
Abstract
Alzheimer’s disease (AD) is a neurodegenerative process characterized by loss of neurons in the hippocampus and cerebral cortex, leading to progressive cognitive decline. Pathologically, the hallmark of AD is accumulation of “senile” plaques composed of amyloid-β (Aβ) protein surrounding neurons in affected regions. Despite extensive research into AD pathogenesis and therapeutic targets, there remains no breakthroughs in its management. In recent years, there has been a spark of interest in the connection between the brain and gastrointestinal tract, referred to as the brain-gut axis, and its potential implications for both metabolic and neurologic disease. Moreover, the gastrointestinal flora, referred to as the microbiome, appears to exert significant influence over the brain-gut axis. With the need for expanded horizons in understanding and treating AD, many have turned to the brain-gut-microbiome axis for answers. Here we provide a review of the brain-gut-microbiome axis and discuss the evidence supporting alterations of the axis in the pathogenesis of AD. Specifically, we highlight the role for the microbiome in disruption of Aβ metabolism/clearance, increased permeability of the blood-brain barrier and modulation of the neuroinflammatory response, and inhibition of hippocampal neurogenesis. The majority of the above described findings are the result of excellent, albeit basic and pre-clinical studies. Therefore, we conclude with a brief description of documented clinical support for brain-gut-microbiome axis alteration in AD, including potential microbiome-based therapeutics for AD. Collectively, these findings suggest that the brain-gut-microbiome axis may be a “lost link” in understanding and treating AD and call for future work.
INTRODUCTION
Neurodegenerative disease is the term to describe a class of conditions, including Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD), characterized by progressive loss of neurons, primarily in the brain, resulting in symptoms ranging from motor ataxias to cognitive dysfunction. Neurodegenerative disease poses a substantial emotional and financial burden on the patient, their family, and society, especially with the increasing aging population [1]. AD is a type of dementia characterized pathologically by cell loss in the hippocampus and cerebral cortex as well as accumulation of insoluble amyloid-β (Aβ) plaques in the interstitium and tau neurofibrillary tangles in neurons. AD is the sixth leading cause of death in the United States, with an estimated incidence of 1–3% and a prevalence of 10–30% of the population > 65 years of age, owing to its mean duration of 10 years [2, 3]. Further, the societal burden of AD is hypothesized to increase tremendously, with the prevalence in the United States increasing from 5.8 million to nearly 14 million and the cost increasing from $290 billion to $1.1 trillion by 2050 [2]. While the pathological hallmarks of AD are well described, the underlying pathogenesis of the disease remains controversial, with multiple hypotheses regarding its etiology proposed [4]. The interaction of the brain and gut microbiome, termed the brain-gut-microbiome axis, has been described in recent years as a model for understanding brain health and disease. Reports of gut microbiome alterations in patients with AD have accumulated a great deal of interest due to their potential to explain various features of the disease process as well as provide a new target for therapeutics [5]. We here provide a review of the evidence supporting a role for alterations along the brain-gut-microbiome axis in the pathogenesis of AD, with a focus on the concordance between these alterations and previously described pathogenic mechanisms. We then conclude with a discussion of the potential for microbiome-targeted therapeutics in the management of AD.
OVERVIEW OF THE BRAIN-GUT-MICROBIOME AXIS
The role of the brain-gut-microbiome axis in health and disease has been a popular source of attention for basic and clinical research, as well as in the media. The concept of an interconnection between the gut and the brain dates as far back as the nineteenth century, with physicians of the time reporting on such conditions as “neurasthenia gastrica” and “autointoxication”, in which it was hypothesized that “nervous weakness” may result in impaired digestion, or conversely that impaired digestion may permit accumulation of toxins from the intestines, including microbes, that disrupt nervous system activity [6]. In 1986, Hegstrand and Hine reported significant differences in brain histamine levels between nephrectomized germ-free (GF) and conventionally housed animals, providing some of the first evidence that changes in microbiome composition can influence brain chemistry [7]. This was further supported by the finding that the hypothalamic-pituitary-adrenal (HPA) stress response is exaggerated in GF mice but can be partly corrected with fecal reconstitution using feces from specific pathogen-free (SPF) animals [8]. Continued study of this gut-brain connection has yielded discovery of three distinct, but parallel, pathways for bidirectional communication along the axis: 1) neural, via direct connection through the enteric nervous system (ENS), 2) endocrine, via hormonal signaling, most notably via the HPA system, and 3) immune, via microbial release of inflammatory factors and modulation of mucosal and systemic immunity [9, 11].
