
Abstract
Hyperbilirubinemia may increase the risk of Alzheimer’s disease (AD) but its mechanistic role in AD pathogenesis remains obscure. Here, we used animal models to investigate the short- and long-term effects of neonatal systemic exposure to bilirubin on brain histology and function as well as the acute effect of lateral ventricle injection of bilirubin in adult rats. We found that three days exposure to bilirubin in newborn rats could induce AD-like pathological changes in late life, including tau protein hyperphosphorylation at multiple sites, increased Aβ production in brain tissues, and spatial learning and memory injury. Bilirubin activated the activities of several protein kinases (GSK-3β, CDK5, and JNK), which were positively correlated with hyperphosphorylated tau; simultaneously increased the expression of AβPP γ-secretase PS2 and decreased the expression of α-secretase ADAM17, which were positively correlated with Aβ production. The above results were well replicated in primary hippocampal cell cultures. These data demonstrate that bilirubin encephalopathy is an AD-like disease, suggesting a potent role of bilirubin in AD.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is the most common form of dementia among the elderly. It is not an independent disease but a heterogeneous group of multiple metabolic disorders caused by a combination of pathogenic factors [1, 2]. AD is a neurodegenerative disorder that is characterized by memory loss and a progressive decline of cognitive function. These cognitive deficits correlate with neuronal and synaptic loss, neurofibrillary tangles formed by hyperphosphorylated forms of tau proteins, and the presence of neuritic plaques including amyloid-β (Aβ) peptides [3, 4].The accumulation of protein aggregates has a fundamental role in the pathophysiology of distinct neurodegenerative diseases [5].
There are two types of AD: the early-onset familial AD (FAD) and late-onset sporadic AD (SAD), although they are clinically indistinguishable. FAD usually has a genetic origin but SAD is the more common form, with unknown etiology [6]. SAD is not caused by a single factor and may rather be the result of many factors [7, 8]. Despite heterogeneous clinical phenotypes, AD is characterized by the accumulation of misfolded proteins and ubiquitinated conjugates in postmortem brains of affected patients. This phenomenon may have a common origin, where disruption of intracellular protein homeostasis during aging may be responsible [9, 10]. Up to now, a large body of evidence supports a link between malfunction of the ubiquitin-proteasome system (UPS) and dementias such as AD [11–14]. The mechanism underlying UPS malfunction and abnormal protein accumulation in AD remains unclear.
Bilirubin is a metabolic product of hemoglobin and has a well-known antioxidant property. Recently we have reported that bilirubin can inhibit UPS function by targeting proteasome-associated deubiquitinating enzymes USP14 and UCHL5, leading to abnormal protein degradation in brain tissues [15]. Normally the unconjugated (free) bilirubin (UCB) in serum is bound by albumin, and only a very small amount of free UCB may pass through the blood-brain barrier (BBB). Once UCB abnormally penetrates into the brain tissues under pathological conditions, it can lead to severe neuronal impairment, clinically known as bilirubin encephalopathy, demonstrating a positive relationship between bilirubin and neuron damage [16–18]. AD patients have significantly higher levels of bilirubin in cerebrospinal fluid (CSF), compared to control groups [19]. Increases in CSF bilirubin were even regarded as a biological indicator of AD pathogenesis [20]. However, it remains unknown how bilirubin promotes AD pathogenesis. Hence, we performed in vivo and in vitro studies to investigate whether bilirubin induces AD-like pathologies, whether bilirubin works as an endogenous pathological factor of AD, and how comparable between bilirubin encephalopathy and AD in terms of brain functioning and pathological alterations at the molecular level.
MATERIALS AND METHODS
Materials
Bilirubin was purchased from Sigma-Aldrich Inc. (St. Louis, MO, USA). Other agents used include NEM (Sigma-Aldrich Inc.), MTS assay kit (CellTiter 96 Aqueous One Solution reagent) (Promega Corporation, Madison, WI, USA). Enhanced chemiluminescence (ECL) reagents were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz Biotechnology, Santa Cruz, CA). All antibodies are described in Supplementary Table 1.
Animals
The protocol for the care and use of all animals in this study was in accordance with the Guangdong Animal Center for the ethical treatment of animals and approved by the Institutional Animal Care and Use Committee of Guangzhou Medical University (SYXK2016-0168, Guangzhou, China). Sprague-Dawley rats (Grade SPF) were obtained from Guangdong Laboratory Animal Monitoring Institute. All rats were maintained in a temperature- and humidity-controlled room (22±2°C) and on a 12-h/12-h light/dark cycle. All animals had access to standard laboratory diet and drinking water ad libitum, and newborn rats were breastfed.
Bilirubin solution preparation and animal model construction
Bilirubin solution preparation for rat’s intraperitoneal injection was as follows: bilirubin (100 mg, Sigma-Aldrich, USA) was dissolved in 0.5 M NaOH solution (1 ml) and diluted in ddH2O to a concentration of 10 mg/ml, and the pH was adjusted to 8.5 with HCl (0.5 M). Bilirubin solution for rat’s lateral ventricle injection was diluted to final concentration of 10 mmol/L. And bilirubin solution for primary culture of rat hippocampal neurons was bilirubin dissolved in DMSO to a final concentration of 10 mmol/L. All solution was stored at –20°C, in the dark [15].
