
Abstract
Mitochondria are important (patho)physiological sources of reactive oxygen species (ROS) that mediate mitochondrial dysfunction and phospholipid oxidation; an increase in mitochondrial content of oxidized phospholipid (OxPL) associates with cell death. Previously we showed that the circulating OxPL 1-palmitoyl-2-(5’-oxo-valeroyl)-sn-glycero-3-phosphocholine (POVPC) increases in patients with Alzheimer’s disease (AD), and associates with lower plasma antioxidant oxocarotenoids, zeaxanthin, and lutein. Since oxocarotenoids are metabolized in mitochondria, we propose that during AD, lower concentrations of mitochondrial zeaxanthin and lutein may result in greater phospholipid oxidation and predispose to neurodegeneration. Here, we have investigated whether non-toxic POVPC concentrations impair mitochondrial metabolism in differentiated (d)SH-SY5Y neuronal cells and whether there is any protective role for oxocarotenoids against mitochondrial dysfunction. After 24 hours, glutathione (GSH) concentration was lower in neuronal cells exposed to POVPC (1–20 μM) compared with vehicle control without loss of viability compared to control. However, mitochondrial ROS production (determined by MitoSOX oxidation) was increased by 50% only after 20 μM POVPC. Following delivery of lutein (0.1-1 μM) and zeaxanthin (0.5-5 μM) over 24 hours in vitro, oxocarotenoid recovery from dSH-SY5Y cells was > 50%. Co-incubation with oxocarotenoids prevented loss of GSH after 1 μM but not 20 μM POVPC, whereas the increase in ROS production induced by 20 μM POVPC was prevented by lutein and zeaxanthin. Mitochondrial uncoupling increases and ATP production is inhibited by 20 μM but not 1 μM POVPC; carotenoids protected against uncoupling although did not restore ATP production. In summary, 20 μM POVPC induced loss of GSH and a mitochondrial bioenergetic deficit in neuronal cells that was not mitigated by oxocarotenoids.
Keywords
INTRODUCTION
Reactive oxygen species (ROS) are produced during mitochondrial oxidative phosphorylation. Increased mitochondrial-derived ROS are associated with mitochondrial dysfunction and have been described as early events in neurodegenerative diseases [1–4]. In a vicious cycle of oxidative damage and ROS production, the oxidation of mitochondrial proteins, DNA, and lipids precedes a decline in mitochondrial integrity. Several phospholipid oxidation products are cytotoxic at the site of production and have the potential to cross biological membranes of neighboring cells to exert distant effects [5–7]. This has been shown in cardiomyocytes, which are sensitive to exposure to cytotoxic oxPL and also accumulate mitochondrial OxPL during ischemia reperfusion and preceding cell death [8].
Carotenoids can quench singlet oxygen and scavenge other ROS without being consumed in the process except when scavenging peroxides [9–11]. While a Cochrane systematic review concluded that beta-carotene supplements confer an increased risk of mortality [12], the oxygen-containing carotenoid hydrocarbons, xanthophylls, which include lutein, zeaxanthin, and meso-zeaxanthin, have shown more promise for positive health outcomes [11]. Lutein and zeaxanthin cannot be synthesized by humans and are obtained from the diet or supplements. The consumption of diet rich in fruits and vegetables as well as the modification of nutritional habits and lifestyle associates with higher oxocarotenoid levels in plasma and a lower risk of age-related diseases and Alzheimer’s disease (AD) [13]. Conversely, in the MARKAGE study, lower levels of β-cryptoxanthin and zeaxanthin were observed in the cognitively frail [14].
The discrete risk/benefit effects of carotenoids that have been observed in vivo may relate to their distinctive metabolism; oxocarotenoids are differentially distributed within subcellular compartments and are associated with specific metabolic enzymes [15]. In contrast to carotene which is metabolized by BCO1 and is retained in the cytoplasm, the enzyme BCO2 is associated with the inner mitochondrial membrane and metabolizes zeaxanthin (which accumulates in the inner mitochondrial membrane) into long-chain apo-carotenoids. BCO2 functions as a key regulatory enzyme that prevents toxicity caused by carotenoid accumulation and loss of BCO2 function is associated with the development of mitochondrial oxidative stress and metabolic diseases [16].
Others have shown that chronic administration of the oxocarotenoid lycopene significantly restores the mitochondrial respiratory enzyme activities in Aβ42 treated rats and attenuated mitochondrial oxidative stress. These observations suggest a role for carotenoids in maintaining mitochondrial integrity in brain [17]. Our own work has shown that lutein, lycopene, and zeaxanthin concentrations were significantly lower in AD patients with vascular co-morbidities compared to healthy subjects [18]. In another study we have shown that the serum phospholipid oxidation product POVPC was higher in AD compared with age-matched control subjects but was not reduced by carotenoid supplementation in extant disease [19]. Taken together these studies indicate an inverse relationship between carotenoid concentration and the oxidized phospholipid POVPC in AD.
We hypothesize that during AD development, lower concentrations of systemic carotenoids are reflected by lower brain mitochondrial zeaxanthin and lutein, greater phospholipid oxidation and contribute to neurodegeneration via oxidative and metabolic stress. To investigate this hypothesis, here we have investigated any protective role for carotenoids against mitochondrial dysfunction induced by POVPC (1-20 μM) in differentiated (d)SH-SY5Y neuronal cells.
MATERIALS AND METHODS
Human neuroblastoma SH-SY5Y cells were purchased from American Type Culture Collections, Manassas, USA. Authentic standards of lutein and beta-apo-8’-carotenal were purchased from Sigma Aldrich (Dorset, UK); zeaxanthin was purchased from Cambridge Biosciences (Cambridge, UK); carotenoid standards were kept in the dark, made up fresh, and used immediately after preparation and confirmation of concentrations. 1-palmitoyl-2-(5’-oxo-valeroyl)-sn-glycero-3-phosphocholine (POVPC) was purchased from Avanti Polar Lipids (Alabaster, AL, USA); MitoSOXTM Red was purchased from Invitrogen (Fisher Scientific, Loughborough, UK); cell titer blue was purchased from Promega Corporation (Hollow Road, USA). All materials for the Extracellular Flux assays were from Agilent Technologies/Seahorse Biosciences (Santa Clara, USA). Carbonyl cyanide p-[trifluoromethoxy]-phenyl-hydrazone (FCCP), oligomycin, and antimycin A were from Sigma-Aldrich. All solvents used were HPLC grade from Fisher Scientific (Loughborough, UK). All other cell culture media and chemicals were also purchased from Fisher Scientific (Loughborough, UK) unless otherwise stated.