Development, structure, and function of the gut microbiome and intestinal barrier
The gut has long been reported to harbor as many as 1014 bacterial cells, accounting for the reputed 10:1 ratio of bacterial to human cells often highlighted in the literature. However, recent calculations have determined the total number of bacterial cells to be approximately 3.8×1013, with 1011 in the gastrointestinal (GI) tract, placing the above ratio at 1.3:1 [12]. In addition to being quite robust, the human gut microbiome is also highly diverse, with one large sequencing study identifying between 1,000 and 1,150 different bacterial species across the cohort and at least 160 different species in each subject [13]. In contrast to previously held dogma, the development of such a robust and diverse microbiome begins as early as the in utero period, supported by the finding that meconium samples, representing material ingested or secreted by the GI tract during fetal life, obtained from neonates demonstrated the presence of conserved microbiota that is unaffected by perinatal, but rather maternal factors [14]. By performing high-throughput sequencing of meconium from term newborns, Gosables et al. identified two distinct types of meconium, the first being enriched with enteric bacteria including the genera Escherichia and Shigella, while the second was enriched with lactic acid bacteria including the genera Leuconostoc, Enterococcus, Lactococcus, Staphylococcus, and Streptococcus. Notably, alterations in this early microbiota are associated with clinical conditions, including neonatal jaundice [16], premature birth [17], and maternal diabetes [18]. Continued development of the gut microbiome occurs throughout the first years of life, with two major life events dictating maturation to an adult-like composition: 1) mode of delivery (i.e., C-section versus vaginal) and 2) feeding pattern (i.e., breast-feeding versus formula) [19]. While vaginal delivery tends to promote an infant microbiome rich in Lactobacillus, Prevotella, and Sneathia species, similar to the maternal microbiome due to vertical transfer of vaginal and perianal microbes, C-section yields a microbiome dominated by Staphylococcus, Corynebacteria, and Propionibacterium, resulting from horizontal transfer of maternal skin flora [20]. The microbiome converges toward its mature adult state by the end of the infant period (3–5 years of life), with factors such as geographical location, home environment, diet, and antibiotic exposure, modifying its final composition [21]. The adult microbiome demonstrates signature diversity along the various regions of the GI tract, and continues to be modified by diet and lifestyle factors [22]. A summary of the changes in the microbiome composition across the lifespan as well as relevant sources is provided in Table 1. The function of the gut microbiome is extensive, with metabolic capabilities including synthesis of vitamins (e.g., vitamin K) [23], synthesis and catabolism of amino acids [24], and formation of short chain fatty acids (SCFAs) which may be used as energy substrates and signaling molecules [25]. Beyond metabolism, the gut microbiome also plays a role in modulating immune system development and function by promoting formation of gut-associated lymphoid tissues (GALT), maintaining the intestinal epithelial barrier, inducing secretory IgA production, and conditioning intestinal mononuclear phagocytes and T-cells [26].
Composition of microbiome across lifespan
Prenatal is subdivided into Types A and B. Infant is subdivided by delivery type (vaginal or Cesarean). Adult is subdivided by region of gastrointestinal tract. *Most common; **High inter-subject variability; *** 70% of all flora.
The GI tract is the largest interface between the external world and the body; while it is primarily considered for its role in food and nutrient processing, this exposure is also critical for modulation of immune function. The intestinal epithelium is a dynamic physical barrier that acts as a component of the innate immune system to prevent exposure to harmful antigens and pathogens. Additionally, the epithelium must be selectively permeable to permit translocation of ingested nutrients into the bloodstream. The barrier is composed of a single epithelial layer lining the GI tract lumen, with cells bound to one another by tight junctions, adherens junctions, and desmosomes [27]. Tight junctions, the most apically located intercellular connections, are composed of multi-protein complexes that regulate paracellular permeability and maintain integrity of the epithelial barrier. Tight junctions are composed of diverse proteins, including occludin, claudin, junctional adhesion molecule (JAM), and tricellulin, which interact with intracellular scaffolds (i.e., zona occludens) to be anchored to the actin cytoskeleton. Disrupted integrity of the intestinal epithelial barrier has been demonstrated in a variety of GI disorders, including inflammatory bowel disease, via alteration of tight junction expression and/or function by inflammatory factors such as IFN- γ, TNF- α, and interleukins [28]. Recent work has demonstrated that intestinal epithelial tight junctions are differentially regulated by the microbiota. Specifically, SCFAs- butyrate and acetate- produced by particular microbiota species are capable of inducing barrier properties in the intestinal epithelium to protect from pathogenic bacteria and toxins [29, 30]. Butyrate has been demonstrated to do so via facilitating tight junction assembly in both AMP-activated protein kinase (AMPK) and hypoxia-induced factor 1 (HIF1)-dependent mechanisms [30, 31]. In addition to the tight junctions between epithelial cells, the layer of mucous produced by goblet cells overlying the epithelium is a critical component of the intestinal barrier. In fact, within the colon, the location with the highest microbiota concentration, the mucous layer is highly complex and organized into three distinct layers: 1) the innermost glycocalyx, composed of membrane-bound mucin proteins, 2) the tightly-linked middle layer, and 3) the low density outer layer formed by middle-layer proteolysis, in which bacteria typically reside [32]. Similar to tight junctions, SCFA-generating microbiota species are protective against pathogenic bacteria via stimulating production of mucus and differentiation of mucus-producing goblet cells [33].
While a healthy microbiome is capable of protecting from pathogenic species via maintenance of the intestinal barrier, dysbiosis, defined as microbial imbalance, may promote barrier disruption and predispose the organism to illness. An important inducer of microbial dysbiosis is antibiotic use, which may cause death of healthy flora and promote the growth of pathogenic species, such as Clostridium difficile. C. difficile results in pseudomembranous colitis through the generation of enterotoxins A and B, both of which have been shown to disrupt the cytoskeleton and promote intestinal permeability [34]. Dietary alterations are also intimately linked with alterations in the microbiome, implicating a role for diet in the pathogenesis of metabolic disease beyond the concept of caloric imbalance [35]. Consistent with this, Desai et. al. recently showed that lack of dietary fiber depletes microbial nutrients, thus forcing these organisms to rely on mucus proteins as a nutritional source, resulting in erosion of the mucus barrier. This barrier disruption thus permitted epithelial access and infection with the mucosal pathogen Citrobacter rodentium [36]. As described, the interaction between microbiota and the intestinal barrier is critically important in regulation of GI tract immunity and may provide insight into pathogenic mechanisms of a variety of disease processes and targets for future therapy.