Animal model A: acute bilirubin treatment
A cohort of neonatal male rats at postnatal day 3 were randomly divided to 4 or 5 experimental groups, and were intraperitoneally injected with vehicle (control group) or 12, 25, 50, and 100 μg/g bilirubin, respectively, twice every day for 3 days [15]. All rats were sacrificed 12 h after the final injection for analyses.
Animal model B: lateral ventricle injection
Adult male rats (280±20 g) were randomly divided into 3 or 4 groups and were deeply anesthetized intraperitoneally with chloral hydrate (30 mg/kg) and placed in a stereotaxic instrument (SR-6 N, Japan). After scalp incision and exposure of the occipital bone, holes were drilled at coordinates of 1.0 mm posterior, 1.5 mm lateral to bregma, and 4.2 mm in depth. Via each hole, a microinjector (10 μl) was slowly implanted into the lateral ventricle of the brain, then a volume of 10 μL solution containing either vehicle or bilirubin (12, 25, 50 μmol/L) saline solution was administered slowly. At 24 h after the intraventricular injection, the hippocampal tissue was collected for further studies.
Animal model C: Long-term observation
Cohorts of male newborn rats subjected to the same treatment as described for animal model A were used for follow-up studies at 1 month or 4 months of age. Both the 25 and the 50 μg/g bilirubin injection regime gave rise to significant hyperbilirubinemia (data not show). Since the rats with neonatal exposure to the 100 μg/g of bilirubin showed an extreme high mortality within 4 months, this dose group is not included for the studies at 4 months of age.
Morris water maze
The spatial memory was blindly evaluated using a Morris water maze (MWM) test. Before each experiment, the rats were brought to the site to allow them to be acclimatized. The temperature of the room and the water were kept at 24±2°C. For spatial learning, the rats were trained to find a hidden platform for 6 consecutive days, 4 trials per day with a 20- to 30-s interval for each rat from 3.00 to 8.00 p.m. On each trial, the rat started from one of the four middle quadrants facing the wall of the pool and ended when the animal climbed on the platform. They were guided to the platform if they could not find the platform within 60 s. Through these training sessions, rats acquired spatial memory about the location of the safe platform. The swimming pathways and the latencies of the rats to find the hidden platform were recorded each day. The pathway and the length that the rat passed through the previous platform quadrant were recorded by a video camera fixed to the ceiling of the room, 1.5 m from the water surface. The camera was connected to a digital-tracking device attached to a computer loaded with the water maze software (Huaibei Zhenghua Biologic Apparatus Facilities). The spatial memory was tested 48 h later after the last training. When the platform was withdrawn, the path, distance, or time was recorded; the longer a rat stayed in the target quadrant where previously the platform had been located, the better it scored for the spatial memory (the distance and the time traveled in the target quadrant and the escape latency were used to indicate the score).
Western blot
Whole cell lysates and rat hippocampal tissue homogenates were prepared in RIPA buffer (1×PBS, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with 10 mM β-glycerophosphate, 1 mM sodium orthovanadate, 10 mM NaF, 1 mM phenylmethylsulfonyl fluoride (PMSF), and 1×Roche Complete Mini Protease Inhibitor Cocktail (Roche, Indianapolis, IN). SDS-PAGE, transferring, and immunodetection were performed as previously described [15]. In brief, equal amounts of total protein extracts were fractionated by 12% SDS-PAGE and electrically transferred onto a polyvinylidenedifluoride (PVDF) membrane. Primary antibodies and appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies were used to detect the designated proteins. The bound secondary antibodies on the PVDF membrane were reacted to the ECL detection reagents (Santa Cruz, CA) and detected via exposing to X-ray films (Kodak, USA). Multiple exposures were taken to select images within the dynamic range of the film. Selected films were scanned and quantified using Image J software. β-Actin/GAPDH bands were used for normalization.
Enzyme-linked immunosorbent assay (ELISA)
The cortex tissue was homogenized in guanidine hydrochloride buffer (GuHCl) (5 M GuHCl, 50 mM Tris-Cl, pH 8.0) containing a protease inhibitor cocktail (KeyGENBioTECH, catalog #KGP603), ultrasonicated, and centrifuged at 13,000 g for 10 min. The supernatant was collected and total protein was measured with a Micro BCA protein assay (Pierce). Aβ40 and Aβ42 were separately measured with ELISA Kits (Cloud-Clone Corp.) by following manufacturer’s instructions.
Quantitative RT-PCR
Total RNA from neonatal SD rat’s cortex was isolated using the RNAiso Plus (Takara Bio Inc, catalog #9108). cDNA was prepared from the RNA using the PrimeScriptTM RT reagent Kit with gDNA Eraser (Takara Bio Inc, catalog #RR047A). The real-time PCR amplification was performed by the SYBR Green method using SYBR Premix Ex TaqTM II (Takara Bio Inc, catalog #RR820A), and according to manufacturers’ instructions.