Cell culture
Routine cell culture
SH-SY5Y cells were maintained routinely in RPMI 1640 media up to passage 20 and supplemented with heat inactivated 10% fetal bovine serum (FBS), 4 mM L-glutamine and 200 U/ml penicillin and 200 μg/ml streptomycin at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Cells were seeded in a T75 flask at a density of 2x105 cells/ml.
Differentiation of neuroblastoma cells
Cells were differentiated into neuronal-like cells for 10 days and medium was replaced at least every 48 h as described previously [1, 20]. Briefly, SH-SY5Y cells were exposed to 10 μM all-trans retinoic acid (RA) on day 1 after seeding. Medium was replaced with fresh RPMI routine culture medium containing RA every 48 h. After 5 days the medium was changed to neurobasal media containing (2M KCl, 100 μg/mL BDNF; 2% B-27® Supplement (20 mL/L) and 400 U/ml penicillin/400 μg/ml streptomycin) for an additional 5 days; medium was replaced every 48 h.
SH-SY5Y cell differentiation was measured by the extension of long neurites and the cessation of proliferation. The length of neurites was measured by image J software and a differentiated cell was defined as a cell with a neurite length greater than the length of the cell body (on average greater than 10 μm in length). More than 80% SH-SY5Y cells displayed extended neurites after 10-day differentiation (Supplementary Figure 1).
Cell treatment with oxidized phospholipid and carotenoids
After 10 days differentiation, dSH-SY5Y cells were treated with POVPC (0, 1, and 20 μM)±carotenoids (0.1, 1, and 10 μM lutein and 0.05, 0.5, and 5 μM zeaxanthin) as indicated for 24 h at 37°C. Cells were washed with phosphate buffered saline (PBS) prior to analysis.
Delivery of carotenoids to cells
Plasma concentrations of lutein and zeaxanthin have been reported to be 0.3 and 0.1 μM, respectively [21]. Following up from our previous observations, we treated cells with lutein (0.1 and 1 μM) and zeaxanthin (0.05, 0.5, and 5 μM) in agreement with Wagener et al. [22]. Carotenoid stock solutions were prepared in tetrahydrofuran (THF) [23] and further diluted (1 : 1000) with neurobasal media. The vehicle treatment had no effect on any analyses. The concentrations of lutein and zeaxanthin were estimated using Beer Lambert’s law (Extinction coefficients in THF: Lutein-145100 l/(mol x cm); Zeaxanthin- 144500 l/(mol x cm).
Extraction of carotenoids from cells
To ascertain the uptake of carotenoids by dSH-SY5Y cells after 24 h exposure, we extracted carotenoids from washed, treated cells, using the protocol adapted from Wagener et al. [22]. SH-SY5Y cells were seeded in 100 mm dishes at 2 x 106 cells/well, differentiated, and then incubated in media containing different concentrations of lutein (0-10 μM) and zeaxanthin (0-5 μM) for 24 h. The medium was then removed and cells were recovered by scraping in 1 ml PBS. An aliquot of the sample was removed for protein assay (BCA). For HPLC, 500 μL THF and 0.5 μM internal standard (trans-β-apo-8’-carotenal) was added to the cells and ultrasonicated for 10 min. 3 mL hexane was used for the extraction of carotenoids and ultrasonicated for another 10 min. After centrifugation at 2000 g, 20°C for 10 min, the organic phase was collected and the solvent was evaporated under nitrogen. The tube wall was rinsed with ether and allowed to dry for another 10 min. Pellets were re-suspended in 50 μl THF and diluted with 100 μl HPLC buffer (54% methanol, 44% acetonitrile, 2% propan-2-ol, 15% water). Extraction was carried out in the dark.
Assessment of metabolic activity
Viability of cells and cytotoxicity of POVPC±carotenoids were estimated using a cell titer blue assay. SH-SY5Y cells were seeded in a 24-well plate at 2×105 cells/well. After 10 days differentiation, cells were treated with POVPC±carotenoids for 24 h. The cells were washed with PBS and incubated with media containing cell titer blue (1 : 5 dilution) in a 37°C incubator for 1-8 h, protected from light. The supernatant was removed into a 96-well plate and the fluorescence was measured at excitation/ emission wavelengths of 560/590 nm in a SpectroMax GeminiXS microplate fluorometer (Molecular Devices, USA); with the diluted dye as the blank. The absolute fluorescent values were calculated and plotted against the concentrations of the treatments. Loss of metabolic activity was evaluated by comparing results to the control sample which was taken to be 100% metabolically active.
RNA isolation and SOD2 mRNA analysis
Total RNA was isolated using the Qiagen RNeasy Mini Kit according to the manufacturer’s protocol. Briefly, dSH-SY5Y cells were pelleted then suspended in RLT lysis buffer (provided) and homogenized by pipetting. To shear genomic DNA, the lysate was transferred to a gDNA spin column and centrifuged at 8000 x g that was primed with 70% (v/v) ethanol. The RNA was eluted adding 30 μL of RNase-free water and purity was assessed by NanoDrop ™ 1000/c (Spectrophotometers, Thermo Fisher Scientific,US). Quantitative PCR was then performed to determine SOD2 expression as previously described and using the following primers (f = forward; r = reverse).