Neuroendocrine interactions along the brain-gut microbiome axis
Neuroendocrine interactions with the GI tract occur via two mechanisms: 1) endocrine, via the HPA system, and 2) neural, directly via the ENS. The HPA axis, often considered the “stress response” system, describes the indirect connection between the hypothalamus, pituitary, and adrenal gland via hormone secretion into the bloodstream. Corticotropin-releasing hormone (CRH) is secreted by the hypothalamus into the hypophyseal-portal blood, where it travels to the anterior pituitary to stimulate secretion of adrenocorticotropic hormone (ACTH) into the bloodstream. At the level of the adrenal cortex, ACTH stimulates synthesis of glucocorticoids (i.e., cortisol) by the zona fasciculata which exert systemic metabolic actions [37]. Activation of the HPA axis is well-described in the setting of chronic stress, depression, and anxiety disorders and is programmed by early life events/stressors. Early life stress is associated with heightened activity of the HPA axis and predisposes animals to subsequent neuropsychiatric pathology [38]. Building off the idea that bidirectional communication between the early nervous and immune systems may alter development of neural systems, Sudo et al. demonstrated exaggerated HPA responses, including elevations in ACTH and corticosterone, in GF mice in response to restraint stress compared to similarly exposed SPF mice. Further, reconstitution of the GF mice with Bifidobacterium infantis reversed the exaggerated response, highlighting the influence of microbiota on postnatal development of the HPA axis [8]. Beyond its influence directly on the HPA axis, the microbiome plays a role in neurotransmitter, most notably tryptophan, metabolism, which is responsible for the generation of the key neurotransmitter serotonin. Absence of such tryptophan metabolizing bacteria is associated with elevated levels of circulating tryptophan, reduced levels of serotonin, and presence of anxiety-like behaviors [39].
Further, the link between the brain, gut, and microbiome is bidirectional. Chronic activation of the HPA axis by stressors can result in altered composition of the microbiome, as demonstrated in a variety of preclinical model of stress such as maternal separation, chronic social defeat, restraint, crowding, heat, and auditory stressors [40]. Alteration of the microbiome by chronic stress, and activation of the immune system, has also been proven to induce colitis; these mechanisms are hypothesized to underly stress-associated gastrointestinal disorders such as inflammatory bowel disease (IBD) and irritable bowel syndrome (IBS) [41, 42].
While the influence of the gut microbiome on the HPA axis is via indirect connection through the bloodstream, its influence on the gut’s nervous system or ENS is more direct and physical. The ENS is comprised of two distinct ganglion within the wall of the GI tract: 1) the myenteric, or Auerbach, plexus between the inner circular and outer longitudinal layers of the muscularis controls GI motility, and 2) the submucosal, or Meissner, plexus within the submucosal layer innervates GI epithelium. These plexuses are connected by nerve fibers which form mesh-like networks traversing the walls of the GI tract [43]. The ENS is innervated by parasympathetic fibers of the vagus nerve and sympathetic fibers of the prevertebral ganglia. The vagus nerve, which emerges from the brainstem and carries efferents to the enteric ganglia and afferents to the nucleus tractus solitarii, provides the primary bidirectional connection between the CNS and ENS, forming the “hardwire” of the brain-gut axis [44]. In fact, the recent identification of enteroendocrine cells within the gut epithelium, which synapse on vagal neurons, provides a mechanism by which the vagus nerve directly interacts with the GI lumen. Physiologically, this provides a mechanism for luminal nutrient sensation, but may also serve as a direct surface for interaction with the microbiome or microbial nutrients [45]. The interaction between the microbiome and ENS begins in development. ENS neurons are derived embryologically from Sox10-expresing neural crest cells which delaminate from the neural tube and migrate throughout the developing gut tube prenatally [46]. However, a niche of enteric neural stem cells remains within the postnatal gut which maintains the potential for modulation of ENS maturation following colonization with gut microbiota at birth [47]. This potential has been proven in both enteric glial cells and neurons in recent years. Enteric glia normally form extensive networks; however, it has been shown that GF mice have reduced number of mucosal enteric glial cells compared with conventionally housed mice. This lack of enteric glia is rescued by postnatal colonization via gavage feeding of microbiota which promotes formation of glial networks [48]. Similarly, GF mice display neuronal anatomical abnormalities and reduced intestinal transit rate, which is corrected by feeding of microbiota via increased production of serotonin and stimulation of enteric neurogenesis [49]. Once mature, homeostatic maintenance of the ENS is reliant on the presence of enteric neural stem cells that may respond to alterations in the surrounding microenvironment or disease processes. Enteric neurons are known to express Toll-like receptors, suggestive of a role for microbial products in maintenance of the mature ENS [50]. Consistent with this, GF mice and mice lacking the TLR4 gene display significant delay in GI motility and reduced numbers of nitrergic neurons, while incubation of immortalized enteric neurons with bacterially-derived TLR4 agonist lipopolysaccharide (LPS) promotes expression of the transcription factor NF-κB and cell survival [51]. Furthermore, in culture, bacterial LPS promotes proliferation and delays differentiation of enteric neural stem cells, providing a mechanism by which microbiota may promote ENS homeostasis [52]. These neuroendocrine interactions are summarized in Fig. 1.