The following primers were used: APP: 5’-TGACAAGAAGGCCGTTATCC-3’ (forward), 5’-TGAGCATGGCTTCAACTCTG-3’ (reverse); PS1 : 5’-CCTCATGGC CCTGGTATTTA-3’ (forward), 5’-CCAGCATACGAAGTGGACCT-3’(reverse); PS2 :5’-AGAACGAGGACGACTGTGAGGAG-3’ (forward);5’-TGACAGGCACGAACAGCATGATC-3’(reverse);BACE1 : 5’-AATCAGTCCTTCCGCATCAC-3’(forward), 5’-GCCTGTGGATGACTGTGAGA-3’(reverse); ADAM17 : 5’-CAAGGTGTGCGGCAACTCCAG-3’(forward),5’-CAGCAGGTGTCGTTGTTCAGGTAC-3’(reverse); GAPDH:5’-AAGGGCTCA TGACCACAGTC-3’ (forward), 5’-GGATGCAGGGATGATGTTCT-3’ (reverse); β-actin was purchased from Sangon Biotech (catalog #B661202). Results were normalized against the internal control using the Δ-ΔCT method.
Histology and immunohistochemistry
At different experimental time point, rats were anesthetized and transcardially perfused rapidly with 200 ml normal saline solution, and fixed in situ by perfusion for 20 min at 4°C with Zamboni’s solution containing 4% paraformaldehyde, 15% saturated picric acid, and 24 mM NaH2PO4/126 mM Na2HPO4 (pH 7.2). The brain was removed from the skull of the fixed animals and post-fixed in the same Zamboni’s solution for another 24 h at 4°C. Then coronal slices (20 μm) were cut with a Vibratome (Leica, VT1000 S, Germany).
Sections were mounted on glass slides and stained with the HE protocol: The slides were immersed in hematoxylin solution for 2–5 min, washed in tap water for 1 min, separately concentrated hydrochloric acid alcohol, washed in tap water for 3–5 min, transferred to eosin for 20–30 s. Conventional dehydration, transparenting with xylene, and mounting with neutral resins were performed. Morphological changes in the stained sections were examined with a light microscope (Olympus BX60, Tokyo, Japan) and photographed. Three consecutive sections from each brain (n = 3) were used for quantification. The integrated optical density (IOD)/Area values of the tissue sections in each group were measured by Image-Pro Plus v. 6.0 software.
The immunohistological procedures were as follows: Sections were permeabilized with 0.3% H2O2 in absolute methanol for 10 min to block endogenous peroxidase, and non-specific sites were blocked with 5% bovine serum albumin (BSA) for 30 min at room temperature and incubated with primary antibodies for 24 h at 4°C. After washing with PBS, sections were subsequently incubated with biotin-labeled secondary antibodies for 1 h at 37°C. The immunoreaction was detected using horseradish peroxidase-labeled streptavidin for 1 h at 37°C and visualized with the DAB tetrachloride system that stains brown. A negative control for every antibody was used with PBS, then observed under a light microscope (Olympus BX60, Tokyo, Japan) and photographed.
Silver staining
In brief, brain sections (20 μm) were mounted on glass slides, hydrated through a series of alcohol solutions, then subject sequentially to the following treatments: 1) 0.3% KMnO4 10 min, then washed to water clarification; 2) 1% C2H2O4•2H2O 2 min, and La(NO3)3 solution (1.15 mM La(NO3)3•6H2O, 14.7 mM CH3COONa•3H2O) 1 h, then washed 3×5 min; 3) alkaline AgI (0.1 M NaOH, 0.06 M KI, 1% AgNO3 35 ml) incubation 45 min in 37°C, avoided light, and the 1% acetic acid washing 3×1 min; 4) the developer solution (I:0.47 M Na2CO3; II: 25 mM NH4NO3, 11.77 mM AgNO3, 3.5 mM H6O39SiW12; III: 25 mM NH4NO3, 11.77 mM AgNO3, 3.5 mM H6O39SiW12, 6.1 ml 40% HCHO, three solutions mixed equal parts, temporarily preparation and avoided light) incubated 30 min, then 1% acetic acid washed; 5) 0.5% AuCl3 30 s, and then washed 2×5 min; 6) 1% NaS3O3•5H2O fixed 1 min, and washed 10 min. Then generally dehydrated, hyalinized, sealed and then observed and photographed using a microscope (Olympus BX60, Tokyo, Japan) equipped with a digital camera.
Thioflavin-S staining
Brain sections (20 μm thickness) mounted on glass slides were hydrated through a series of alcohol solutions, soaked in 0.3% KMnO4 for 4 min, followed by a wash with 1% oxalic acid for 1 min. Next, the brain sections were incubated with 0.05% aqueous Thioflavine-S (SIGMA, catalog #T1892) dissolved in 50% ethyl alcohol for 8 min at room temperature in dark, then washed in 50% ethyl alcohol twice and subsequently in three exchanges of distilled water. The slides were then incubated in a high concentration of phosphate buffer (411 mM NaCl, 8.1 mM KCl, 30 mM Na2HPO4, 5.2 mM KH2PO4, pH 7.2) at 4°C for 30 min, washed, and finally sealed in 10% glycerin with coverslips. The stained sections were stored in a dark box at 4°C before they were observed with a fluorescence microscope (Leica, Germany) and photographed. Plaques were quantified using Image J software by measuring the area of Th-S staining in a well-defined selected area of the cortex.
