
Abstract
Epidemiological, preclinical, and clinical studies have suggested a role for microdose lithium in reducing Alzheimer’s disease (AD) risk by modulating key mechanisms associated with AD pathology. The novel microdose lithium formulation, NP03, has disease-modifying effects in the McGill-R-Thy1-APP transgenic rat model of AD-like amyloidosis at pre-plaque stages, before frank amyloid-β (Aβ) plaque deposition, during which Aβ is primarily intraneuronal. Here, we are interested in determining whether the positive effects of microdose lithium extend into early Aβ post-plaque stages. We administered NP03 (40μg Li/kg; 1 ml/kg body weight) to McGill-R-Thy1-APP transgenic rats for 12 weeks spanning the transition phase from plaque-free to plaque-bearing. The effect of NP03 on remote working memory was assessed using the novel object recognition task. Levels of human Aβ38, Aβ40, and Aβ42 as well as levels of pro-inflammatory mediators were measured in brain-extracts and plasma using electrochemiluminescent assays. Mature Aβ plaques were visualized with a thioflavin-S staining. Vesicular acetylcholine transporter (VAChT) bouton density and levels of chemokine (C-X-C motif) ligand 1 (CXCL1), interleukin-6 (IL-6), and 4-hydroxynonenal (4-HNE) were probed using quantitative immunohistochemistry. During the early Aβ post-plaque stage, we find that NP03 rescues functional deficits in object recognition, reduces loss of cholinergic boutons in the hippocampus, reduces levels of soluble and insoluble cortical Aβ42 and reduces hippocampal Aβ plaque number. In addition, NP03 reduces markers of neuroinflammation and cellular oxidative stress. Together these results indicate that microdose lithium NP03 is effective at later stages of amyloid pathology, after appearance of Aβ plaques.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is the leading cause of dementia in older adults and is currently without a cure or disease-modifying therapy [1]. Pathological hallmarks of AD include the accumulation of extracellular plaques comprised of aggregated amyloid-β (Aβ) peptides, intraneuronal neurofibrillary tangles consisting of hyperphosphorylated tau, extensive neuron loss, and widespread brain atrophy [2, 3].
Lithium is first-line therapy for bipolar affective disorder and has been used clinically for more than 60 years due to its mood stabilizing properties [4]. Epidemiological studies indicate that prolonged lithium treatment is associated with a reduction in the incidence of dementia [5–8]. Clinical studies on lithium effects on dementia have yielded conflicting results [7]. Short term treatment of 10 weeks failed to show positive effects [9]; however, studies with longer treatment have shown stabilized cognition in subjects receiving lithium compared to placebo [10–12]. The discontinuation rates of lithium in elderly populations are high [13], owing to its narrow therapeutic window and toxic side effect profile [14]. Therefore, if effective, microdose lithium formulations circumventing toxicity would offer an appealing alternative to conventional lithium. In two clinical studies, targeting subtherapeutic concentrations of lithium stabilized performance on memory and attention tests in individuals with amnestic mild cognitive impairment [11, 15]. Moreover, observational studies have found that long-term exposure to microdose lithium in drinking water is associated with lower incidence of AD [16].
NP03 is a novel microdose lithium therapeutic formulation consisting of lithium encapsulated in reverse water-in-oil microemulsion composed of self-assembled specific polar lipids, surfactant and co-surfactants lecithin, and ethanol [17–19]. The transmucosal route of administration avoids acid hydrolysis in the gastrointestinal tract, bypasses hepatic metabolism, and leads to high bioavailability and enhanced central nervous system (CNS) uptake. As a result, a significantly lower amount of lithium may be administered as compared to conventional lithium [20]. Using the McGill-R-Thy1-APP transgenic rat model of AD [21] (hereafter, AD rats) in a previous study we reported that NP03 was protective against cognitive impairment at the earliest stages of amyloidosis, before the onset of Aβ plaque deposit [22]. NP03 also reduced BACE1 hyperactivity, Aβ42 production, Wnt/β-catenin and GSK-3β signaling, and restored impaired CRTC1 signaling required for learning and memory as well as neurogenesis. A subsequent study, also in the McGill-R-Thy1-APP transgenic rat model, revealed that pre-plaque administration of NP03 quelled neuroinflammation and markers of cellular oxidative stress [23].
Further to the above, we are now interested in determining whether NP03 might have beneficial effects during the early Aβ post-plaque stage of the amyloid pathology. Investigating this period would allow us to examine the impact of NP03 on additional features of AD pathology—foremost being Aβ plaque deposition and cholinergic impairment—as these pathologies only appear at this later time point. For example, basal forebrain cholinergic neurons are selectively vulnerable to AD pathology [24–26] and McGill-R-Thy1-APP rats display a loss of cholinergic boutons at the post-plaque stage [27]. Accordingly, we administered NP03 and measured its effects on cognition, cholinergic boutons density, amyloid plaque pathology, oxidative stress, and neuroinflammation.
MATERIALS AND METHODS
Animals
All procedures were approved by institutional animal care committee at McGill University and were carried out under strict accordance to the rules set out by the Canadian Council of Animal Care. McGill-R-Thy1-APP transgenic rats and their wild type littermates were obtained from our animal colony. McGill-R-Thy1-APP transgenic rats express the human amyloid precursor protein gene (APP) with Swedish double and Indiana mutations [21]. The Swedish double and Indiana genetic mutations were identified in kindreds with an aggressive form of early onset AD [28, 29]. Rats were socially housed and maintained under 12-h light-dark schedule at 21°C, with free access to food and water. Male and female homozygous transgenic rats and their wild type littermates were used for this study. The number of animals used in each experiment is indicated in the figure legends.
Drug treatment
Animals were treated starting at 13 months of age for 3 months with vehicle (1 mL/kg) or NP03 (40μg Li/kg; 1 mL/kg) by deposit on the rectal mucosa. Drug administration was performed daily, 5 days per week. Experimental groups included wild-type littermates treated with vehicle, wild-type littermates treated with NP03, homozygous McGill-R-Thy1-APP Alzheimer transgenic rats treated with vehicle, and homozygous McGill-R-Thy1-APP Alzheimer transgenic rats treated with NP03. NP03 and empty vehicle were kindly provided by Medesis Pharma, Montpellier, France.
Behavior testing
For testing novel object recognition, rats were first habituated in a 220 cm×300 cm×500 cm black box covered with 2.5 cm of bedding material for a period of 5 min. Following at 30 min intertrial interval, rats were placed inside the box to explore 5 objects during the 2 min Familiarization Phase. Exploration was defined as the snout being in contact or within 2.5 cm of the object with clear whisking motion of the whiskers. We recorded the time exploring objects, and none of the rats showed a spontaneous preference to any of the 5 objects. Rats were returned to their home cage following the Familiarization Phase. Objects were mixed with soiled bedding material between trials to saturate odor cues. In the Test Phase, one of the familiar objects was replaced with a novel object. Thirty minutes later, the rats were replaced into the box for 2 min and a recognition index (RI) was calculated such that RI = (time exploring novel object)/(total time exploring all objects) × 100 and where chance performance was equal to an RI = 20%.