Human SOD2-f GTTCAATGGTGGTGGTCATATCA
Human SOD2-r GCAACTCCCCTTTGGGTTCT
DNA isolation
Total DNA from dSH-SY5Y cells was obtained using Qiagen DNeasy kit according to manufacturer’s instructions. Briefly, the cell pellet was resuspended in 200 μl of PBS, followed by proteinase K (provided). For efficient lysis, buffer AL was added to the cells at 56°C for 10 min. To precipitate DNA, 96-100% ethanol was added, mixed by vortexing then transferred into a QIAamp mini column. After washing, samples were eluted from the column by centrifugation of the samples. DNA purity was detected by NanoDrop ™ 1000/c (Spectrophotometers, Thermo Fisher Scientific, USA). DNA copy number was analyzed as described previously here using the primers for tRNA-Leu(UUR) relative to B2-microglobulin as housekeeper (https://currentprotocols.onlinelibrary.wiley.com/doi/abs/10.1002/0471142905.hg1907s68).
Mitochondrial isolation from cells
Mitochondria were isolated from dSH-SY5Y by cell disruption followed by differential centrifugation using a method adapted from Lampl et al. [24]. First, mitochondrial isolation buffer (MIB) (70 mM sucrose, 210 mM mannitol, 5 mM HEPES, 1 mM EGTA, pH 7.4) was prepared and stored at 4°C. All procedures were performed at 4°C. For the isolation of mitochondria, two T75 flasks of cells per treatment were used. Cells were washed in cold PBS. Following, cells were scraped in cold PBS using a cell scraper and collected in 15 mL falcon tubes. Tubes were centrifuged for 5 min at 700 x g, 4°C. Supernatants were aspirated and cells were resuspended in 500 μL of MIB. Cell suspensions were transferred to a Precellys tube containing silica beads. Following, the tissue was homogenized using a VelociRuptor V2 Microtube Homogenizer (Scientific Laboratories Supplies, UK) for 5 s. The homogenized solution was transferred to a 15 mL falcon tube and the solution was draw into a 5 mL syringe using a 19-gauge inch needle and expelled back into the tube using a 28 gauge inch syringe. This process was repeated 5 times. The solution was transferred to a 1.5 mL Eppendorf tube and centrifuged for 5 min at 600 x g, 4°C. The supernatant was carefully removed and transferred to a new 1.5 mL Eppendorf tube. Supernatant was centrifuged for 10 min at 11,000 x g, 4°C. The resulting supernatant was collected for cytosolic subcellular fractionation. The resulted pellet containing mitochondria was kept on ice.
Mitochondria were resuspended in 1X RIPA buffer containing protease and phosphatase inhibitors (Roche). Next, the mitochondria was homogenized by needle (10 times). Homogenate was centrifuged for 10 min at 10,000 x g, 4°C. Finally, the supernatant was collected and protein concentration was measured by BCA protein assay (Bio-Rad). A concentration of 30 μg of protein was used to evaluate the protein carbonyl formation and α-tubulin.
Determination of cellular glutathione (GSH and GSSG) levels
Total GSH and GSSG levels were estimated as an index of oxidative stress by the GSH recycling assay according to the method described by [25]. Briefly, dSH-SY5Y cells were seeded in 12-well culture plates at 2 x105 cells/well. After 24 h incubation, cells were scraped from the plate and washed twice with PBS. Sulfosalicylic acid (SSA; 3.33 μl of 100% made up in distilled water) was added to the cell pellet and vortexed for 10 s. Stock buffer (96.6 μl of 125 mM sodium phosphate, 6.3 mM disodium EDTA, pH 7.5) was added to each tube, vortexed and centrifuged at 6,600 g for 2.5 min and supernatants were collected carefully and stored immediately at – 80°C prior to GSH analysis by 6 mM DTNB (5,5’-dithiobis-(2-nitrobenzoic acid)) recycling assay. The yellow color was measured at 410 nm at 0, 1, 2, 5, and 10 min using the spectrophotometer.
HPLC method development for carotenoid analysis
Carotenoid analysis was carried out isocratically on an Agilent 1200 series high performance liquid chromatography (HPLC) instrument using a method adapted from [22]. Carotenoids were separated using a reversed phase Suplex pKb-100 HPLC Column 250x4.6 mm, 5 μm (Sigma Aldrich). The mobile phase was 54% methanol, 44% acetonitrile, and 2% (propan-2-ol, 15% water) at a flow rate of 1 mL/min. Extracted carotenoids (99 μL) were injected onto the HPLC.
The HPLC system comprised of a thermostatically controlled autosampler, a quaternary pump and a variable wavelength detector (VWD). Lutein and zeaxanthin were detected at 450 nm and identified by their retention time and absorption spectrum compared with the characteristics of pure standards (>95%). Quantification was performed using the Agilent chemstation (Version B.03.01-SR1) in relation to the internal standard (ISTD). Concentrations were calculated by comparing the peak heights with standard reference curves. Standard chromatograms are shown in (Supplementary Figure 2).
Determination of linear dynamic range, LOD, and LOQ of lutein and zeaxanthin
An external calibration curve was prepared for lutein and zeaxanthin for the determination of their linear range, LOD, and LOQ. Calibration curves were produced by injecting authentic solutions between 0.01 and 40 μM. The concentration ranges selected for the calibration curves were based on preliminary data on the dynamic ranges. LOD and LOQ were calculated using the visual evaluation method from the ICH guidelines [26] and are reported in Supplementary Table 1.
HPLC method precision, accuracy, and recovery
Percentage recovery analysis was determined according to [27]. The recovery percentages were estimated by comparing the level of the carotenoid standards (lutein and zeaxanthin) with or without spiking into cells. Carotenoids were extracted as described earlier. We calculated recovery percentages for low, medium, and high concentrations of lutein (0.1, 1, 10 μM) and zeaxanthin (0.05, 0.5, 5 μM). The accuracy of the method was determined for the optimum concentrations (1 μM lutein and 0.5 μM zeaxanthin) by calculating the mean of the experimental values with the actual concentrations of the standards. Recovery was calculated for six replicates of each compound and presented as a percentage of initial material. Precision is required to be within±15% and accuracy between 85-115% [28].