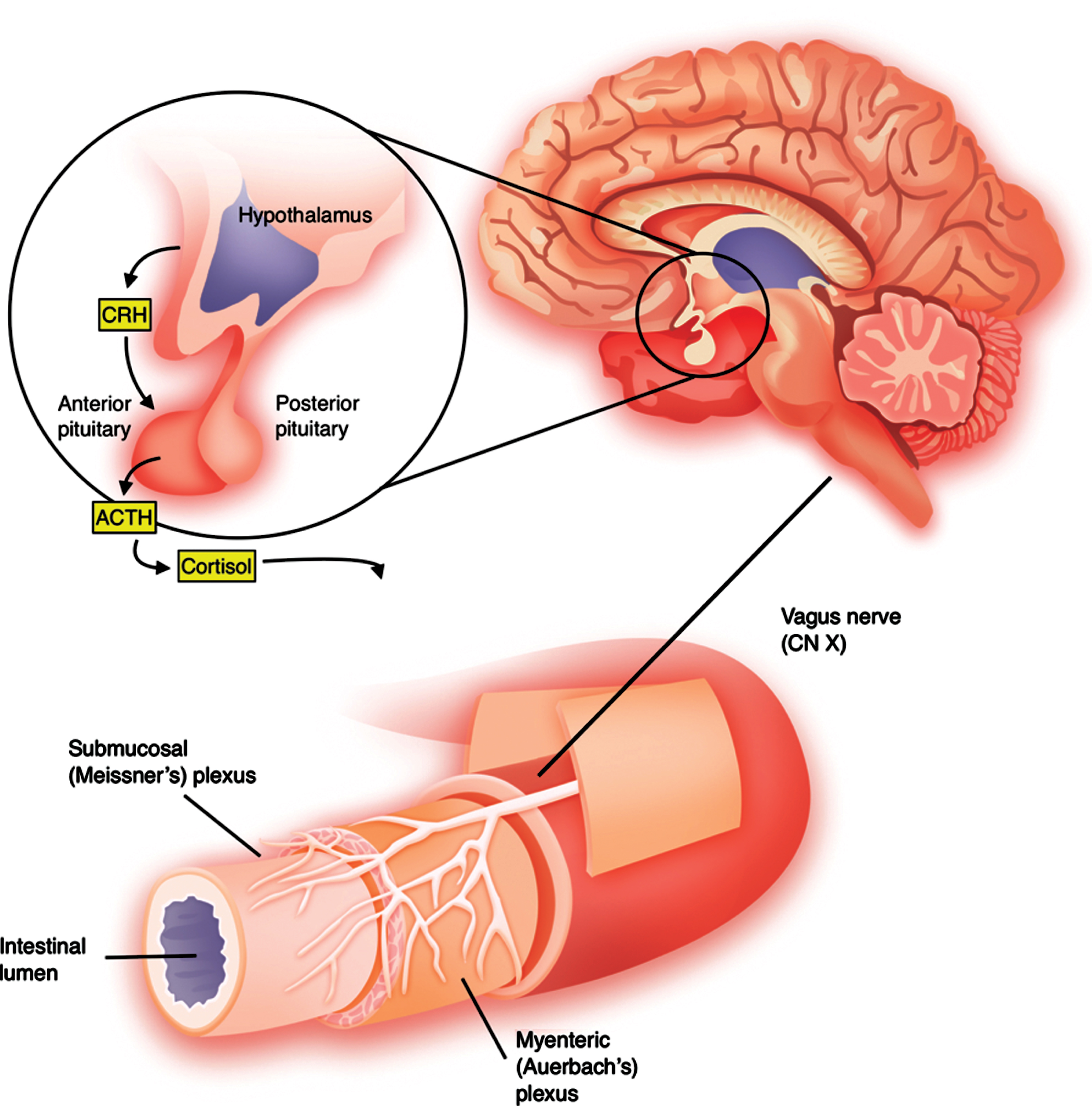
Neuroendocrine interactions between the brain and gastrointestinal tract. The vagus nerve (CN X) projects from the nucleus and provides parasympathetic innervation to foregut and midgut derived structures. The hypothalamic-pituitary-adrenal axis, classically referred to as the “stress axis”, is the primary endocrine connection between the brain and gastrointestinal tract.
MECHANISMS OF BRAIN-GUT-MICROBIOME ALTERATION IN ALZHEIMER’S DISEASE
The direct and indirect connections comprising the brain-gut-microbiome axis described above, and their alteration in a variety of pathologies, raise the question: What is the influence of the axis on the development and/or progression of AD? Despite extensive research into the neuropathology of this devastating disease, little progress has been made in identifying novel targets for therapy. In the subsequent pages we provide a discussion of the evidence for brain-gut-microbiome disturbance in AD and consider the axis as a missing link in AD pharmacotherapy. These mechanisms are summarized in Fig. 2.

Summary of bidirectional interactions between the brain, immune system, and gut microbiota via neuroendocrine, immune, and metabolic crosstalk.
Microbial modulation of amyloid-β accumulation and clearance
The amyloid-β (Aβ) hypothesis, based on the neuropathologic findings of amyloid plaques within the brains of AD patients, has been the classic explanation of AD pathogenesis since early reports suggesting altered Aβ metabolism in AD. This is supported by genetic findings of mutations in the amyloid-β precursor protein (AβPP) on chromosome 21 in some AD patients, as well as the increased incidence of AD in individuals with trisomy 21 [53]. A complete review of the Aβ hypothesis is beyond the scope of review. However, to briefly summarize Aβ metabolism: 1) after synthesis of AβPP, it is cleaved into a C-terminal fragment by β- or α-secretase; 2) the C-terminal fragment is then cleaved to various forms of Aβ, most notably Aβ40 and Aβ42 [54]. Aβ42 is considerably more hydrophobic than its counterpart and, despite being the less predominant form, has greater tendency to aggregate and form amyloid plaques [55]. Under normal circumstances, as in younger individuals, Aβ is rapidly degraded or removed preventing accumulation and plaque formation. However, with increased age or in pathologic states, the ability to clear these abnormal proteins is reduced [56]. The Aβ plaques are then thought to induce neuronal and synaptic damage, as well as stimulate inflammation via microglia, leading to the characteristic neurodegenerative and cognitive changes [57, 58].