Brain tissue preparation
Briefly, rats were deeply anesthetized by intraperitoneal administration of a mix of chloral hydrate and sodium pentobarbital, (6.5 mg and 3 mg, respectively, per 100 g of body weight) and transcardially perfused with ice-cold 0.1 M phosphate buffer. The intact left hemisphere was post-fixed for 48 h in a 4% formalin solution in 0.1 M phosphate buffer, pH 7.4, then moved to 30% sucrose at 4°C until cryosectioning, while the remaining right hemisphere was dissected and tissue was frozen at –80°C until biochemical analysis.
Blood plasma preparation
Aortic blood was collected into K2 EDTA Microtainer tubes (Cat. # 365974; Becton, Dickinson and Company, NJ). 1 volume of prepared balanced salt solution was added into the blood tube. Ficoll-Paque PLUS (Cat. # 17-1440-02) was added to the bottom of a clean Falcon tube and the diluted blood sample was carefully layered on top. The sample was centrifuged at 400 g for 30 min at room temperature. The upper layer of plasma was collected and stored at –80°C.
VAChT bouton quantification
For vesicular acetylcholine transporter (VAChT) bouton quantification, tissue sections were washed 3×10 min in PBS, blocked in a 10% normal rabbit serum (Jackson ImmunoResearch, Cat# 011-000-120, 1:10,000) solution for 1 h before incubating in polyclonal goat anti-VAChT primary antibody (Millipore, Cat# ABN100) diluted in a 5% normal serum in PBS-T overnight at 4°C. The following day, tissue sections were incubated in goat anti-rabbit antibody (MP Biochemicals), followed by a rabbit anti-peroxidase monoclonal antibody complex (MAP/HRP complex, MediMabs) and developed using DAB as the chromogen (Vector Laboratories, Inc.). Sections were then processed through a graded alcohol series, defatted and cleared in xylenes, and coverslipped using Permount mounting medium (Fisher Scientific). Images were acquired under oil immersion using the 63X objective on an Axio Imager 2 microscope (Carl Zeiss Canada, North York, ON) running Zen Blue software (Carl Zeiss) by an experimenter blinded to treatment group. In order to quantify cholinergic bouton density, digital images from the CA1 region of the hippocampus were acquired in Z-stack 1 um each to span 11μm in total. Three images were captured per region from three sections per animal. Images were analyzed in an automated fashion using ImageJ (National Institutes of Health, USA) and a custom-designed macro. Briefly, immunoreactive signal was separated from background using optical density and size criteria. More precisely, the optical intensity threshold was established using the triangle algorithm [30]. Following thresholding, the watershed and despeckle filters were applied. Immunoreactive structures of a size above 0.1μm2 were included in the varicosity count. The varicosity counts are expressed as a density of boutons (number of boutons in an imaging field of 30,000μm2).
Thioflavin-S staining
Coronal sections of 40μm were first washed in distilled water. Sections were incubated in 0.1% thioflavin-S in 50% ethanol for 5 min at room temperature and then differentiated in 50% ethanol, rinsed in distilled water, and washed in PBS. Sections were coverslipped using Aqua/Poly Mount (Polysciences, Inc., Warrington, PA) aqueous mounting media. Photomicrographs were acquired at 10x magnification using the Axio Imager 2 microscope (Carl Zeiss Canada, North York, ON) using Zen Blue software (Carl Zeiss) by an experimenter blind to treatment group. Images were analyzed using ImageJ software (National Institutes of Health, USA). The subiculum was manually delineated as the region of interest (ROI), and color threshold adjusted using the Renyi’s entropy thresholding with black as a color threshold. The integrated density of the delineated ROI on the filtered imaged was measured using the Analyze Particles function and average across three sections spanning the anterior portion of the dorsal hippocampus per animal.
Amyloid electrochemiluminescent assay (ECLIA)
Levels of human Aβ38, Aβ40, and Aβ42 were measured in brain-extracts and plasma collected at the time of perfusion using the Aβ Peptide Panel 1 from Meso Scale Discovery (MSD, Rockville, MD; Cat: N45197A). Tris-soluble and insoluble fractions were prepared from cortical homogenates. First, approximately 20–25 mg of cortex tissue was homogenized in a Dounce homogenizer containing an 8 × volume of ice-cold 50 mM Tris-HCl buffer (150 mM NaCl, 50 mM Tris-HCl, 5 mM EDTA), pH 6.0, supplemented with protease and phosphatase inhibitors. Homogenates were spun at 100,000× g at 4°C for 1 h and the supernatant represented the soluble fraction. The resulting pellet was weighed and resuspended in 5 × volume of ice-cold 5 M guanidine-HCl solution (5 M guanidine-HCl, 50 mM Tris-HCl), also supplemented with protease and phosphatase inhibitors. Insoluble fractions were sonicated on ice 2×2 s at 40% amplitude with a 5-s pause between each sonication. Samples were incubated on ice for 30 min at 4°C before centrifuging 100,000× g at 4°C for 1 h. The collected supernatant represented the insoluble fraction. Soluble and insoluble cortex and plasma samples from each rat were analyzed. One NP03-treated soluble Aβ42 sample was excluded as an outlier as defined by more than 3 SD outside the mean. Samples were diluted 1:2 in Diluent 35 (MSD, Rockville, MD). Plates were processed following directions provided by the manufacturer, and were read on a Sector Imager and analyzed using Discovery Workbench software (MSD, Rockville, MD). For cortex samples, analyte concentration was corrected for protein concentration loaded, as determined by the Lowry assay, and are expressed as pg analyte per μg protein.
Immunofluorescent staining and confocal microscopy
Tissue sections containing the subiculum region of the hippocampus were washed with PBS and incubated at RT in 50% ethanol for 20 min. Following PBS-T wash, sections were blocked with 10% NGS in PBS-T for 1 h at room temperature (RT) and incubated at 4°C overnight with primary antibody (anti-4-hydroxynonenal (4-HNE): Abcam, ab46545; anti-IL-6: Abcam ab9324; Anti-CXCL1: US Biological, 141648) diluted in 5% NGS with PBS-T. Sections were washed and incubated with secondary antibody (ThermoFisher Scientific, A11037) conjugated to Alexa Fluor 594 diluted in PBS-T and 5% NGS for 2 h at RT. Following another wash (PBS-T), sections for 4-HNE staining were incubated with an anti-NeuN antibody (EMD Millipore, MAB377X) conjugated to Alexa Fluor 488 for 2 h at RT. All sections were washed in PBS-T, incubated with 0.3% Sudan Black for 5 min and washed in PBS-T then PBS before mounting and coverslipping with Aqua-Poly/Mount (Polysciences, Inc., Warrington, PA) aqueous mounting media. Photomicrographs were acquired at 20x magnification using a Zeiss AxioObserver Z.1 LSM800 inverted microscope (Carl Zeiss Canada, Toronto, ON) or LSM710 Confocal Laser Scanning Microscope (Carl Zeiss AD, Germany) running ZEN imaging software (Carl Zeiss v2.3 Black). The subiculum of the dorsal hippocampus was imaged in z-stack to acquire three images spanning 5μm total. Maximum intensity projection (MIP) images were created using Zen Black. To quantify neuron-specific 4-HNE staining, FIJI ImageJ software (National Institutes of Health, USA) was used to define neuron specific ROIs guided by the NeuN signal. To the NeuN channel, the despeckle function was applied followed by the Default threshold. Only particles equal to or larger than 10μm2 were included in the ROI mask by employing the size-exclusion function. This mask was applied to the 4-HNE channel, and the sum of the integrated densities (IntDen) for this ROI was then divided by the total area of the ROI selected, representing neuron-specific 4-HNE intensity. For IL-6 and CXCL1 mean fluorescence intensity of the total image area was quantified using the maximum intensity projection and measured using ImageJ.