Primary cell culture
Hippocampal neurons were prepared from one-day-old rats for primary cell cultures [15]. In brief, the rats were rapidly decapitated and the brains were isolated in calcium-free Hanks’ balanced salt solution (HBSS-2; Life Technologies Inc., Grand Island, NY); the hippocampus was mechanically fragmented, transferred to a 0.025% trypsin in HBSS-2 solution, and incubated with 0.25% trypsin at 37°C for 15 min. Following trypsinization, cells were washed twice in HBSS-2 containing 10% fetal calf serum, and resuspended in the Neurobasal medium (Life Technologies Inc.), and triturated with a Pasteur pipette, then the cell suspension was filtered through a nylon mesh (200 μm), and centrifuged at 210×g for 5 min. The cells were suspended in the Neurobasal medium supplemented with 2% B27 (Life Technologies Inc.), GlutaMAX, 100 U/ml penicillin, and 100 μg/ml streptomycin, and plated on poly-D-lysine (0.1 mg/ml) pre-coated 12-well tissue culture plates, and maintained at 37°C in a humidified atmosphere of 95% air and 5% CO2. Medium was changed after 24 h, and every three days 0.5 ml of old medium was removed by aspiration and replaced by the same volume of fresh medium during the culturing period. Cells were used after 8 days in culture. Neurons were morphologically characterized by phase contrast microscopy, and by indirect immunocytochemistry for neurofilaments.
Immunofluorescence
Primary hippocampal neurons were seeded in 12-well plates containing PDL coverslips. Cells were treated and fixed with a 4% paraformaldehyde solution at room temperature for 15 min and washed 3 times in PBS (pH7.4) for 5 min each. Cells were then permeabilized with 2% Triton PBS (2 ml Triton/100 ml PBS) for 10 min and blocked with 5% BSA in PBS for 1 h at room temperature in a humid chamber, then incubated overnight at 4°C with primary antibodies. Removal of unbound primary antibodies via washing with buffer was followed by incubation with appropriate fluorescent dyes conjugated secondary antibodies at room temperature for 1 h. The cells were then incubated with 4,6-diamidino-2-phenylindole (DAPI) (10.5 μg/ml, molecular probes by life technologies) for 15 min before they were observed using a fluorescence microscope (Leica, Germany) and photographed [15].
Statistical analysis
All the results were expressed as Mean±SD where applicable. GraphPad Prism 4.0 software (GraphPad Software) was used for statistical analysis. Differences between two groups were evaluated for statistical significance using two-tailed unpaired Student’s t-test. A difference among 3 or more groups, one-way ANOVA or when appropriate, 2-way ANOVA, followed by the Holm-Sidak test for pair-wise comparisons were performed. p value of < 0.05 was considered statistically significant.
RESULTS
Short term of exposure to bilirubin induces AD-like cognitive impairment in late life in rats
Animal Model C was used for cognitive impairment test. Newborn rats were treated with bilirubin (12, 25, 50, or 100 μg/g, i.p, twice/day) for 3 consecutive days, 1 or 4 months later spatial learning and memory were tested with MWM. One month after the bilirubin injections, all rats were trained for 6 consecutive days to remember the location of the platform. The MWM data showed that the latency (the time to find location of the platform) was significantly increased for bilirubin-treated rats relative to the control rats, which indicates a dose-dependent decrease of learning capability with bilirubin treatment (Fig. 1A).When the platform was removed, rats’ memory was tested, and the results showed that bilirubin-injected rats stayed less time in the target quadrant and traversed fewer times through the target quadrant than the control group (Fig. 1B, C), which unraveled the fact that bilirubin could also induce a dose-dependent decrease in rats’ memory. When grown up to 4 months of age, the bilirubin treated rats (12, 25, 50 μg/g, i.p, twice/day) yielded the similar results to 1-month-old rats as shown in Figure 1D-F. Overall, the data from the water maze tests demonstrate that neonatal short exposure to elevated bilirubin can lead to impairment in the spatial learning and memory in the later life.

Bilirubin treatment impairs spatial learning and memory. Morris water maze tests were performed on rats at 1 month (A, B, C) and 4 months (D, E, F) after they were subject to intraperitoneal injections with either vehicle control (Con.) or the indicated doses of bilirubin twice per day for 3 consecutive days starting at postnatal day 3. A) The effect of bilirubin treatment on the escape latency of the rats to find the hidden platform at the 1-month time point. B) The effect of bilirubin treatment on the time traveled in the target quadrant. C) The times of passing through the target point in 60 s after the platform was withdrawn. D) The results of swim training at 4 months after bilirubin injection, illustrating rats’ learning ability. E) The effect of bilirubin treatment on the distance at the 4-month time point. F) The effect of bilirubin treatment on the times traversed through the target point in 60 s after the platform was withdrawn. Representative trajectory charts, and the summary of the escape latency, the traveling distance and duration of time are shown. Results from one of two sets of independent experiments are shown. Results are expressed as means±SD (n = 8). *p < 0.05, **p < 0.01 versus the control group (Con.), and #p < 0.05, ##p < 0.01 versus the 12 μg/g dose group.
Since short exposure to bilirubin induces long-lasting characteristic of neuronal damage, which triggers the decrease in learning and memorizing abilities similar to AD, we then carried out further examinations to detect the major AD-associated pathological characteristics including tau hyperphosphorylation and Aβ production.