Targeting Aβ, in an effort to prevent formation of neurotoxic plaques within the brain, has been a pharmacological strategy for treatment of AD for over 20 years. Compelling evidence from animal studies have demonstrated that removal of Aβ, typically through immunotherapeutic approaches such as immunization or antibody administration, prevents plaque formation and is correlated with alleviation of the associated cognitive decline [59, 60]. However, to date, there has been no phase III clinical trial that has demonstrated success in achieving primary outcomes using therapy targeting the Aβ pathway [61]. This track record of failure has drawn a great deal of criticism toward the Aβ hypothesis. While it remains a central focus of AD research, and must continue to be studied, there is a general consensus throughout the field that new ideas into the mechanisms and significance of Aβ deposition in AD must be considered. Emerging evidence from the brain-gut-microbiome axis may provide a link understanding Aβ metabolism in AD.
As described earlier, the brain-gut-microbiome axis has a “hard wire” connection in the form of the vagus nerve. This direct connection may permit retrograde neuronal transport of substances from the gut to the brain, bypassing the protection of the blood-brain barrier (BBB). This has recently been demonstrated in an animal model in which α-synuclein, the protein component of Lewy bodies that are hallmarks of PD, was transported within the vagus nerve from the gut to the dorsal motor nucleus within the brainstem in a mechanism dependent on microtubules [62]. This provides evidence for the spread of abnormal, potentially pathogenic, proteins via the ENS. While there is no direct evidence of this exact mechanism for Aβ, previous work has demonstrated that Aβ may spread between connected brain regions, thereby accumulating within nodes such as the hippocampal dentate gyrus. Lesioning the connections was sufficient to reduce intra-neurite AβPP accumulation and extracellular Aβ burden [63]. If this mechanism is clinically significant in the pathogenesis of AD, the microbiome may be a source of amyloid protein. Various studies have demonstrated that microbiota species secrete functional amyloid into the GI tract. These proteins were thought to play a role in evasion of the immune system, thus promoting pathogenicity of the secreting organism. For example, the protein Curli, secreted by Enterobacteria, including E. Coli, is a particularly well described bacterial amyloid which functions in formation of biofilms [64]. Exposure to Curli has recently been shown to enhance accumulation of α-synuclein within the gut and brain of C. elegans fed a diet rich in Curli-producing bacteria, suggesting that this bacterial amyloid protein may facilitate α-synuclein aggregation via cross-seeding and priming of the immune response [65]. Similarly, transport of bacterial amyloid and subsequent exposure in the brain may facilitate accumulation of Aβ in AD. In particular, Aβ is known to induce accelerated Aβ deposition and plaque formation in a prion-like seeding mechanism [66]. Recent work has demonstrated that cross-seeding, accelerated aggregation induced by an unrelated misfolded protein, occurs following inoculation of transgenic AD mice with prion protein [67]. As such, in AD, bacterial amyloid might act in a similar fashion to induce the aggregation of Aβ. Another mechanism underlying enhanced Aβ pathology in AD may be related to the release of metabolites from gut microbiome that alter production and/or deposition. For example, it was recently demonstrated that the bacterial metabolite trimethylamine-N-oxide (TMAO) increased with age in both wildtype and transgenic AD mice and that depletion of TMAO decreased hippocampal levels of Aβ42 and β-secretase [68].
In addition to its production of amyloid protein that may be transported to the brain and facilitate subsequent Aβ aggregation, microbial factors may alter clearance of amyloid within the brain. Microglia, the brain’s resident macrophages, play an important role in homeostatic clearance of Aβ, thus preventing accumulation in physiological states. This response can be stimulated by cytokines such as macrophage colony-stimulating factor (M-CSF), which promote the phagocytosis and lysosomal degradation of Aβ protein in animal models [69, 70]. While this pro-inflammatory response appears to have a protective role in AD pathogenesis, chronic inflammation may actually perpetuate Aβ aggregation. Qiao et al. demonstrated that long-term administration of the bacterial endotoxin LPS markedly accelerated deposition of Aβ [71]. Subsequent work has demonstrated that this may occur through impaired efflux of Aβ via LPS-induced reduction in cerebrospinal fluid, central and peripheral clearance of Aβ, and increased vascular sequestration of Aβ [72]. Closely related to Aβ plaque formation and clearance is hyperphosphorylation of tau protein which contributes to formation of neurofibrillary tangles, a hallmark of AD pathology. In a recent study, Wei et al. demonstrate that vesicles produced from the outer membrane of gut microbiota induce activity of glycogen synthase kinase 3β (GSK-3β) leading to tau phosphorylation and cognitive dysfunction. Notably, their methodology involved extracting these outer membrane vesicles (OMVs) from human AD patients’ feces and injecting them into mice in comparison with OMVs from healthy controls, substantiating the clinical relevance of this pathway significantly [73].
Overall, there is significant support for the role of the microbiome in contributing to alterations in Aβ metabolism in AD, including via 1) direct production of amyloid proteins, which may accumulate or cross-seed, and 2) inhibition of mechanisms required for homeostatic Aβ clearance in the brain. While the role and significance of Aβ in AD pathogenesis is still yet to be elucidated, it is apparent that the microbiome may contribute to the neuronal damage of AD in an Aβ-dependent mechanism.
