absence of substrates showed significant difference between NC group and antagomir-195 group (Fig. 5b). Comparing with NC group, the levels of ETS max exhibited higher activity in SAMP8 administrated with antagomir-195 (Fig. 5e). Treatment mice with antagomir-195 enhanced the maximum respiratory capacity following damage in the SAMP8 mice (Fig. 5f). We also measured changes in ATP levels after antagomir-195 treatment (Fig. 5g) and the results showed that antagomir-195-treated SAMP8 mice had higher ATP levels compared to control mice. Thus, the treatment with antagomir-195 significantly improved the mitochondrial respiratory capacity in SAMP8 mice.
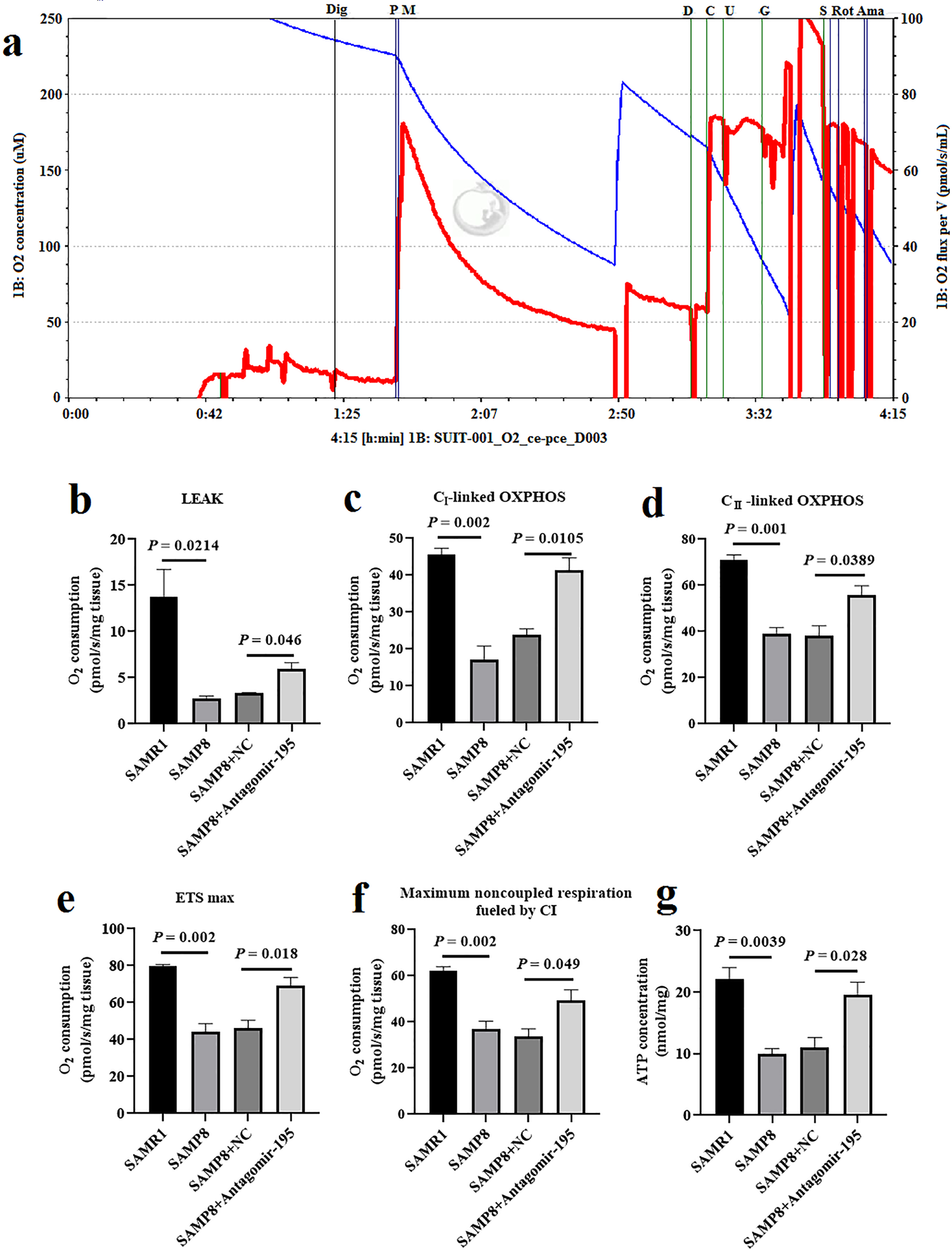
Administration of miR-195 inhibitor improved the mitochondrial respiratory function in hippocampal neurons of SAMP8 mice. a) Substrate-uncoupler-inhibitor titration (SUIT) protocols representative oxygraph traces of oxygen flux relative to tissue mass, vertical solid lines show the introduction of substrates or inhibitors. Blue line indicates the level of oxygen (μmol/l, left Y-axis) and red line indicates oxygen consumption rates (OCR (pmol*s-1*mL-1), right Y-axis). The addition of substrates/inhibitors: Dig: Digitonin (10μg/mL); P: Pyruvate (5 mmol/L); M: Malate (2 mmol/L); D: ADP (2.5 mmol/L); C: Cytochrome (10μmol/L); U: Uncoupler (0.5μmol/L); G: Glutamate (10 mmol/L); S: Succinate (10 mmol/L); Rot: Rotenone (0.5μmol/L); Ama: Antimycin A (2.5μmol/L). b-f) Treatment with antagomir-195 significantly improved the mitochondrial complex of hippocampal neurons of SAMP8 mice, including LEAK (b), OXPHOS CI (c), OXPHOS CII (d), ETS max (e), and Maximum noncoupled respiration fueled by CI (f). g) The effect of antagomir-195 on ATP levels in cortex neurons of SAMP8 mice measured by ATP Bioluminescence Assay Kit. The values are the mean±SD (n = 6).