Pro-inflammatory electrochemiluminescent assay (ECLIA)
Brain-extracts and plasma collected at the time of perfusion were analyzed for CXCL1 using the Proinflammatory Panel 2 (rat) from Meso Scale Discovery (MSD, Rockville, MD; Cat: N05059A). For cortical samples, approximately 30 mg of tissue was manually homogenized in 10 × volume of ice-cold RIPA buffer. Extracts were sonicated at 40% amplitude 2 × for 5 s twice, and were then incubated on ice for 15 min prior to centrifugation step of 13,000 rpm at 4°C to remove cellular debris. Plasma samples were prepared as described above. Samples were diluted 1:2 using Diluent 42 provided by the manufacturer. Plates were processed following directions provided by the manufacturer, and were read on a Sector Imager and analyzed using Discovery Workbench (MSD, Rockville, MD). For cortex samples, analyte concentration was corrected for protein concentration loaded, as determined by the Lowry assay, and are expressed as pg analyte per μg protein.
Statistical analysis
The effects of APP transgene and NP03 drug treatment were evaluated by performing 2-way analysis of variance (ANOVA) with Tukey’s post hoc tests. Means of two groups were compared by performing unpaired Student’s t-tests. Non-normally distributed data were analyzed using the non-parametric Mann-Whitney U test. All analyses were performed using GraphPad Prism software (GraphPad Software Inc., La Jolla, CA) and statistical significance was defined as p < 0.05.
RESULTS
NP03 reverses object recognition impairments
To determine whether NP03 might have beneficial effects during Aβ post-plaque stages of the amyloid pathology, we administered NP03 or vehicle to control wild-type and littermate AD transgenic rats spanning the preplaque to early post-plaque period (Fig. 1). First, we investigated the effect of NP03 on remote working memory as assessed using the novel object recognition (NOR) task. We observed equal exploratory behavior across all groups, with rats spending an average 74.2 s (±16.25 se), approximately 60% of allotted time during the familiarization phase, actively exploring a set of 5 unique objects (Fig. 2a). There was no spontaneous preference for or aversion to any of the objects. 30 min following the familiarization phase, one object out of five was replaced with a novel object, and rats were returned to the area to explore. While the wild-type vehicle- and NP03-treated groups displayed a clear preference for the novel object (mean recognition index = 38.9% and 37.0%, respectively, with chance recognition index equal to 20%), the AD transgenic rats were clearly impaired (2-Way ANOVA: Treatment×Transgene interaction: F (1,32) = 5.661; p = 0.0235; Tukey’s post hoc test: WT Veh versus TG Veh: p < 0.05), demonstrating only near-chance preference (Fig. 2b). In contrast, however, AD transgenic rats that had received NP03 performed significantly better than those that received vehicle (39.6%, TG Veh versus TG NP03: p < 0.05), and performed as well as wild-type rats, indicating that NP03 reverses the Aβ-driven cognitive deficits in these rats.

Schematic representing the experimental design. McGill-R-Thy1-APP transgenic and wild-type rats were treated for 3-months beginning at 13-months of age with NP03 (40μg Li/kg; 1 mL/kg) or vehicle formulation (1 mL/kg). The treatment period extended from the pre-plaque phase to the early Aβ plaque stage of the amyloid pathology.
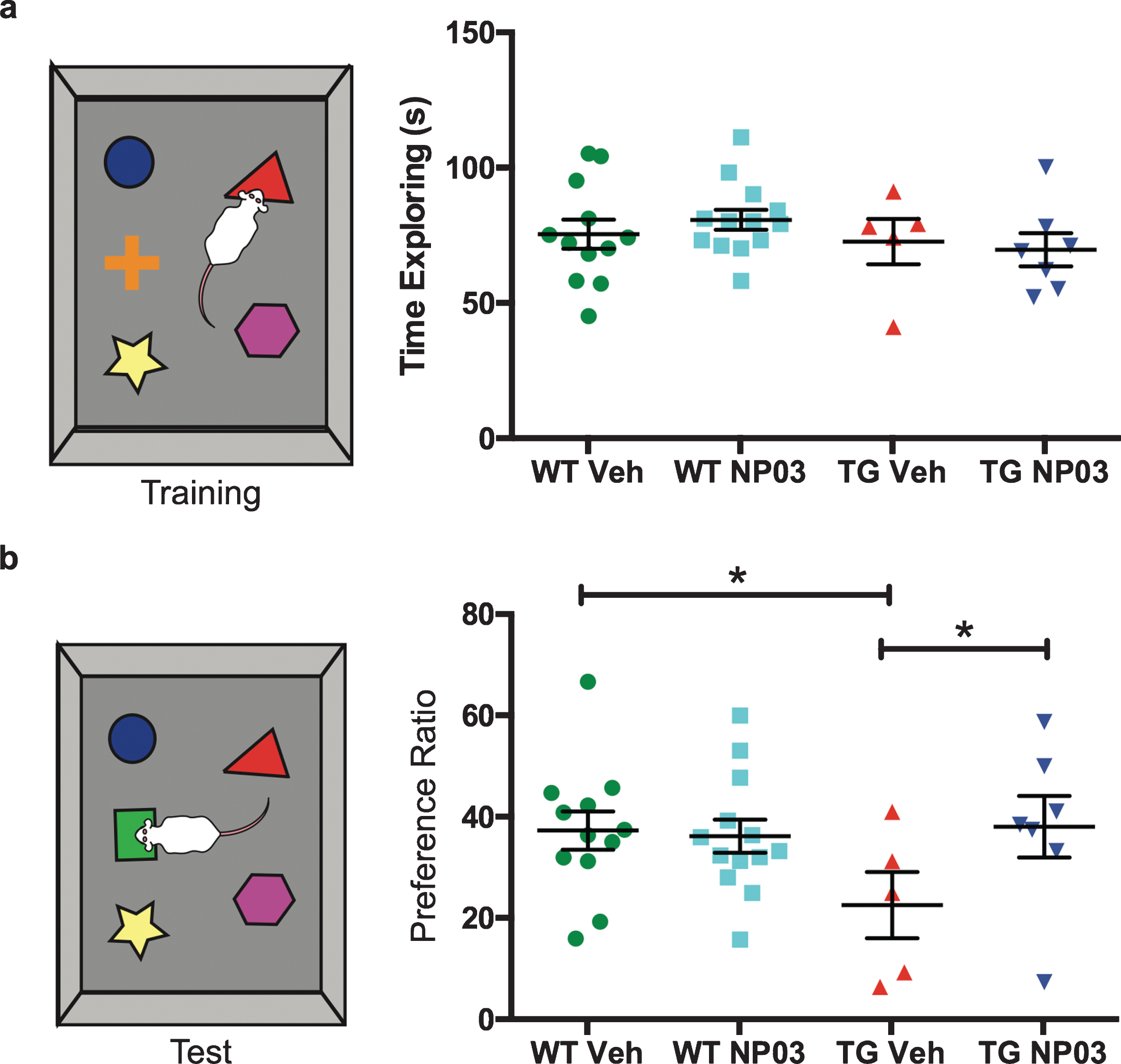
NP03 restores remote working memory in AD rats. a) In the Novel Object Recognition (NOR) task, all groups displayed a high level of exploratory behavior, spending approximately 60% of their time during the Familiarization Phase actively exploring the objects. b) Wild-type vehicle-treated rats displayed a clear preference for the novel object, with a recognition index of ∼40%. In contrast, Alzheimer transgenic vehicle-treated rats were significantly impaired, and showed a near-chance level preference for the novel object, ∼20% (green dashed line). NP03 treatment completely restored novel object recognition in the Alzheimer transgenic rats. Data analyzed using 2-way ANOVA with Tukey’s post hoc for multiple comparisons. *p < 0.05.