Microbial-mediated alteration of blood-brain barrier permeability and neuroinflammation
Given the lack of evidence supporting the Aβ hypothesis as the sole explanation for AD pathogenesis, more recent research has focused on other mechanisms that may contribute to neurodegeneration in the AD brain. The BBB is present at the level of brain capillaries and is composed, structurally, of tight junctions between endothelial cells. Associated cells, including astrocytes, pericytes, and microglia contribute to integrity of the BBB via regulation of the endothelial barrier [74]. It has long been known that the BBB, which normally acts to protect the brain from exposure to harmful substances in blood (i.e., pathogens, inflammatory mediators, toxins, etc.) is disrupted in AD [75]. In fact, radiological evidence has identified leakage through the BBB in the early stages of AD, suggesting that disruption may contribute to the cascade of pathologic events characteristic of the disease process [76]. Various studies have demonstrated impairment of BBB integrity by Aβ protein in AD models. One study showed that administration of Aβ42 to endothelial cells isolated from rat cerebral cortex altered the expression and cellular localization of tight junction proteins, notably promoting cytoplasmic translocation of claudin-5 and zona-occludens-2 [77]. Under normal circumstances, astrocytes contribute to integrity of the BBB by stimulating expression tight junction proteins by endothelial cells via secretion of factors from their characteristic endfeet [78]. However, secondary to β42, astrocytes secrete matrix metalloprotease 9, leading to disruption of the BBB likely through degradation of claudin-5 in endothelial cells [79]. However, we must acknowledge that recent evidence challenges the conception of BBB disruption as a hallmark of AD. In particular, Bien-Lyn et al. demonstrated that impairment of BBB integrity was not observed in various mouse models of AD, including PS2-APP, tau transgenics, and APOE4 knockin mice. Importantly, they also observed no increase in the incidence of infarcts in human AD brains that may be suggestive of widespread BBB disruption [80]. Overall, we believe that there is sufficient evidence to support a role for BBB disruption in AD, although this may not be as widespread as previously thought.
Given the ability for Aβ to induce permeability of the BBB, it is reasonable to hypothesize that a similar effect may occur from secretion of bacterial amyloids. In fact, the concept of microbes and/or microbial factors inducing alteration of the BBB is not new. Some bacteria are capable of directly traversing the BBB, giving rise to CNS infection and/or abscess, via specialized components such as pilli, while others do so via stimulation of an inflammatory response [81]. Specifically, LPS is an endotoxin and pro-inflammatory agent produced by gram-negative bacteria that can potently stimulate BBB disruption in infectious states [82]. However, subclinical alterations in the gut microbiome can also result in variable BBB integrity. For example, in a study comparing GF and pathogen-free mice, it was found that GF mice displayed increased permeability of the BBB due to reduced expression of tight junction proteins occludin and claudin-5. Additionally, treatment of the GF mice with the pathogen-free microbiome rescued the BBB defect, resulting in increased tight junction protein expression and reduced permeability [83]. One mechanism by which altered microbiome composition may influence the BBB is differential secretion of factors by distinct strains of bacteria. LPS is typically associated with pathogenic strains, while commensal or normal flora produce SCFAs. Consistent with this, recent work showed that the bacterial metabolite propionate, an SCFA, protected the BBB from damage via inhibiting oxidative stress in an NFE2L2-dependent mechanism [84].
While this data confirms the ability of varied microbiota composition to influence permeability of the BBB, the significance of this in relation to the neurodegenerative changes characteristic of AD must also be considered. The BBB contributes to isolation of the brain from circulating amyloid protein, thus preventing accumulation of potentially pathogenic amyloid. In pathologic states in which the BBB is disrupted, however, circulating amyloid may enter the CNS. For example, using monkeys, Mackic et al. demonstrated that circulating Aβ can cross the BBB and contribute to formation of amyloid plaque aggregates consistent with AD pathology [85]. In fact, the mechanisms governing transport of Aβ appear to be highly regulated. Two transporters are responsible for cerebrovascular transport of Aβ: 1) low-density lipoprotein receptor related protein-1 (LRP1); and 2) receptor for advanced glycation end products (RAGE). The former is responsible for brain-to-blood efflux, while the latter facilitates blood-to-brain influx [86, 87] Implicating a role for microbial and inflammatory modulation of Aβ transport across the BBB, LPS has been shown to increase Aβ influx and decrease Aβ efflux via decreased expression of LRP1 [88]. This alteration can thus permit accumulation of Aβ, which may act as a seed for subsequent deposition and plaque formation [66]. Similarly, secreted bacterial amyloid in serum may now be able to enter the CNS via the blood, in addition to via the retrograde neural transport above described.
Until recently, the brain was considered to retain immune privilege, a term describing its apparent lack of exposure to the inflammatory milieu of circulating blood. In addition to the BBB, which isolates the brain from the periphery, the concept of immune privilege was derived from findings that there is a lack of professional antigen presenting cells (i.e., dendritic cells, macrophages) and low expression of MHC molecules within the CNS. However, in recent years, this concept has been brought to question by the discovery of a meningeal lymphatic system and the presence of mechanisms permitting immune cell entry into the CNS [89]. It seems that, rather than serving as rigid barrier to immune cell entry, the BBB acts in a regulated manner to selectively permit entry of inflammatory cells and mediators. As such, in inflammatory states, which tend to promote BBB disruption (i.e., systemic LPS), entry of inflammatory cells may be permitted [90]. In fact, such systemic inflammation has been associated with increased risk of developing an AD-like phenotype in mice exposed to the viral mimic polyriboinosinic-polyribocytidilic acid [91]. Inflammatory mediators such as complement proteins, chemokines/cytokines, radical oxygen species, and inflammatory enzymes are produced by the brain’s resident immune cell, microglia, and have been observed in abundance within postmortem AD brains [92]. Oxidative stress has been extensively studied in neurodegenerative diseases, including AD, and the role for microglia in production of reactive oxygen species provides a potential mechanistic link between inflammation and neurodegeneration [93]. Furthermore, inflammatory infiltrate with bone-marrow derived microglia has been observed in transgenic mouse models of AD, suggesting a contribution of the peripheral immune system, although its role and significance in AD pathogenesis is still being elucidated [94]. Administration of LPS, a potent proinflammatory factor, resulted in increased levels of inflammatory factors, notably IL-1β, IL-6, and TNF-α, as well as Aβ within the hippocampus of rats [95]. Implicating a role for the microbiome in initiation of an innate immune response in AD, Zhao et al. recently reported immunohistochemical evidence of microbiome derived LPS within the perinuclear region of human AD brains. This data suggests that LPS secreted by gram-negative bacilli of the microbiome may leak across an impaired BBB and accumulate within the brain, stimulating an inflammatory response and possible degradation [96].