DISCUSSION
AD is known as a complex progressive neurodegenerative disease amongst the elderly [21]. In recent years, while the pathogenesis of AD is still not confirmed, the continuous investigations have identified that the aberrant regulation of miRNAs have been implicated in the development of AD. Epidemiological studies have revealed that AD-specific miRNA changes consistent with their role as potential biomarkers of disease [22]. miRNAs have emerged as crucial regulators in several physiological processes involves the progression of pathological brain aging [23]. Among all the regulatory molecules, miRNAs represent key regulators of disease-associated pathways. Our previous study found that increased miR-34c mediated synaptic and memory deficits by targeting SYT1 through ROS-JNK-p53 pathway and the miR-34c/SYT1 pathway could be considered as a promising novel therapeutic target for patients with AD [24]. Moreover, we found the abnormal expression of miR-195 played a potential role in mitochondrial disorder by targeting mfn2 in hippocampus of SAMP8 mice [4]. The discovery of miRNAs has opened a new window to look at cellular and molecular mechanisms of neurodegeneration disease and provide a new strategy for developing gene-based therapeutics of AD [25–28]. So, in this study we focused on the role of the inhibition of miR-195 to improve to the mitochondria disorder and cognitive impairments in worked mice.
Previous evidence suggested that Aβ impairs mitochondrial electron transport chain, enhances ROS production in neurons [29–31]. To elucidate how miRNAs exert its roles in Aβ-induced oxidative stress injury, the expression of miR-195 was detected and the results showed that the expression of miR-195 was increased in Aβ-treated primary hippocampal neurons and HT-22 cells.
Mitochondria play an important role in oxidative stress modulation [32]. As is known, Aβ1–42 disrupts the transport of mitochondrial proteins and metabolites, decreases ΔΨm, and causes mitochondrial damage [33]. Park et al. conducted a correlation study between Aβ oligomers (AβOs) and mitochondrial morphological changes in N2a cells. The results showed that intracellular AβOs not only caused mitochondrial fragmentation and reduced the expression of mitochondrial MFNs, but also resulted in mitochondrial dysfunction [34]. In this study, our results showed that miR-195 inhibitor had significantly exhibited a certain protective effect on HT-22 cells, including improve mitochondrial morphology, increasing ΔΨm and ATP levels. It was further proved that inhibiting miR-195 could reduce the effects of Aβ1–42 on cell mitochondrial damage. Purohit et al have revealed that miR-195 exerts pro-apoptotic effects in breast cancer cells, and regulates mitochondrial function by targeting mitofusin2 in breast cancer cells [35], which is consist with our previous report [4].
Mitofusin2 (MFN2), a mitochondrial fusion protein, is expressed mainly in tissues with high energetic requirements, such as brain, skeletal muscle, and heart [36]. Several evidence have demonstrated that MFN2 was not only involved in the control of proliferation and apoptosis of various cells but also convinced to participate in modulating of mitochondrial shape and mitochondrial metabolism [37]. Present studies have indicated that loss of MFN2 caused mitochondrial dysfunction. Their findings indicate that both basal and maximal oxygen consumption rate along with spare respiratory capacity were significantly reduced in the synaptic mitochondrial from MFN2 cKO mice as compared with control [32]. Then, another study observed a decrease in respiration capacity under uncoupled conditions in MFN2 knockout heart mitochondria [38]. Mourier et al observed a clear decrease in respiration capacity under phosphorylating and uncoupled conditions in MFN-2 knockout heart mitochondria [38]. We found that HT-22 cells incubated with miR-195 inhibitor have significantly increased the respiratory capacity of mitochondria following stimulated cells with Aβ1–42. Then we found that inhibiting miR-195 could improve LEAK of hippocampal neurons mitochondria in SAMP8 mice, but it has no effect in HT-22 cells. The reason may be that the AD cell model currently only used a single factor to induce, which does not conform to the complexity of the etiology and pathogenesis of AD. The antagomir-195 might play a role in improving mitochondrial function in other ways in vivo. We will continue to explore this question in follow-up research. Taken together, these results demonstrated that inhibition of miR-195 improved the mitochondrial dysfunction in HT-22 cells induced by Aβ1–42.