Bilirubin treatment induces hippocampal tau protein hyperphosphorylation in rats
Three in vivo models were used for hippocampal tau protein assays. In
As shown in Figures 2A (Model A), 2B (Model B), and 2 C and 2D (Model C, 1 month and 4 months, respectively), the total amount of tau protein (Tau 5) remained unchanged by the treatment of bilirubin in all three models; however, decreases in the staining density of Tau-1 (non-phosphorylation site of tau at serine-199/202) and increases in tau hyperphosphorylation as reflected by the staining densities of pS396, pS214, pS262, or pT231(specific phosphorylation of tau at Serine396, 214, and 262 or at Threonine 231) were observed not only in the acute models (Model A and B; Fig. 2A and 2B) but also in the chronic model (Model C; Fig. 2 C and 2D).These findings indicate that the bilirubin treatments led to hyperphosphorylation at multiple epitopes of tau proteins in the hippocampus, a biochemical hallmark of AD.

Bilirubin treatment induces hyperphosphorylation at multiple epitopes of tau proteins in the hippocampus and cortex tissue. Total tau proteins and phosphorylated tau proteins at Tau-1, Ser396, Ser262, Ser214, and Thr231 epitopes in hippocampus were detected with western blot analyses. Representative images and summary data were shown in the various models: (A) and (a) newborn rats after bilirubin treatment for consecutive 3 days; (B) and (b) adult rats treated with bilirubin via lateral cerebral ventricle injection; (C) and (c) 1 month old and (D) and (d) 4-month-old rats who had received bilirubin treatment for 3 consecutive days initiated at their postnatal day 3 (n = 5). In (a), (b), (c), and (d), results are expressed as means±SD (n = 5). *p < 0.05, **p < 0.01, ***p < 0.001, versus the control group. (E) and (e) immunostaining of tau protein and T22, and silver staining in hippocampus and cortex tissues. Rats were treated as described in (D). Immunohistochemical images of tau-1 (non-phosphorylation site), pS396 (Tau protein phosphorylated at Ser396), and T22 (oligomeric tau), as well as the images of silver staining in the hippocampus were shown in E (upper). Immunohistochemical staining of tau protein phosphorylated at Ser-262 epitope (pS262) and tau oligomers (T22) in the neurons, as well as pS262 and silver staining in the nerve fibers of the cortex are shown in E (middle). Scale bar = 100 μm. In (e), results are expressed as means±SD (n = 3). *p < 0.05, **p < 0.01, versus the control group.
To further test whether bilirubin exposure at the neonatal stage leads to brain pathological changes in adulthood, we also performed immunochemical staining for phosphorylation of tau at S396 or S262 and for the oligomeric tau (detected by T22 antibody) as well as silver staining in the hippocampus or cortex of the brain from the 4-month-old rats as described in Model C. Immunochemical results showed that bilirubin treatment induced hyperphosphorylation of tau proteins at multiple epitopes (Ser199/202, Ser396) in the hippocampus, at Ser262 epitopes in the neuron and fibers of cortex. Oligomeric tau proteins in both the hippocampus and cortex were increased by bilirubin treatment in a dose-dependent manner (Fig. 2E), which was further confirmed by the silver staining (Fig. 2E). All the morphological detections demonstrate that neonatal short-term exposure to bilirubin induces long-lasting brain tau hyperphosphorylation, a pathological change also seen in AD brains.
Effects of bilirubin on tau phosphorylation related kinases and phosphatasesin rat hippocampal tissues