The role of the microglia in AD pathogenesis has been more widely explored and reported. Original hypotheses suggest that the inflammation seen within the AD brain is merely a secondary response triggered by the neuronal death and degeneration pathologically occurring due to the disease process. However, more recent work indicates that inflammation may be an important driver of AD pathology. As described earlier, microglia contribute to Aβ clearance via phagocytosis and degradation of the abnormal Aβ42 protein [69, 97]. Microglia are described as having two activated states: the M1, or classical, is the proinflammatory state characterized by production of neurotoxic proinflammatory mediators; the M2, or alternative, state inhibits inflammation and promotes tissue repair [98]. Stimulation of the M2 state is also associated with enhanced clearance of Aβ and reduced cognitive deficit in an animal model of AD [99]. However, as with most cases of inflammation, microglia act as a double-edged sword. Microglia may be activated into the M1 state by binding of extracellular Aβ42 protein to the TLR2 receptor with subsequent release of the proinflammatory mediator IL-8 and associated neuronal damage. Further, deficiency of TLR2 promotes a shift of the microglia to the M2 state and ameliorates the damage [100]. Interestingly, mutations in myeloid cell molecules CD33, a regulator of innate immunity, and TREM2, an innate immune receptor, have been reported to increase risk of AD [101, 102]. Interaction between the microbiome and microglia is considered an important link in the brain-gut-microbiome axis [103]. Microglia are macrophages that localize to the brain during embryogenesis prior to formation of the BBB. Recently, it has been shown that GF mice display altered patterns of microglial development in a sexual dimorphic and, further, treatment of adult mice with antibiotics produce sex-specific perturbations [104]. This suggests that microbial alterations throughout the lifespan may predispose individuals to altered microglial responses and may be important in the pathogenesis of a variety of neurological diseases, including AD. The mechanism for microbial influence on microglial development and function is likely through the secretion of metabolites, including LPS and SCFAs. Supporting this, microglia from mice deficient for the SCFA receptor FFAR2 display a similar immature phenotype as those obtain from GF mice [105]. One mechanism by which microbial dysbiosis may alter microglial activation in the AD brain is through modulation of peripheral immune cells. Supporting this, a recent report demonstrated that alteration of the gut microbiome in a mouse model of AD led to peripheral accumulation of the amino acids phenylalanine and isoleucine, which are potent stimulators of differentiation and proliferation of pro-inflammatory T-helper 1 (Th1) cells. These cells can then activate microglia into the M1 phenotype above described. Supporting the clinical relevance of this finding, elevated phenylalanine and isoleucine levels were associated with increased Th1 levels in patients with mild cognitive impairment and treatment with a sodium oligomannate that is currently in Chinese clinical trials attenuated neuroinflammation and reduced associated cognitive decline [106].
Overall, the inflammatory contribution to AD appears to be significant and warrants further study. The impact of the microbiome and microbial factors on the inflammatory cascade is particularly interesting in this respect, as it may provide a foundation for understanding variability in the progression of AD across diverse populations as it relates to exposure, comorbid inflammatory disease, antibiotic use, diet, etc.
Regulation of adult hippocampal neurogenesis
The conventional dogma has long been that development of new neurons is confined to the embryonic and perinatal periods and does not occur in the adult brain. However, this has been largely refuted by the anatomic and molecular findings of life-long neurogenesis within the hippocampus of mammals, including humans [107–109]. Despite the recent report by Sorrells et al. that hippocampal neurogenesis occurs rarely, if at all, past adolescence [110], there remains a great deal of support that neurogenesis proceeds throughout the life span. Most recently, three separate studies demonstrated that neurogenesis, while declining with age, occurs as late as the tenth decade of life, and reduction in neuroblasts and immature neurons is associated with cognitive decline and/or neuropsychiatric/AD [111–113]. This correlates with evidence from animal models that has demonstrated decreased levels of neurogenesis in the pathogenesis of AD, in some cases even occurring prior to the onset of the classical AD pathology [114, 115].
These data suggest that alterations of neurogenesis are critical, potentially even causal events in the development and progression of AD. In accordance with the Aβ hypothesis, soluble Aβ has been shown to impair neuronal proliferation, while oligomeric Aβ promotes the formation of microglia [116]. Brain-derived neurotrophic factor (BDNF) is a growth factor produced in the brain that acts as a key mediator of adult neurogenesis and, notably, facilitates exercise-induced neurogenesis [117]. Levels of BDNF have been observed to be decreased in the brain, blood, and cerebrospinal fluid of individuals with AD; in fact, lower levels of BDNF even correlate with cognitive ability as assessed in the Mini-Mental Status Examination [118]. The mechanism of BDNF reduction may be dependent on exposure to Aβ, as Aβ is capable of reducing mRNA transcripts of BDNF in human neuroblastoma cells [119]. Alterations in the intestinal microbiota are also associated with variation in hippocampal levels of BDNF, with GF mice demonstrating reduced BDNF and exploratory behavior that is rescued following colonization with microbiota from SPF mice [120]. With the known ability of bacteria to produce LPS and amyloids, it is possible that these molecules may mitigate a reduction in BDNF in a manner similar to that of Aβ. Utilizing antibiotic treatment, Möhle et al. showed that alteration in the gut microbiota reduced hippocampal neurogenesis and memory retention, which could be rescued by reconstitution with SPF microbiota, supplementation of probiotics, and BDNF. Interestingly, depletion of Ly6Chi monocytes abrogated the rescue effect of these measures suggesting that this population of monocytes is critical in preservation of brain homeostasis [121]. Macrophages derived from Ly6Chi monocytes have previously been shown to promote functional recovery following stroke in mice [122], and this data may provide a link between microbial alteration and neurogenesis that is crucial to the pathogenesis of AD.