NP03 protects against cholinergic bouton loss in McGIll-R-Thy1-APP rats
McGill-R-Thy1-APP rats at the post-plaque stage have been shown to have a loss of cholinergic boutons at the post-plaque stage [27]. Accordingly, we investigated possible cholinergic dysfunction and the effect of NP03 at the early post-plaque stage by quantifying expression of the synaptic vesicular acetylcholine transporter (VAChT) in the CA1 layer of the hippocampus, a region strongly innervated by ascending cholinergic projections [31, 32]. This analysis revealed that at the early Aβ post-plaque stage, the number of VAChT-immunoreactive boutons was significantly reduced in AD transgenic rats (Fig. 3a,b; 2-Way ANOVA: Treatment × Transgene interaction: F (1,34) = 10.93; p = 0.0022; Tukey’s post hoc test: WT Veh versus TG Veh: p < 0.0001; WT NP03 versus TG Veh: p < 0.001). The application of NP03 reduced this loss in AD transgenic rats. However, while varicosity number in AD NP03-treated rats reflected a loss compared to the wild type control rats (WT Veh versus TG NP03: p < 0.05), it was nevertheless significantly improved compared to AD rats receiving vehicle (TG Veh versus TG NP03: p < 0.05), indicating that NP03 diminishes cholinergic boutons loss in these rats.
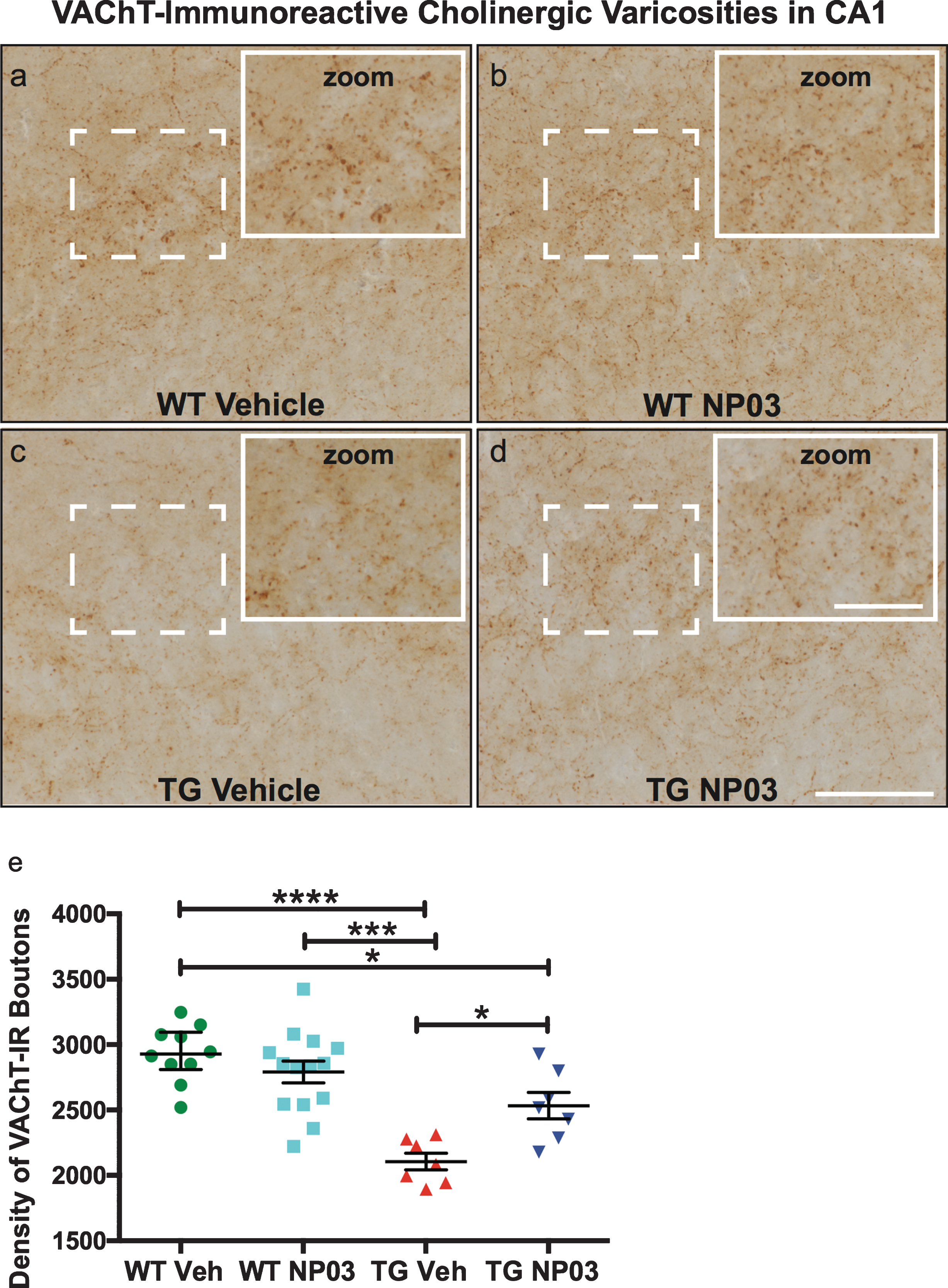
NP03 protects cholinergic varicosities in CA1 region of the hippocampus. a-d) Photomicrographs showing expression of VAChT in the CA1 regions of the hippocampus of NP03-treated and control rats. Scale = 50μm (inset 25μm). e) In the CA1 layer of the hippocampus the density of VAChT-immunoreactive varicosities was significantly reduced in AD rats. Although remaining lower compared to the WT, cholinergic varicosity density was significantly higher in AD rats receiving NP03 compared to vehicle. Data were analyzed using 2-way ANOVA with Tukey’s multiple comparisons post hoc test. *p < 0.05, ***p < 0.001, ****p < 0.0001.
NP03 reduces soluble and aggregated Aβ42 in brain and plasma
The McGill-R-Thy1-APP transgenic rat expresses the mutated human APP gene associated with aggressive early-onset AD, to mimic the amyloid-driven aspects of AD pathogenesis [21]. In this model, regulated sequential cleavage by the β-site amyloid precursor protein cleaving enzyme 1 (BACE1) of mutated AβPP leads to the misproduction of toxic Aβ42 amyloidogenic peptides, which are prone to aggregation [33, 34]. Rats therefore accumulate Aβ plaques beginning in the subiculum of the hippocampus, then in layers II and III of the entorhinal cortex, before depositing throughout the hippocampus and neocortex [21, 35–39]. We investigated whether the protective effects of NP03 on NOR and cholinergic boutons was related to a reduction of Aβ pathology at the early Aβ post-plaque stage. Compared to vehicle-treated rats, we found NP03 significantly reduced thioflavin-S-positive Aβ aggregates in the subiculum region of the hippocampal formation (Student’s t-test, t (13) = 3.001; p = 0.0051), one of the first regions in these AD transgenic rats to show plaque deposition (Fig. 4a,b). On exploring further the contents of soluble and insoluble Aβ peptide pools, we found that while not affecting levels of Aβ40 or Aβ38, NP03 significantly reduced levels of both TBS- and guanidine-soluble Aβ42 peptides (Student’s t-test, TBS-soluble: t (10) = 2.130; p = 0.0295); Guanidine-soluble: (t (10) = 1.881; p = 0.0433; Fig. 4b,d,f). We also observed a significant reduction in Aβ42 in plasma collected from NP03-treated AD rats (t (13) = 1.913; p = 0.0390; Fig. 4c), but no significant changes were observed in plasma Aβ40 or Aβ38 (Fig. 4c,e,g). Together, these data support that when administered during the early Aβ plaque-deposition stage, NP03 diminishes Aβ pathology in the brain and in the periphery.