The phosphorylation level of tau protein mainly depends on the relative activity of protein kinases and protein phosphatases [4]. Next the main kinases, including glycogen synthase kinase-3 (GSK-3), mitogen-activated protein kinase (MAPK), c-Jun amino terminal kinase (JNK), and cyclin-dependent kinase 5 (CDK5) that can hyperphosphorylate tau proteins, as well as phosphatasePP-2A, were further assessed in the brain tissue samples from the various animal models [21–25]. As described in various animal models, the hippocampal tissues were collected for western blot analyses. In
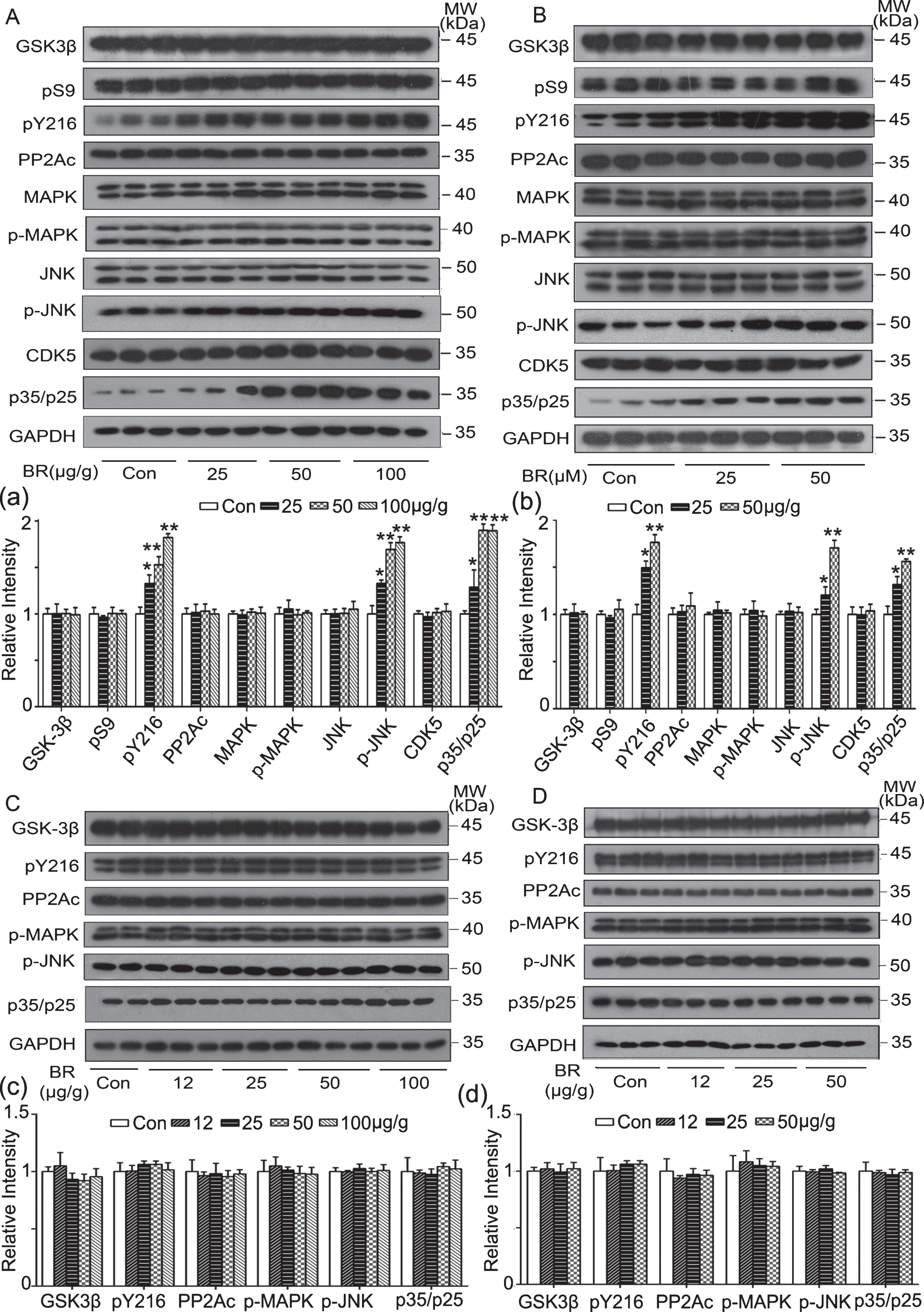
Effect of bilirubin on the tau-phosphorylation kinases and phosphatase in brain tissues. Tau phosphorylation-associated kinases (GSK-3β, MAPK, JNK, and CDK5) and phosphatase (PP-2Ac) in the hippocampus were detected with western blot. Representative images and summary data in various models are shown: (A) and (a) newborn rats after bilirubin treatment for 3 consecutive days; (B) and (b) adult rats treated with bilirubin via lateral cerebral ventricle injection; (C) and (c) 1 month; and (D) and (d) 4-month-old rats after bilirubin treatment for 3 consecutive days initiated at postnatal day 3 (n = 5). In (a), (b), (c), and (d), results are expressed as means±SD (n = 5). *p < 0.05, **p < 0.01, versus the control group.
Bilirubin treatment increases Aβ production in rat brain
To test Aβ production in brain tissues,

Bilirubin treatment increases Aβ production in rat brains. Rats subjected to bilirubin treatment for 3 consecutive days initiated at postnatal day 3 were used at 1 month or 4 months of age. Aβ40 relative content was detected with ELISA and immunostaining, and Th-S was detected with immunostaining in rat’s cortex, respectively. A, B) Changes of Aβ40 in the cerebral cortex of 1-month-old rats and 4-month-old rats. Aβ40 and Aβ42 contents are expressed as means±SD (*p < 0.05, **p < 0.01 versus control group; #p < 0.05, ##p < 0.01 versus 12 μg/g group; ΔΔ p < 0.01 versus 25 μg/g group, n = 5). (C, D) and (c, d) Th-S-staining (left) and Aβ40 expression (right) detected with immunostaining in the cerebral cortex of 1-month-old rats and 4-month-old-rats (scale bar = 100 μm). In (c) and (d), relative level of OD are expressed as means±SD (n = 3). *p < 0.05, **p < 0.01, versus the control group; # p < 0.05, ##p < 0.01, versus 12 μg/g group.
Effects of bilirubin treatment on AβPP in hippocampal tissue of SD rats
Because the increased production of Aβ can be caused by the increased metabolic cleavage of the amyloid-β protein precursor (AβPP), we next explored the effect of bilirubin on AβPP expression in the three animal models (

Effect of bilirubin treatment on the amyloid precursor protein AβPP. Total AβPP and Thr-668 phosphorylated AβPP proteins in the hippocampus were detected with western blot. Representative western blot images and summary data were shown in various models: (A) and (a) newborn rats after bilirubin treatment for 3 consecutive days; (B) and (b) adult rats treated with bilirubin via lateral cerebral ventricle injection; 1-month-old (C) and (c) and 4-month-old rats (D) and (d) who had been treated with bilirubin treatment for 3 consecutive days at their neonatal stage. In (a), (b), (c), and (d), relative intensity is expressed as means±SD (n = 5). *p < 0.05, **p < 0.01, versus the control group. E, F) AβPP mRNA levels in the hippocampus detected with qPCR. The left side is the data from the neonatal rats as in (A) and the right side is from rats subjected to the intracerebroventricular injection as in (B). mRNA is expressed as means±SD (n = 5).