The impact of the microbiome on neurogenesis is critical, not only to AD pathogenesis, but to the regulation of behavior in physiologic settings. More research in this topic is certainly warranted, especially in the context of antibiotic use and its impact on the flora.
CLINICAL CORRELATIONS AND POTENTIAL THERAPEUTIC APPROACHES
The majority of the above described work is taken from in vitro or in vivo animal studies, which are important, but may not correspond completely to patients at the bedside. As such, we will briefly discuss clinical work that supports the notion that the microbiome is altered in patients with AD. First off, the natural biodiversity of microbiota is well known to decline with increased age, with a relative reduction in commensal species, such as bacteroides, bifidobacteria, and lactobacilli and relative increase in opportunists/potentially pathogenic species such as enterobacteria, C. perfringens, and C. difficile [123]. The variation in microbial composition is also dependent on diet and lifestyle; for example, among the elderly, individuals residing in long-term care facilities maintain a microbiome that is significantly different from those living at home [124]. It is likely that the effects are exerted through changes in production of bacterial metabolites, such as LPS and SCFAs. Variation in microbiota provides an additional source of background variation, in addition to genetics and exposures, that may account for the variable courses seen among a multitude of diseases, including AD. Even in familial cases of AD, which are characterized by younger age of onset and typically heightened severity, there remains a considerable deal of variation in the progression that is not accounted for in current models [125]. Background differences in the microbiome, and its influence on Aβ metabolism, barrier permeability, inflammation, and neurogenesis may help provide an understanding of the variable disease course. Perhaps one of the most significant findings in human samples supporting a microbial hypothesis in AD is the finding of a large bacterial load within the cerebral cortex of AD patients using 16 s ribosomal next generation sequencing, a technique considered state-of-the-art for microbiome analysis. In particular, the authors reported enrichment of Firmicutes species and P. acnes [126]. To the best of our knowledge, there is limited data regarding the variations in the microbial composition of AD patients, with only one study reporting significant differences in gut microbial genotypes of AD patients and controls [127].
With the evidence supporting microbial alteration, an obvious question is: Can the microbiome be targeted in AD therapy? Probiotics, defined as live microbial food ingredients that exert beneficial effects on health, are most notably used in the treatment of GI disease processes including lactose intolerance, diarrhea, and antibiotic-induced side effects. However, future use in other pathologies is promising based on current research [128]. Probiotics have been used with benefit to alleviate stress and improve cognitive function in adults experiencing stress, suggesting a potential use in neurocognitive and/or psychiatric disease [129, 130]. In the setting of AD, probiotics have been used limitedly, with most studies evaluating the effect in animal models. One renowned example is the work of Bonfili et al. which demonstrated that oral supplementation with the probiotic formulation SLAB51 resulted in altered microbiome composition, as assessed by 16 S ribosomal RNA sequencing, as well as reduced inflammatory cytokines. Most notably, this probiotic actually attenuated cognitive decline via reduced Aβ aggregation and restoration of neuronal proteolytic pathways [131]. More recently, our laboratory demonstrated that a novel probiotic formulation (BIOCG) displayed similar neuroprotective effects in a transgenic mouse model of AD through several processes including attenuation of microglial activation, reduction in Aβ load, and preservation of dendritic spine structure and function. This work, which is currently unpublished and in submission, is to our knowledge the first evidence that probiotic administration, and subsequent alteration in microbial composition can alter neuronal physiology via improved long-term potentiation and preserved synaptic spine morphology in a mouse model of AD. As described, clinical data regarding probiotic administration to AD patients is limited. One small exploratory study demonstrated that supplementation with a multispecies probiotic influenced serum tryptophan metabolism, an important modulator of neurotransmitter synthesis which may be disrupted in AD [132]. A larger randomized controlled trial showed that consumption of milk containing Lactobacillus acidophilus, Lactobacillus casei, Bifidobacterium bifidum, and Lactobacillus fermentum produced significant improvement in Mini-Mental Status Examination scores and improvement on various metabolic measures [133]. This is particularly significant as recent evidence supports the association between metabolic diseases, such as obesity and type 2 diabetes, with the occurrence of AD. In particular, a recent publication has demonstrated that diet-induced perturbation in the gut microbiome alters the shikimate pathway leading to elevated levels of the cytotoxic amine tryptamine [134]. These studies demonstrate that probiotics may be helpful supplements for symptomatic control in AD and management of comorbid metabolic disease. Future research identifying specific strains that are altered in AD or may exert beneficial effects is warranted. At present, there are two registered clinical trials seeking to assess the effect of probiotic supplementation on AD or mild cognitive impairment, one of which is currently recruiting patients.
CONCLUSION AND FUTURE DIRECTIONS
Overall, our review provides a great deal of evidence, in both preclinical and clinical studies supporting the role of the brain-gut-microbiome axis in AD. A summary of the above described interactions is provided in Fig. 2. Future directions for clinical work include utilizing sequencing to identify AD-specific alterations in the gut microbiome that may provide insight into the mechanisms by which alteration influences AD and potential therapeutic targets. Additionally, large-scale analyses correlating microbial diversity with cognitive status and disease progression in patients with AD may provide helpful prognostic data.