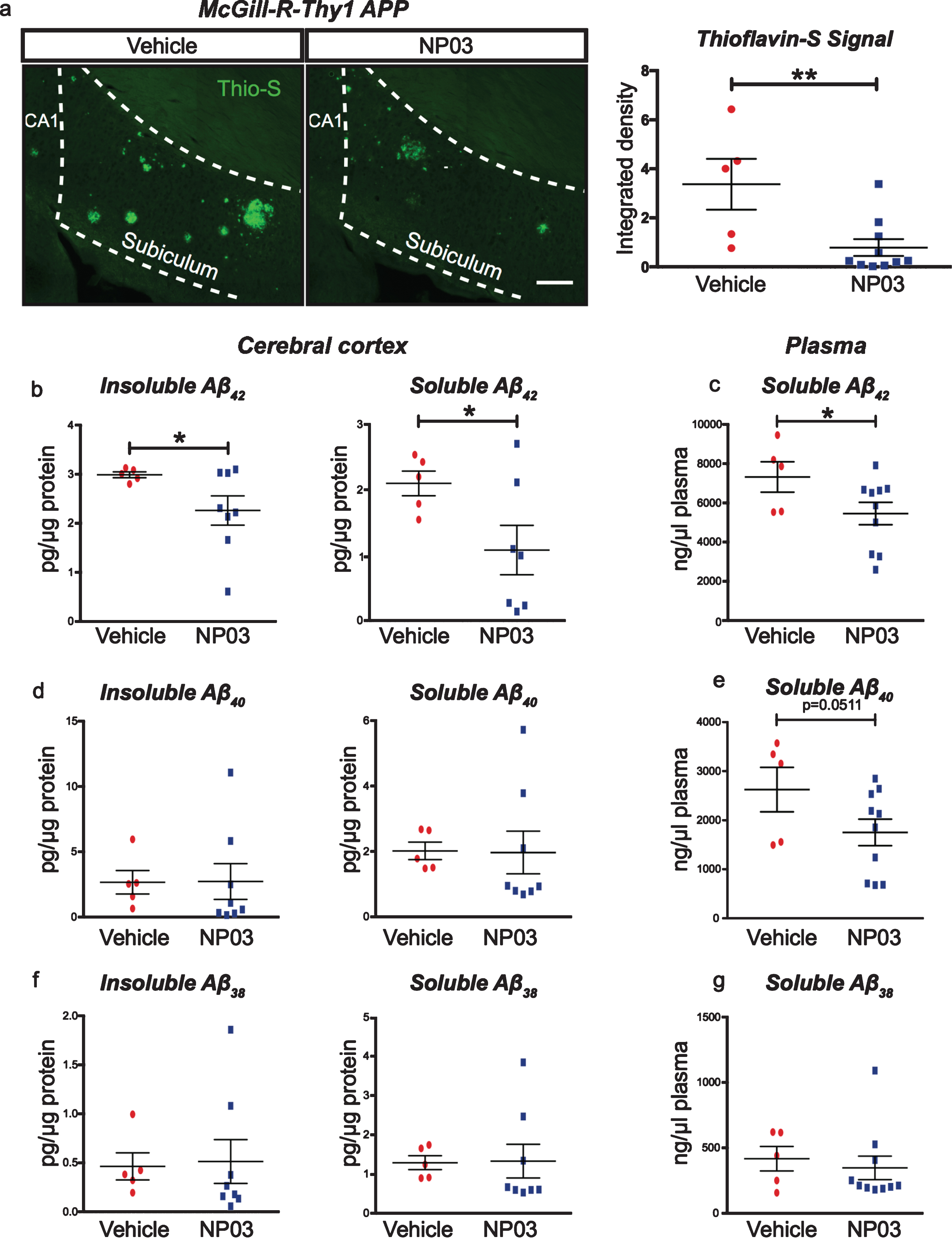
NP03 reduces mature Aβ plaques, insoluble and soluble Aβ42. a) Representative photomicrographs and quantification of Thioflavin-S staining. Thioflavin-S signal intensity in the subiculum was measured as integrated density, and was significantly reduced by NP03. Scale = 200μm. b) Cortical insoluble Aβ levels were also reduced with NP03 treatment, as were levels of soluble Aβ42. c) A similar reduction in Aβ42 was observed in plasma after NP03 treatment. d-g) No significant differences were observed in Aβ40 or Aβ38 in cortex or plasma, indicating the action of NP03 is specific to processing events affecting only Aβ42 production. Data were analyzed using unpaired Student’s t-test. *p < 0.05, **p < 0.01.
NP03 lowers oxidative stress marker 4-HNE in pyramidal neurons of the CA1
To assess whether NP03 modified oxidative stress in AD rats, we quantified levels of the oxidative stress marker 4-HNE, a major product of lipid peroxidation. 4-HNE-protein adducts are detected in the brains of AD patients [40, 41], and we previously reported protein-bound 4-HNE levels to be significantly increased in cortical tissue of pre-plaque AD transgenic rats and reduced with NP03 treatment [23]. By co-fluorescent immunolabeling using a neuron-specific antibody, we found that neuronal 4-HNE in the dorsal CA1 deep pyramidal layer was significantly increased in vehicle-treated AD transgenic rats compared to the vehicle-treated wild-type rats (Fig. 5a,b; 2-Way ANOVA: Main effect of Transgene: F (1,36) = 14.37; p = 0.0006) and NP03 treatment (2-Way ANOVA: Main effect of Treatment: F (1,36) = 7.02; p = 0.0119; Tukey’s post hoc test: WT Veh versus TG Veh: p < 0.05; WT NP03 versus TG Veh: p < 0.001). NP03 reduced the elevation of 4-HNE in AD transgenic rats to normal levels (Tukey’s post hoc test: WT Veh versus TG NP03: p > 0.05), indicating that NP03 reduces oxidative damage during the early Aβ post-plaque stage of the amyloid pathology in these rats.