Effect of bilirubin on the secretion enzyme related to AβPP metabolism in SD rat brain tissue
Based on the above results, we speculated that bilirubin-induced AβPP reduction might result from the metabolic degradation of AβPP. The classic pathways for metabolic cleavage of AβPP are the α-secretase pathway (non-amyloid metabolic pathway) and the β-secretase pathway (amyloid metabolic pathway). Therefore, the relevant enzymes in these two metabolic pathways, including α-secretase (ADAM17, main component), β-secretase (BACE1), and γ-secretase (PS1, PS2) [28–30], were examined in the three animal models. The results showed that, with the increase of the bilirubin dose, the content of ADAM17 gradually decreased and the expression level of PS2 increased, while PS1 and BACE1 remained unchanged (Fig. 6A) in
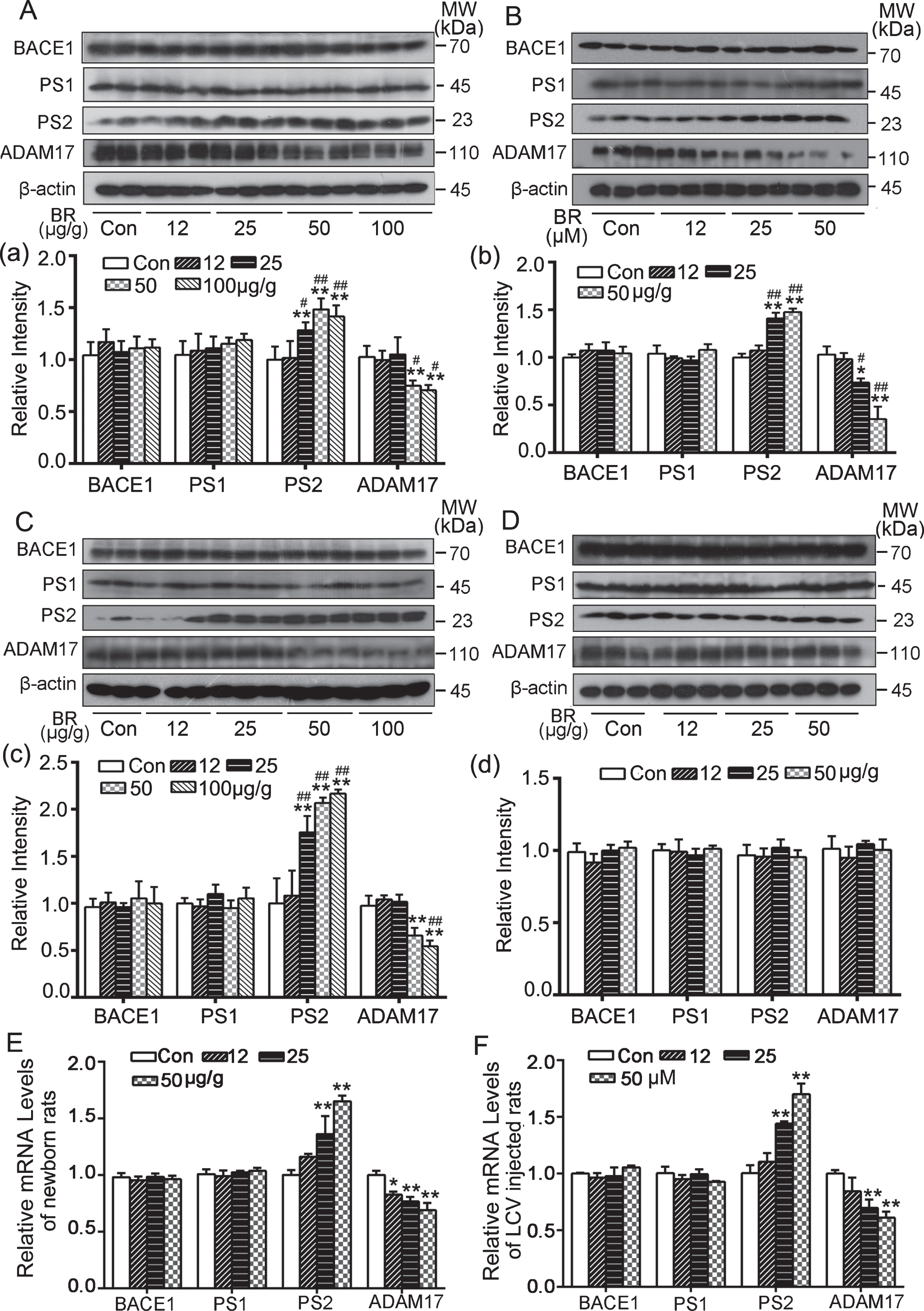
Effect of bilirubin treatment on AβPP cleavage enzymes in rat brain tissues. AβPP cleavage enzymes (BACE1, PS1, PS2, ADAM17) in hippocampus were detected with western blot analyses. Representative western blot images and summary data were shown in various models: (A) and (a) newborn rats after bilirubin treatment for 3 consecutive days; (B) and (b) adult rats treated with bilirubin via lateral cerebral ventricle injection; 1-month-old rats (C) and (c) and 4-month-old rats (D) and (d) who had received bilirubin treatment for 3 consecutive days at their neonatal stage. In (a), (b), (c), and (d), relative intensity is expressed as means±SD (n = 5). *p < 0.05, **p < 0.01, versus the control group; #p < 0.05, ##p < 0.01, versus 12 μg/g group. E, F) mRNA levels of AβPP cleavage enzymes in hippocampus were detected with qPCR. The left side is the data from the neonatal rats as described in (A) and the right side is from the rats subjected to intracerebroventricular injection of bilirubin as described in (B). mRNA is expressed as means±SD (*p < 0.05, **p < 0.01, versus control group, n = 5).