Analysis of POVPC levels
Cellular POVPC levels were determined by mass spectrometry as detailed in Ademowo et al. [19]. Lipid extracts (10 μl) were separated on an Acclaim C18 column (internal diameter 2.1 mm, column length 150 mm, particle size 3 μm, Thermo Scientific, UK) using the mobile phases consisted of (A) 10 mM ammonium formate in methanol:water:formic acid (20 : 80 : 0.1, v/v/v) and (B) 2 mM ammonium formate in 2-propanol:methanol:formic acid (90 : 10 : 0.1, v/v/v) at 45°C. Flow rate was maintained at 100 μl/min with the gradient as follows: 30% B from 0 to 5 min, 30– 70% B from 5 to 20 min, 70– 100% B from 20 to 35 min, 100% B 35– 40 min, 100– 30% B from 40 to 41 min, 30% B 41– 51 min. The POVPC analyte eluted at 20.8 min while the ISTD, dPOPC eluted at 30.8 min.
Mitochondrial function assay
Mitochondrial function of dSH-SY5Y cells was measured using Agilent Seahorse XF24 Extracellular Flux Analyzer that allows the determination of oxygen and proton concentrations in real time [29]. dSH-SY5Y cells were seeded in the XF24 culture plates at 80,000 cells/well, a day prior to treatment. Cells were treated with POVPC±carotenoid for 24 h as indicated. On the day of assay, cells were incubated in assay medium containing (1 mM sodium pyruvate, 2 mM glutamine, 10 mM glucose) in a CO2-free incubator for 1 h prior to the assay in order to de-gas solutions. The Seahorse protocol consisted of calibration/equilibration step, followed by the synchronized injection of drugs and reagents in each of three different ports. Ports were loaded with oligomycin– ATP synthase inhibitor, FCCP– oxidative phosphorylation uncoupler; and a mixture of rotenone/antimycin A– inhibitors of respiratory complexes I and III respectively [30]. 3 min mix, 2 min wait, and 3 min measure cycles were used; three baseline measurements were taken before and a subsequent three measurements were taken after drug additions. Data were expressed as the rate of oxygen consumption (OCR) in pmol/min. Cell numbers (80,000 cells/well), Oligomycin (1 μM), FCCP (1 μM), and antimycin A/Rotenone (0.5/0.5 μM) concentrations were optimized prior to assay. Our optimum Seahorse reagent concentrations agree with Schneider et al. [1].
Respiratory parameters were measured by calculating respiration rates before and after the addition of electron transport chain inhibitors. The parameters calculated included ATP production (baseline respiration minus oligomycin post injection respiration); and proton leak (oligomycin respiration minus rotenone / antimycin A post injection respiration) [30]. Optimal concentrations of oligomycin, FCCP, and antimycin A/Rotenone were diluted in assay media and 75 μL of each stock solution was loaded onto an XF24 cartridge. The microplate was loaded into the Seahorse XF24 analyzer according to the manufacturer’s instruction. Experiments were carried out at 37°C. The OCR were automatically calculated, recorded, normalized with protein concentration and plotted by Seahorse XF24 software (Wave Desktop Version 2.4).
MitoSOX oxidation analysis
dSH-SY5Y cells were seeded in a 24-well plate at 2x105 cells/well. After 10 days of differentiation, cells were treated with POVPC and carotenoids; POVPC (1 μM, 20 μM, 50 μM) and carotenoids (lutein and zeaxanthin). 24 h after the cells were POVPC±carotenoid treated, the cells were incubated with media containing 5 μM MitoSOX Red Probe in a 37°C incubator for 10 min, protected from light. The media was removed and replaced with 0.5 mL measurement buffer (PBS with Ca2 +/Mg2 +). Fluorescence was measured with a microplate reader (510 nm excitation/595 nm emission); with the measurement buffer as the blank. MitoSOX oxidation values were normalized against protein concentration and calculated as a percentage of the control.
Statistical analysis
Results are expressed as mean±standard error of mean (SEM). Statistical analysis was performed using Graphpad Prism software (version 7). One-way analysis of variance (ANOVA) was used to compare the effects of different concentrations of POVPC. The effects of carotenoid on cells were measured using two-way ANOVA. Post hoc comparisons between groups were made using Dunnett’s multiple comparison test/Tukey’s test. All results are means of three independent experiments performed in triplicates (except where otherwise stated) and p < 0.05 was considered significant.
RESULTS
POVPC toxicity in dSH-SY5Y cells is associated with oxidative stress
dSH-SY5Y cells were exposed to increasing concentrations of POVPC (1, 20 μM, and 50 μM) for 24 h. Lower concentrations of POVPC did not affect viability, but significant cell death ( 25%) was observed after co-incubation for 24 h with 50 μM POVPC (p < 0.05; Fig. 1A). To confirm that POVPC is taken up by neuronal cells, we analyzed the POVPC levels in cells before and after carotenoid supplementation by our multiple reaction monitoring (MRM) mass spectrometry method. Figure 1B confirms that POVPC is detectable in cells at different concentrations according to exposure, confirming that POVPC was delivered into cells. Despite the lack of toxicity following 20 μM POVPC exposure, significantly increased concentrations of the oxPL were recovered in neurons after 24 h compared to untreated control cells (Fig. 1B). At non-toxic concentrations of POVPC, a concentration-dependent increase in oxidative stress was observed, determined as a significant decrease in total glutathione levels after exposure to > 1 μM POVPC and a significant 50% increase in mitochondrial ROS production at 20 μM POVPC (Fig. 1C, D). Confirmation of MitSOX oxidation was achieved by microscopy (Supplementary Figure 3).