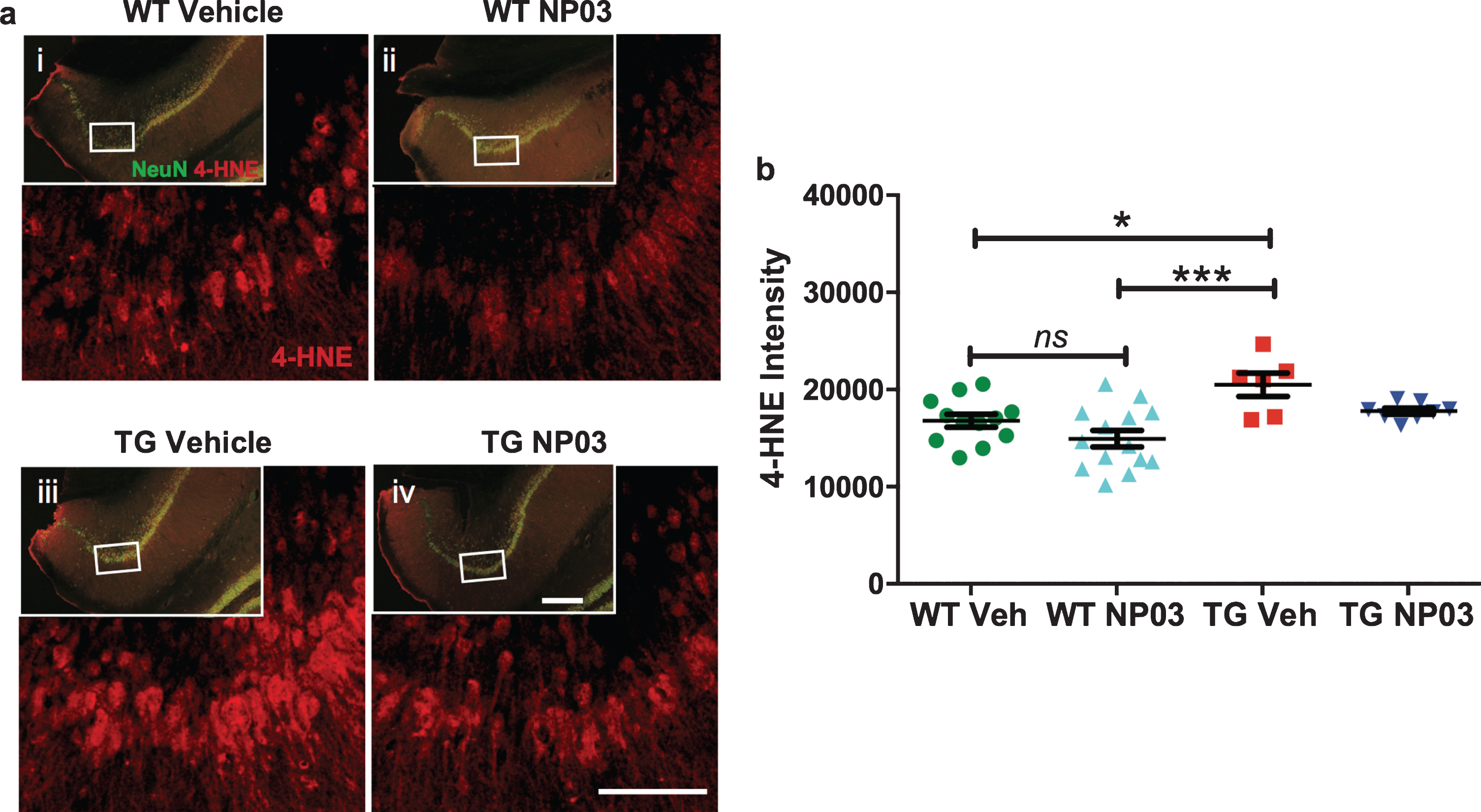
NP03 lowers elevated oxidative stress marker 4-HNE in neurons of the CA1 region of the hippocampus. a) Photomicrographs showing immunofluorescent staining and 4-HNE in the subiculum region of the hippocampus. Neuronal 4-HNE levels were determined by co-labeling with the neuron-specific antibody NeuN (insets). Scale = 100μm (inset = 200μm). b) Graph showing neuronal 4-HNE increased in vehicle-treated APP rats compared to the wild-type receiving vehicle and NP03. NP03 reduced the elevation of 4-HNE in AD rats to normal levels. Data were analyzed using 2-way ANOVA with Tukey’s multiple comparisons post hoc test. *p < 0.05, ***p < 0.001.
NP03 reduces pro-inflammatory mediators in CA1
We next measured expression of innate immune markers interleukin-6 (IL-6) and the chemokine (C-X-C motif) ligand 1 (CXCL1) [42]. IL-6 transcriptionally upregulates CXCL1, which is expressed by neurons and endothelial cells [43, 44]. Increases in CXCL1 precede peripheral monocyte and neutrophil brain incursion and contributes to Aβ-mediated transendothelial migration of monocytes [45]. We observed significantly elevated IL-6 protein expression in AD transgenic rats compared to the wild-type rats (Fig. 6a,c,e; 2-Way ANOVA: Main Effect Treatment: F (1,31) = 12.93; p = 0.0011, Main Effect Transgene: F (1,31) = 18.27; p = 0.0002; Tukey’s post hoc test: WT Veh versus TG Veh: p < 0.05; WT NP03 versus TG Veh: p < 0.0001). Furthermore, NP03 significantly reduced IL-6 production in AD transgenic rats (Fig. 6d; TG Veh versus TG NP03: p < 0.01), restoring it to baseline levels (WT Veh versus TG NP03: p > 0.05). Similarly, we observed a significant 36% increase in CXCL1 production in CA1 in AD transgenic rats (Fig. 7a,b; 2-Way ANOVA: Treatment×Transgene interaction: F (1,30) = 13.88; p = 0.0008; Tukey’s post hoc test: WT Veh versus TG Veh: p < 0.01). This increase was reversed with NP03 treatment (TG Veh versus TG NP03: p < 0.01; WT Veh versus TG NP03: p > 0.05). Moreover, CXCL1 was significantly increased in plasma obtained from AD rats (Fig. 7c; Kruskal-Wallis: H (4) = 8.492; p = 0.0369; Tukey’s post hoc test: WT Veh versus TG Veh: p < 0.05). NP03 treatment prevented this increase in plasma CXCL1 levels. Taken together, these experiments reveal that NP03 reduces hippocampal IL-6 expression in CA1 and CXCL1 levels both in the hippocampus and in circulation.

NP03 lowers elevated pro-inflammatory cytokine IL-6 in the subiculum. a-d) Immunofluorescent staining of the CA1 region of the hippocampus. Scale = 100μm. e) We observed significantly elevated IL-6 protein expression in AD rats (TG Veh) compared to the wild-type rats (WT Veh and WT NP03). NP03 significantly reduced IL-6 production in AD rats, restoring it to baseline levels. Data were analyzed using 2-way ANOVA with Tukey’s multiple comparisons post hoc test. *p < 0.05, **p < 0.01, ****p < 0.0001.