The formation and accumulation of extracellular Aβ plays the key role in the pathogenesis of AD [39]. The Aβ1–42 represents the most expressed form in several types of AD cases. Therefore, Aβ1–42 induced cellular AD model was constructed in many studies [40–42]. The in vivo environment is more complicated than the in vitro cell model, and the results of the miR-195 inhibitor acting on the AD cell model in vitro may not be confirmed at an overall level. The SAMP8 strain is a mouse model that exhibits early onset of learning and memory deficits during aging process [43].Many studies have shown that SAMP8 mouse is a useful model for studying age-related cognitive impairments and AD [44, 45]. It has been reported that there are similarities with the pathology features of aging and early cognitive decline and other unique hallmarks of AD, such as extracellular Aβ accumulation, neurofibrillary tangles and oxidative stress-induced mitochondrial dysfunction in the hippocampus [46]. And we performed the MWM test to evaluate the learning and memory abilities of the SAMP8 mice in this study. So SAMP8 mouse model which were evaluated using the MWM test is a known and acceptable model of AD. Therefore, in this study the miR-195 inhibitor was injected into the third ventricle of AD model mice as previous described [24] to observe the improvement of cognitive dysfunction and the effects on hippocampal neuron mitochondrial structure and synaptic morphology.
Learning and memory abilities are related to the hippocampal area and depend on hippocampal synaptic plasticity [47]. Studies have found that the length of the synaptic active area and the thickness of PSD in AD model mice are reduced [48]. Xu et al. showed miR-132 can regulate the expression of synaptic proteins through the key initiating protein of classical complement cascade—C1q. The protein deacetylase 3 signaling pathway can also be directly targeted and inhibited by miR-132 to prevent Aβ1–42-induced synaptic dysfunction [49]. In this study, our experiments confirmed that inhibition of miR-195 improves spatial learning and memory ability in SAMP8 mice.
Our study indicated that inhibition of miR-195 can improve synaptic structural damages in SAMP8 mice. The morphological changes of hippocampal neuron mitochondria have been found in AD patients and animal brain tissues, including uneven mitochondrial size, huge differences, and short crests [19, 51]. Earlier studies confirmed that inhibition of miR-195 could increase the expression of MFN-2 [4]. MFN-2 is one of the important proteins of mitochondrial outer membrane fusion, which precisely regulates the balance of mitochondrial fusion and division [52, 53]. After inhibiting MFN-2, the mitochondrial varied in size and shape, and the degree of fragmentation was more serious [54]. The results of TEM showed that inhibiting miR-195 can significantly improve hippocampal mitochondrial morphology in SAMP8 mice. Also in animal experiments, we observed that the mitochondrial respiratory function of SAMP8 mice treated with antagomior-195 was improved compared to the control group.
In summary, this study provided evidence that miR-195 inhibition can effectively improve cognitive impairment and alleviate neuronal damage in hippocampus tissues in SAMP8 mice. miR-195 might be considered to function as a potential therapeutic target for the treatment of AD. Although our understanding of the etiology of AD is still in its infancy, miRNAs, its biology, and potential applications bring great hope to individuals suffering from this irreversible disease. However, further studies are required to fully understand the mechanisms of miR-195 involved in AD, and this study will provide an experimental basis for further exploring the treatment strategy of AD.
Footnotes
ACKNOWLEDGMENTS
Hebei Provincial Natural Science Foundation (H2018206358, H2020206105 and H2020206224), International Cooperation Project of Hebei Province (18397789D), Projects of introducing foreign intelligence in Hebei Province (2019YX007A), Special Funding for Local Science and Technology Development Guided by the Central Government (206Z7701G), The Science and Technology project of the People’s Livelihood in Hebei Province (20377707D), Projects of outstanding talents funded by the Government (LS201904, LS201908 and LS202107), Prevention and control project of geriatric diseases in Hebei Province (LNB201810, LNB201810, LNB201910 and LNB201903). Key Program of traditional Chinese medicine in Hebei Province (Z2022015).