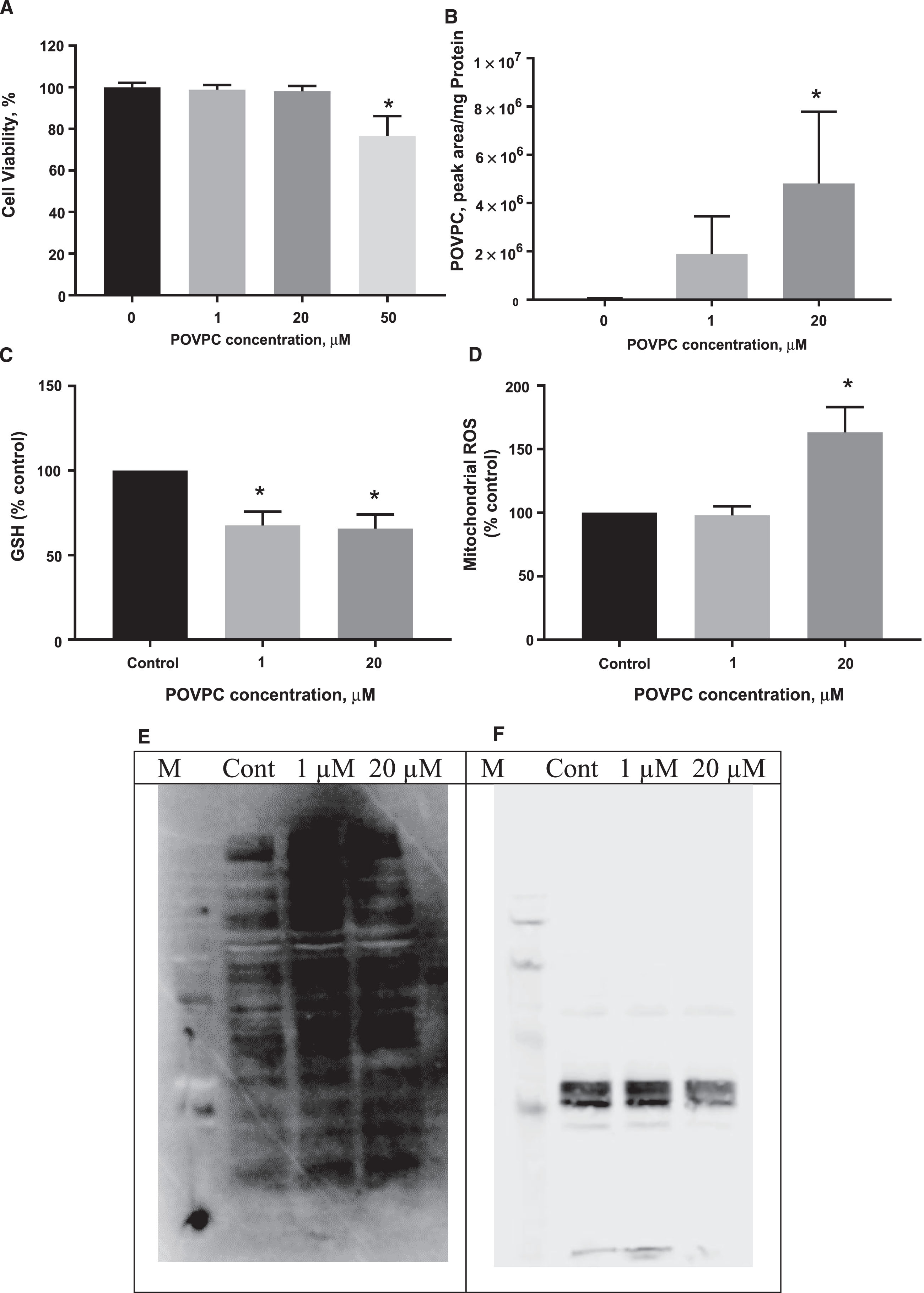
Exposure of dSH-SY5Y cells to POVPC decreases cell viability, causes toxicity and oxidative stress in a concentration-dependent manner over 24 hours. Data (A-D) are expressed as means±SEM of 3 independent assays and in triplicates and differences were evaluated by ANOVA. *p<0.05 compared to untreated control. Panels E and F are representative of two independent blots of mitochondrial proteins stained for protein carbonyls (E) with mitochondrial tubulin as a loading control (F).
To understand whether mitochondria were oxidatively modified by POVPC after 24 h treatment, we isolated mitochondria by centrifugation and analyzed mitochondrial protein carbonyls by western blot. There was a dose-dependent increase in protein carbonyl content as determined by Image J analysis that was more evident in some lower molecular weight bands than others (Fig. 1E). In both experimental replicates, the tubulin housekeeping band intensity was lower after 20 μM POVPC treatment, for equivalent protein loading (Fig. 1F).
Carotenoid uptake and antioxidant activity in dSH-SY5Y
To confirm that carotenoids were taken up by neuronal cells, cells were lysed after 24-h incubation in the presence of increasing concentrations of lutein or zeaxanthin. We were not able to detect lutein in cells treated with low concentrations of the carotenoid (0.1 μM) possibly reflecting its consumption to below detectable levels over 24 h; however, at 1 μM lutein recovery was estimated at 72.8% (Table 1). Similarly, at the lowest concentration of zeaxanthin investigated (0.05 μM) we were not able to detect any uptake into cells, however, at 0.5 and 5 μM zeaxanthin, 80 and 43.6% uptake were measured.
Precision, accuracy and % recovery of carotenoids from SHSY5Y cells
n=6.
HPLC chromatograms and calibration curves are shown in Supplementary Table 1. The accuracy was 90–106% and % CV of 5–7% [28]. The limit of detection (LOD) for lutein, zeaxanthin, and the internal standard are 0.1, 0.1, and 0.05 μM respectively while the limit of quantification (LOQ) for lutein, zeaxanthin, and the internal standard are 0.5, 0.5, and 0.2 μM, respectively.
Following coincubation of dSH-SY5Y cells with lutein (0.1 and 1 μM), there was no loss of viability (Fig. 2A). In addition, there was a significant decrease in mitochondrial ROS production (Fig. 2C) without effect on total GSH (Fig. 2B). Interestingly, the presence of POVPC increased expression of the mitochondrial antioxidant enzyme, SOD2 (Fig. 2D).