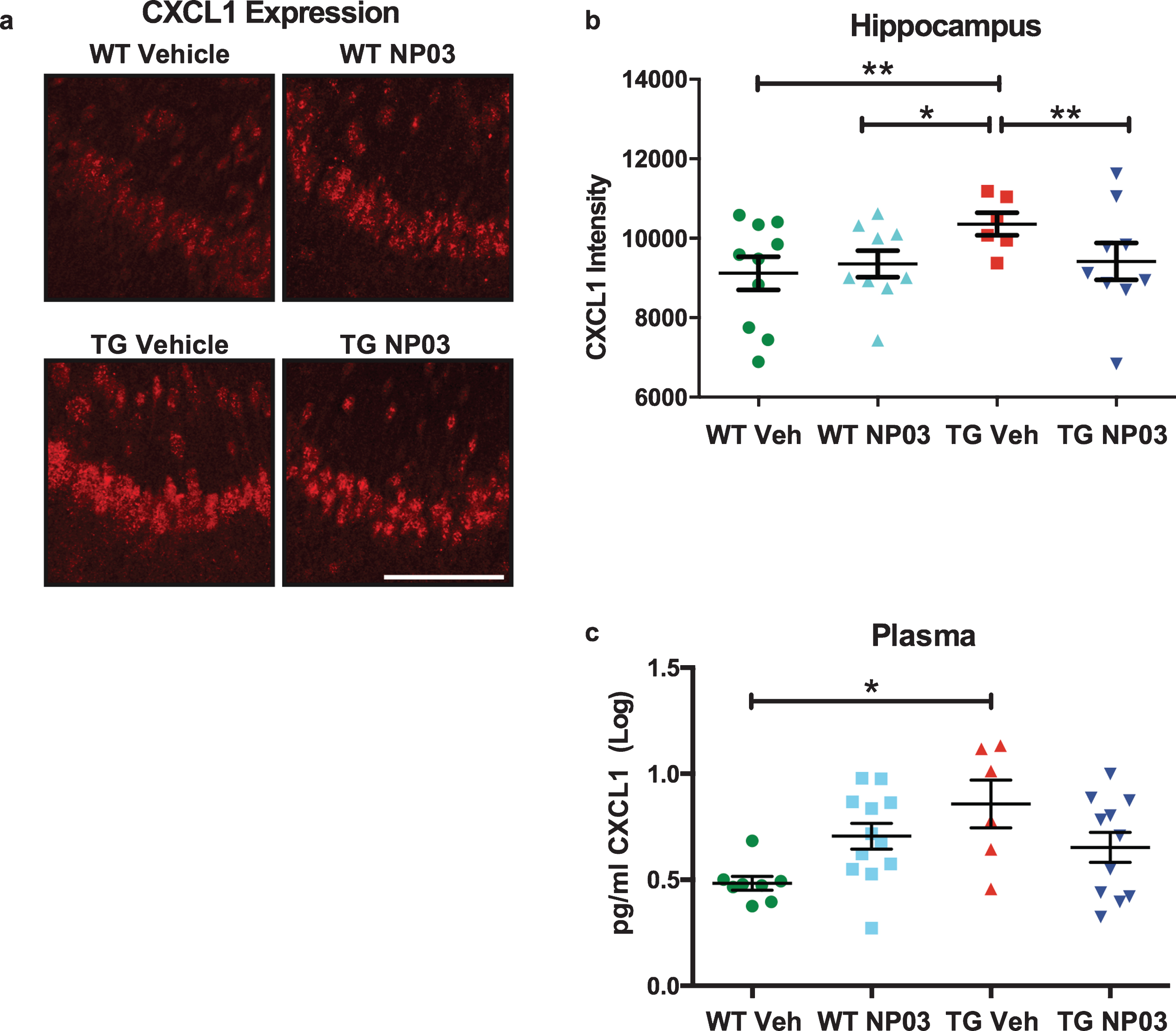
NP03 reduces pro-inflammatory chemokine CXCL1 protein levels in hippocampus and blood. a) Immunofluorescent staining of the pro-inflammatory chemokine C-X-C motif ligand 1 (CXCL1) in the CA1 region of the hippocampus. Scale = 100μm. b) We observed significantly elevated CXCL1 expression in the subiculum of AD rats (TG Veh) compared to the wild-type rats (WT Veh and WT NP03). NP03 significantly reduced CXCL1 production in AD rats, restoring it to baseline levels. c) Plasma CXCL1 was measured by ECLIA and revealed a significant elevation over wild-type levels, while plasma CXCL1 was not significantly elevated in the AD rats receiving NP03. Data were analyzed using 2-way ANOVA with Tukey’s multiple comparisons post hoc test. *p < 0.05, **p < 0.01.
DISCUSSION
In a series of studies we have been documenting the impact of microdose lithium NP03 on the sequential stages of the AD-like amyloid pathology in McGill-R-Thy1-APP rats [22, 23]. Initial findings indicated that, when applied at the early pre-plaque pathological stages where the amyloid accumulation is restricted to the intraneuronal compartment, NP03 blunts the amyloid-driven AD-like pathology and may have potential as a preventive therapy [22, 23]. In the present study, we examined the effect of NP03 at later stages of the AD-like pathology, when extracellular amyloid deposition has started and cognitive deficits have progressed. We found here that at the early Aβ post-plaque stage, NP03 reversed memory deficits, slowed the loss of cholinergic boutons, reduced levels of soluble Aβ and mature plaques, and reduced markers of oxidative stress and neuroinflammation. Together these results indicate that the positive effects of microdose lithium NP03 extend into the early Aβ post-plaque stage of the amyloid pathology. They also point to a possible translational use of NP03 to rescue AD-like cognitive deficits at pathological stages where mature amyloid plaques are detectable in the subiculum and plasma Aβ42 levels are decreased [46, 47].
The relief of cognitive decline by NP03 is accompanied by the rescue of the central features of AD. Specifically, our results suggest that microdose lithium NP03 is capable of preserving cholinergic function by reducing the loss of cholinergic boutons. The cholinergic hypothesis of AD holds that the progressive loss of limbic and neocortical cholinergic innervation contributes significantly to the cognitive decline associated with AD [48, 49]. In transgenic models, cholinergic neurites appear to be most vulnerable to Aβ, followed by the glutamatergic terminals and GABAergic terminals [50]. The protection of cholinergic boutons is encouraging, given that cholinergic failure in AD is linked with a large number of other pathological processes, including Aβ and tau deposition [51–55], AβPP processing [56, 57], blood-brain barrier integrity [58], and lymphatic drainage [59], among others. Protection of cholinergic synapses should have a beneficial effect on cognitive outcomes independent of co-morbidities.
NP03 significantly reduced soluble and insoluble Aβ42 in cortex and thioflavin-S-positive mature Aβ plaques in the hippocampus. No differences were observed in Aβ40 or Aβ38 in cortex, suggesting that the action of NP03 in this context appears as being specific to processing events affecting preferentially Aβ42 production. BACE1 inhibitors decrease cerebrospinal fluid (CSF) and plasma levels of Aβ40 and Aβ42 in mice, guinea pigs, and non-human primates [60–64], and we have previously shown that NP03 inhibits BACE1 activity through Wnt/β-catenin signaling [22]. Thus, it is likely through its action on BACE1 that NP03 diminished production of Aβ42. We have also previously investigated the phosphorylation status of GSK-3β, a multifunctional serine/threonine kinase widely expressed in brain and linked to AD-like amyloid pathology by assessing the relative phosphorylation of GSK-3β at serine 9 compared to total GSK-3β as an indicator of GSK-3β inactivation [65]. Using quantitative western blotting, we reported that the ratio of phospho-GSK-3βSer9 to total GSK-3β reflected diminished inhibitory GSK-3β phosphorylation in AD-like transgenic rats which is restored to normal levels in the hippocampus of NP03-treated transgenic rats. Alternatively, given its effects on β-catenin and GSK-3β, NP03 may decrease Aβ42 levels by directly impacting γ-secretase activity, a process yet to be examined. Indeed, Aβ peptides are derived from the sequential cleavage of AβPP by BACE1 and then by presenilin-dependent γ-secretase. Presenilins interact with α-catenin, β-catenin, and GSK-3β and it has been shown that lithium interferes with the cleavage of AβPP by γ-secretase [66].
We also observed a reduction in plasma Aβ42 in AD rats receiving NP03. Emerging evidence supports the putative use of plasma Aβ42 as a biomarker in AD. It was recently reported that plasma Aβ42 and Aβ40 levels were lower in AD and amnestic mild cognitive impairment (MCI) than in nonamnestic MCI individuals, and were correlated with cognitive status and CSF biomarkers [46]. Further to it, Nakamura and colleagues [67] used the ratios of APP669–711:Aβ42 and Aβ40:Aβ42 as well as the composite of these ratios as biomarkers in two cohorts comprising cognitively healthy individuals, individuals with MCI and individuals with AD. All peptide ratios significantly correlated with brain Aβ burden, as measured by PIB PET, and CSF Aβ42 level, with the composite of the two peptide ratios rendering the best biomarker performance overall. It has also been reported that plasma Aβ40:Aβ42 ratio predicts cerebral Aβ PET status in cognitively normal individuals at risk for AD [68]. Nevertheless, a measure of caution must be exercised when interpreting plasma Aβ peptides levels as several lines of evidence suggest a dynamic bimodal regulation of plasma Aβ42 levels as pathology evolves. For instance, it was reported that concentrations of plasma Aβ40 and Aβ42 are significantly higher in adults with Down’s syndrome (DS) who inevitably develop early AD pathology because of APP gene triplication [69] compared to adults with sporadic AD and controls age-matched to the DS group [70]. Further to it, our group has shown an early rise in Aβ in plasma of DS subjects which later declines, coinciding with advancing preclinical pathology and cognitive decline [71]. This early rise in Aβ is reflected in DS CSF [72], which is similarly followed by a decline in mid-life [73–75]. Together, the timeline of plasma Aβ dynamics seems to suggest that plasma Aβ42 would first increase in early pathological stages inherent to the accruing levels of Aβ42 in the brain, followed by a gradual decrease in plasma Aβ42, likely reflecting sequestration of Aβ monomers into plaques as the pathology begins to accelerate (‘sink phenomenon’). An experiment using 3xTgAD mice also suggested an age-dependent inverse correlation between plasma and CSF Aβ42 levels with advancing pathology up to the point of the pre-plaque to post-plaque transition stage [76].
It is likely that NP03 might have an effect on tau pathology. In this regard, it is worth noting that the application of NP03 in a model of Huntington disease resulted in a reduction of tau phosphorylation at GSK-3 residues Thr205 and Ser396, which coincided with GSK-3β inhibition [20].
The oxidative stress marker 4-HNE is a major product of lipid peroxidation with 4-HNE-protein adducts a prominent feature in AD brains [40, 77]. While HNE reacts with various cellular components, including DNA and other molecules, proteins appear to be the preferential target of modification caused by HNE [78]. Redox proteomic studies have identified a series of proteins as being modifiable by HNE, a growing list that includes plasma membrane transporters, growth factor receptors, neurotransmitters, mitochondrial electron transport chain proteins, molecular chaperones, proteasomal proteins, and cytoskeletal proteins [79, 80]. Interestingly, Aβ is also modified by HNE adducts, with evidence suggesting this may be an alteration that accelerates Aβ protofibril formation [81]. Beyond amyloid, studies have identified HNE-modified proteins in AD brain hippocampus and cortex, including glutamine synthase, aconitase, aldolase, peroxiredozin 6, and α-tubulin at late stage AD [41], and lactate dehydrogenase, phosphoglycerate kinase, heat shock protein 70, α-actin, elongation factor Tu, translation initiation factor α, malate dehydrogenase, triose phosphate isomerase and F1 ATPase at early onset AD and MCI stages [82, 83]. Together, these early and late AD modifications appear to implicate an impairment in energy metabolism. We previously reported that NP03 reduced elevated cortical protein-bound 4-HNE levels in cortical pre-plaque McGill-R-Thy1-APP rats [23]. Here we show that NP03 is also capable of reducing hippocampal neuron 4-HNE levels to baseline levels at early post-plaque stages. This indicates that NP03 remains capable of reducing oxidative stress from the initial intraneuronal pathology [23], as well as in the stage of early Aβ plaque pathology.
Similarly, we measured levels of the proinflammatory cytokine IL-6 and found it to be reduced with NP03 treatment. Furthermore, it is promising that NP03 reduced, both in the brain and blood, CXCL1, a chemokine associated with transendothelial migration of monocytes in AD [45]. Preventing monocyte migration from blood to brain in AD may reduce maladaptive, late CNS inflammation [84], and promote resolution of the immune response. CXCL1 also leads to the activation of GSK-3β, prompting caspase-3 activation and tau cleavage [85]. Therefore, NP03 reduction of CXCL1 should have beneficial effects not only by possibly reducing infiltration of peripheral monocytes, but also through inactivation of multi-target GSK-3β and caspase-3, key mediators of AD pathogenesis.
In summary, we confirm that microdose lithium formulation NP03 is effective in reducing components of the AD-like pathology in the McGill-R-Thy1-APP rat model of AD during the early Aβ post-plaque stage. NP03 rescues functional deficits in object recognition, reduces loss of cholinergic boutons in the hippocampus, reduces cortical Aβ42 production and hippocampal Aβ plaque burden. NP03 reduces markers of neuroinflammation and cellular oxidative stress in the CA1 region of the hippocampus. The effectiveness of microdose lithium at early Aβ plaque-forming stages supports further preclinical investigation into the prophylactic effect of NP03 at still later pathological stages, and supports its investigation as a possible therapeutic agent at late preclinical or at the earliest clinical stages of AD.
Our findings also illustrate that, beyond what is currently known regarding the pharmacology of high doses of lithium, as currently clinically applied, there is a new scenario regarding the pharmacology of microdoses of lithium. It took over 30 years to define the multiplicity of targets modulated by lithium at such clinical concentrations which are likely to recruit both low and high receptor sites. From our studies, one can signal that lower doses of lithium engaged targets of high relevance such as inflammation and neurogenesis, Wnt/β-catenin, GSK-3β, and CRTC1 signaling pathways and BACE-1, the latter resulting in lower Aβ pathology, and can likely affect tau pathology given its effects on GSK-3β [22, 23]. In short, future pharmacological research should decipher the specific (high affinity) targets responding to microdose lithium.
Footnotes
ACKNOWLEDGMENTS
We are grateful to the McGill University Life Sciences Complex Advanced BioImaging Facility (ABIF) for use of their facilities. This work was funded by CIHR grants (285643MOP126012 and 201603PJT364544) to ACC. ACC acknowledges the Charles E. Frosst/Merck endowed Chair in Pharmacology and SDC the Charles E. Frosst/Merck Research Associate Position. LAW is the recipient of a doctoral training fellowship from the Fonds de Recherche du Québec-Santé. HH is the past recipient of a Fonds de Recherche du Québec-Santé postdoctoral fellowship. LFA is the recipient of a PBEEE Doctoral Merit Scholarship from the Fonds de Recherche du Québec–Nature et technologies and a doctoral scholarship granted by CONACyT, Mexico. MKF is the holder of a Frederick Banting and Charles Best CIHR doctoral award. MFI was supported by a Doctoral Award from the Alzheimer’s Society of Canada. ACC is a member of the Canadian Consortium on Neurodegeneration in Aging (CCNA).