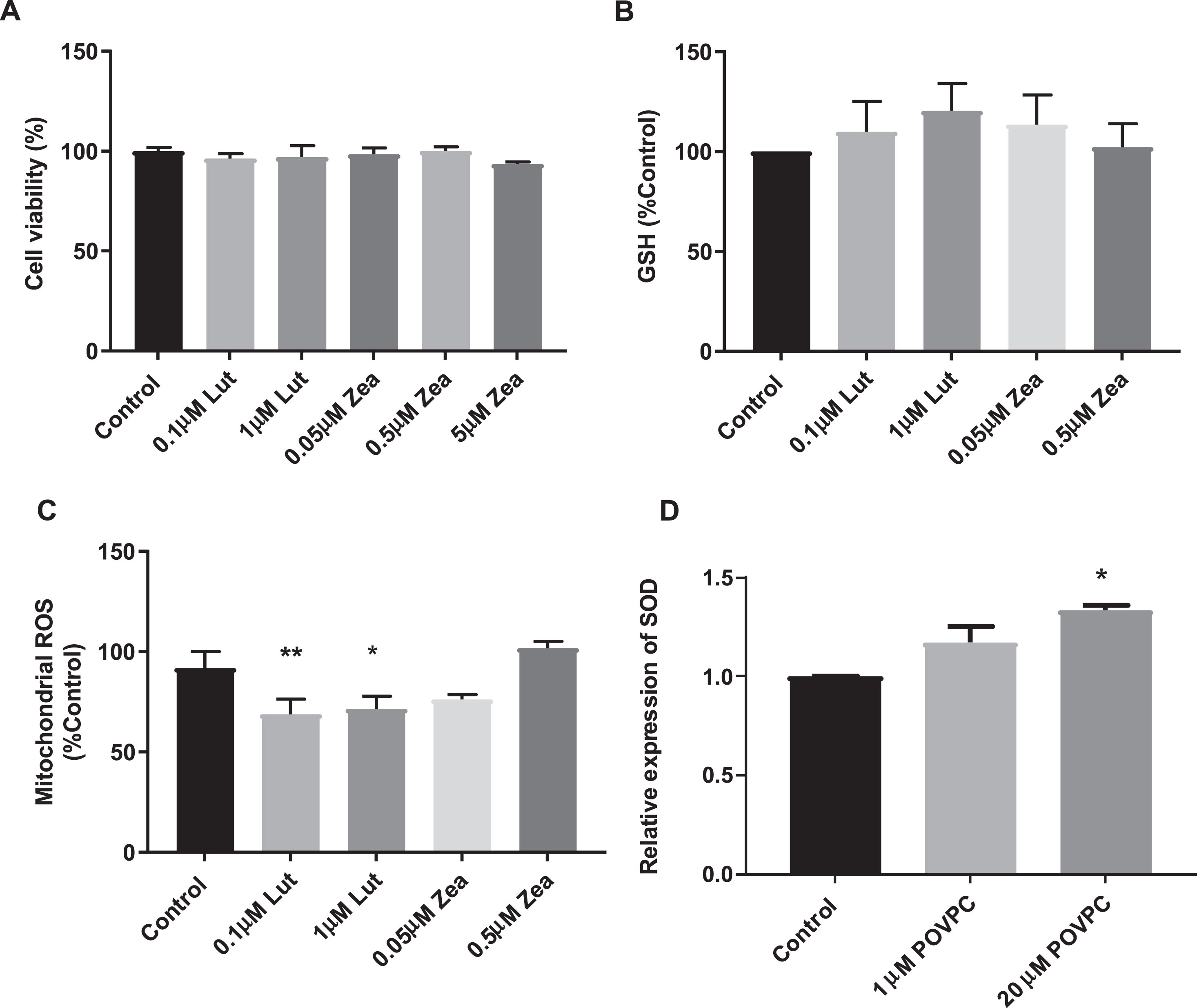
Exposure of dSH-SY5Y cells to carotenoid reduced oxidative stress over 24 hours. Data are expressed as means±SEM of 3 independent assays and in triplicates and differences were evaluated by one-way ANOVA. * p < 0.05 compared to untreated control.
Carotenoids partially protect against toxicity and oxidative stress induced by POVPC in dSH-SY5Y cells
In order to investigate any protective effect of carotenoids against POVPC-induced oxidative stress, we determined cell viability and intracellular total GSH in dSH-SY5Y cells treated with POVPC in the presence or absence of carotenoids for 24 h. The combination of POVPC and carotenoids did not affect cell viability over the concentration ranges used here (Fig. 3A, B). Both carotenoids were protective against the loss of GSH induced by 1 μM POVPC but had no effect on the changed cellular GSH levels due to 20 μM POVPC. GSSG concentration was almost doubled by 20 but not 1 μM POVPC and in the presence of lutein or zeaxanthin (0.05 μM) alone, GSSG concentration was halved (Fig. 3D).

Effect of carotenoids on POVPC-induced loss of total GSH, GSSG levels, and viability. Data are expressed as means±SEM of 3 independent assays and in triplicates apart from GSSG that was determined in duplicate.
Effects of carotenoids on POVPC-induced mitochondrial ROS
To ascertain whether carotenoids protected against POVPC-induced mitochondrial ROS production, MitoSOX red, a mitochondrial superoxide indicator was used. Figure 4 confirms that the presence of carotenoids reduced mitochondrial stress by 50% in the presence of 20 μM POVPC compared to controls. The increase in MitoSOX oxidation induced in 20 μM POVPC treated cells was mitigated by the presence of any carotenoid treatment whereas no significant effect was seen at lower POVPC concentrations.

Carotenoids protect against of POVPC (20 μM) induced MitoSOX oxidation in dSH-SY5Y cells. Data are expressed as means±SEM of 3 independent assays and in triplicates and differences were evaluated by ANOVA. ***p < 0.001 compared to no carotenoid control.
Effects of POVPC and carotenoids on mitochondrial respiration in neuronal cells
Mitochondria serve as important stores for intracellular GSH which in turn protect from uncoupling and apoptosis. From earlier experiments, we have confirmed oxidative stress was induced with 1 and 20 μM POVPC concentrations and showed some protective effects of carotenoids against POVPC. We therefore analyzed mitochondrial respiration using the Seahorse Extracellular Flux Analyzer in neurons treated with different concentrations of POVPC and carotenoids. Figure 5A shows that 1 μM POVPC treatment did not significantly affect mitochondrial function. However, 20 μM POVPC-treated cells showed significant proton leak which associated with POVPC concentration dependent loss of ATP production. While apparent proton leak induced by 20 μM POVPC was prevented by zeaxanthin and lutein (Fig. 5A), ATP production was not significantly rescued with carotenoid supplementation Fig. 5B. In order to evaluate whether this effect was due to mitochondrial number, we determined copy number. There was no effect of POVPC treatment due to change in mitochondrial content in cells (Fig. 5C).
DISCUSSION
Cross-sectional studies have shown that plasma oxocarotenoid concentrations correlate positively with cognitive function in older adults [9, 32]. In addition, dietary patterns of higher oxocarotenoid dietary intake associate with lower risk for cognitive decline in cohort studies. It is known that oxocarotenoids can act as antioxidants in vitro; however, few have investigated the underlying mechanism in detail. Here we have shown for the first time that lutein and zeaxanthin are taken up by neuronal cells and protect neuronal mitochondria from loss of function induced by the OxPL, POVPC. These findings suggest that mitochondrial function merits investigation as a biomarker of oxocarotenoid benefit.
Previous studies showed that exogenous POVPC is toxic as it is readily internalized, migrates to and damages the mitochondria [5]. Neurons rely on mitochondria to meet energy demands, and dysfunction of oxidative phosphorylation associates with several neurological disorders. In addition, impaired mitochondrial function is frequently observed in neurodegenerative diseases such as AD [33–35]. Others have shown that POVPC could induce oxidative stress and impairs mitochondria function in a dose-dependent manner; 50 μM oxidized phospholipid (PGPC and POVPC) induced apoptosis in macrophages [6, 36].
Our study shows for the first time that the oxPL POVPC (1 μM) induced neuronal oxidative stress while preserving metabolic function in vitro; however, a higher sublethal concentration of POVPC (20 μM) caused mitochondrial proton leak, loss of GSH, an increase in SOD2 expression and inhibition of mitochondrial ATP production. The decrease in metabolic activity that we observed with > 20 μM POVPC agrees with previous studies of Stemmer et al. [6].
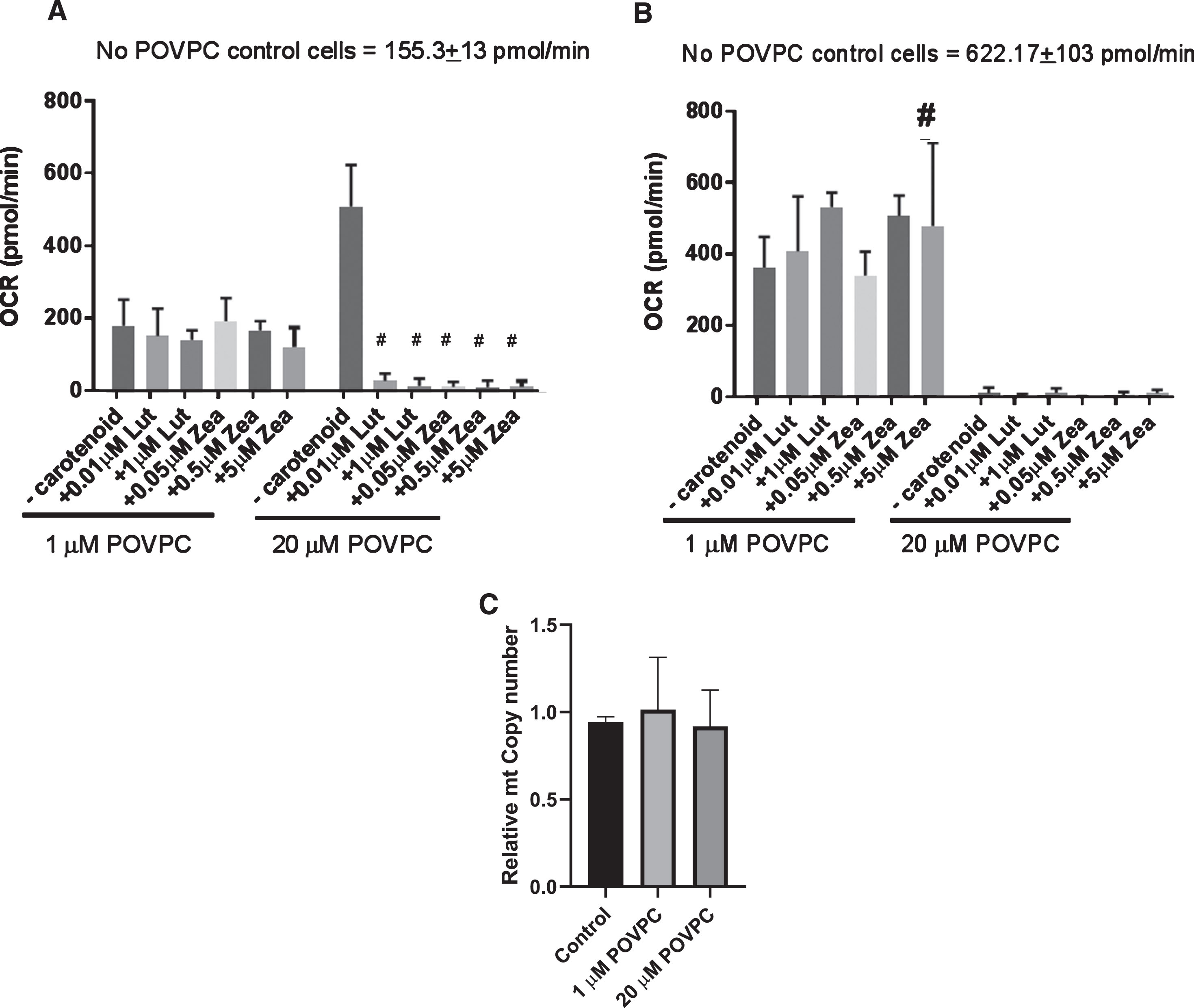
Mitochondrial stress test of SH-SY5Y cells treated with POVPC and carotenoids. Data are expressed as means±SEM of 3 independent assays and differences were evaluated by ANOVA #p≤0.0001 compared to no carotenoid control; # p < 0.05 compared to no carotenoid control.
The uptake, distribution, and metabolism of carotenoids have been attributed to the activities of different protein transporters (SR-BI, CD36, NPC1L1), digestion enzymes (PNLIP, CES), cleavage enzymes (BCO1/2), intracellular transporters (FABP2), and receptors (LPL, APOC/E, LDLR). It is predicted that carotenoids are bioavailable for many cell types [37]. Using recovery studies, here we have confirmed the uptake of carotenoids by (d)SHSY5Y cells.
The differential distribution and specificity of carotenoid cleavage enzymes, BCO 1 and 2 led us to speculate that mitochondria may be an important target for neuronal protection by oxocarotenoids, because lutein and zeaxanthin are metabolized by BCO2 which is located within mitochondria.
The Seahorse Extracellular flux analysis has emerged as a powerful tool to study mitochondrial bioenergetics [30] suggesting possible causes of mitochondrial dysfunction and an in-depth understanding of metabolic pathways, signals, and phenotypes and enables the calculations of mitochondrial and non-mitochondrial respiration. Exposure of cells to POVPC with and without carotenoids inhibited mitochondrial respiration. Proton leak and ATP production efficiency were significantly impaired in a dose-dependent manner. Proton leak and non-mitochondrial respiration (data not shown) was higher in 20 μM POVPC treated cells compared to 1 μM POVPC treated cells. This observation was independent of copy number suggesting the absence of mitophagy at the concentrations of POVPC studied here. However, we did observe an increase in protein carbonyl formation in mitochondria isolated from POVPC treated cells, suggesting they may be susceptible to oxidative damage.
Here, we showed that delivering zeaxanthin and lutein at concentrations similar to those seen in plasma was able to protect against mitochondrial proton leak and ROS production but did not mitigate against loss of glutathione or loss of mitochondrial ATP production by higher POVPC (20 μM) concentration. Despite preventing proton loss, carotenoids could not restore mitochondrial ATP production in cells exposed to higher POVPC concentrations, probably due to sustained GSH depletion. Oxidized GSH is depleted from cells by transport extracellularly when its capacity for reduction is exceeded. It is possible that the loss of ATP may reflect a switch of cell metabolism toward the pentose phosphate pathway, away from mitochondria, to produce NADPH and enable effective reduction of GSH. Both lutein and zeaxanthin have been reported to exert protective effects through the activation of the Nrf2 transcription factor in retinal epithelial cells, potentially acting as indirect antioxidants in the closely related disease of age-related macular degeneration [38–40]. Nrf2 activation leads to increased expression of antioxidant enzyme genes such as the SOD family and glutathione biosynthetic enzymes which may prevent depletion of GSH. Further studies are needed to investigate whether preincubation and then removal of oxocarotenoids from neuronal cells can offer protection against metabolic dysfunction and cell death via induction of Nrf2.
Neurons have high energetic requirements, primarily using glucose as the energy substrate. They are highly susceptible to alteration of mitochondrial function leading to glucose hypometabolism [41]. It has been proposed that maintaining mitochondrial metabolism may support neuronal survival. In support of this, metformin administration improves neuronal survival and mitochondrial function in preclinical studies of AD [42].
Previous studies have shown that mitochondrial-targeted antioxidants are capable of maintaining blood flow, mitochondrial function and physiological redox state in mice [43], again supporting the hypothesis that ROS may arise from and lead to mitochondrial dysfunction. The mechanism by which lutein and zeaxanthin protect neuronal tissue against degenerative diseases has been suggested to be due to their lipid-soluble singlet-oxygen quenching antioxidant property which is related to their localization and orientation within the lipid membrane [44, 45].
Carotenoids such as lutein and zeaxanthin are able to penetrate membranes due to their lipophilicity and may be able to restore mitochondrial function under low stress conditions [11]. Carotenoids are effective modulators of the physical properties of both natural and model membranes, increasing their rigidity and thermostability [46]. Oxo-carotenoids stabilize membranes to a greater extent than β-carotene; they are incorporated into bilayers and span the membrane with their lipophilic core, being further anchored in the aqueous area with – OH groups, thus functioning like a molecular rivet [46, 47]. Our observation that oxocarotenoids counteract POVPC-dependent proton leakage presents the possibility that POVPC may otherwise target and disrupt physical properties of the mitochondrial membrane causing loss of membrane stability and function.
Bohn et al. [31] reviewed the strength of evidence between carotenoid intake and changes in oxidative stress in human observational studies and intervention trials in health and diseases, proposing that circulating levels of carotenoids can be used as markers for total mortality. We and others have shown inverse correlations between plasma concentrations of lycopene, lutein, zeaxanthin, meso-zeaxanthanin but not carotene with circulating levels of peroxidized lipids in dementia patients. In older adults, lutein and zeaxanthin supplementation have been shown recently to facilitate the brain capacity for cognitive compensation in a functional MRI study of healthy aging; this showed the integration between different networks that are functionally segregated earlier in the lifespan. This study lends some credence to the concept that any protective effect of oxocarotenoids on cognitive function should be evaluated in prodromal stages of disease [48].
Conclusion
Physiological concentrations of oxidized phospholipid induced neuronal oxidative stress, but not a bioenergetic deficit, that can be mitigated by the xanthophyll carotenoids in a dose dependent manner. However, delivery of carotenoids did not protect neuronal cells from elevated oxidized phospholipid concentrations, probably due to GSH depletion and mitochondrial dysfunction.