To explore the potential cause for the protein level changes of the above metabolic enzymes in Model A and Model B, we used qPCR assays to assess their transcript levels. We found that the transcript level of the PS2 gene dose-dependently increased while the expression level of ADAM17 decreased as the bilirubin dose increased; PS1 and BACE1 mRNA showed no significant changes in either
Effect of bilirubin on primary hippocampal neurons
Based on the in vivo results above, we performed in vitro assays to confirm further those changes similar to AD-like pathological characteristics. At 3 days after plating, primary hippocampal neurons in culture were treated for 12 h with different concentrations of bilirubin (3, 6, 12 μM), then western-blotting, immunofluorescence and qPCR were carried out. The results showed that following bilirubin treatment, tau protein was hyperphosphorylated at Tau-1and Ser396 sites (Fig. 7A, B); tau phosphorylation-associated kinases p-Tyr216-GSK3β, p-JNK and the active form of CDK5 p25/35 were increased (Fig. 7B); the immunofluorescence results showed a dose-dependent accumulation of Aβ in hippocampal neurons (Fig. 7 C); PS2 was increased and ADAM17 decreased by bilirubin treatment while AβPP, pT668, BACE1, and PS1 were stable during this process (Fig. 7D). As shown in Figure 7E, bilirubin induced an increased expression of PS2 mRNA and decreased expression of ADAM17 mRNA in the cultured neurons.

AD-like changes in cultured primary hippocampal neurons. Cultured primary hippocampal neurons were treated for 12 h with different concentrations of bilirubin (3, 6, 12 μM), then the protein and mRNA levels of the indicated proteins were assessed. A) Tau-1and phosphorylated tau at Ser396 site detected with immunofluorescence (n = 3). B) Tau protein phosphorylation and associated kinases measured with western blot analyses (n = 6). C) Aβ40 level revealed with immunofluorescence (n = 3). D) AβPP and its related secretases assessed with western blots. E) mRNA level of AβPP and its related secretases measured with qPCR (*p < 0.05, **p < 0.01 versus control group, n = 6).
DISCUSSION
AD is the most common neurodegenerative disorder and a major driver of dementia syndromes around the globe. Despite great advances in neurodegenerative research over the past decade, AD remains a significant diagnostic and treatment challenge, and imposes tremendous socioeconomic burdens [31]. The etiology of AD is complicated. The upstream activators lead to tau hyperphosphorylation and Aβ increases but the underlying mechanisms are not fully understood [32]. In addition, abnormal protein aggregation in brain tissues in AD is far from being fully understood. Several studies have revealed that early abnormalities in brain glucose and insulin metabolism might be etiological events in the pathogenesis of SAD and free radicals and oxidative stress may play key roles; however, none of these elements can explain the full spectrum of AD pathological and clinical characteristics [33–35]. Based on our results, bilirubin toxicity could potentially explain most of these phenomena.
Firstly, we have previously reported that bilirubin could inhibit protein degradation by directly inducing UPS malfunction in neurons, thus leading to neurotoxicity in vitro and impairment of spatial learning and memory in vivo [15]. The in vivo results have also been confirmed in this study (Fig. 1). Both ubiquitinated protein accumulation and cognitive impairment induced by bilirubin strikingly resemble changes in patients with AD.
Secondly, tau phosphorylation was tested in various bilirubin exposure animal models and in vitro. These results demonstrate that bilirubin can not only acutely induce AD-like tau-pathological changes in neonatal and adult rat brains but also, strikingly, the tau-pathological changes induced at the neonatal stage are long-lasting and clearly detectable after the rats fully grow up into their adulthood (1 month and 4 months). Positive staining for oligomeric tau and silver staining further revealed that bilirubin encephalopathy replicates the in vivo pathological condition of AD [36]. It was further found that the enhanced tau phosphorylation was associated with tau-related kinase activation. Bilirubin showed no effect on the gene expression of the kinases related to tau phosphorylation, but the phosphorylation levels of three kinases, including GSK-3, CDK5, and JNK, were significantly increased by bilirubin treatment, indicating that bilirubin can lead to the activation of the kinases responsible for AD-associated tau hyperphosphorylation in the brain. Meanwhile, there was no change in the expression and enzyme content of the phosphatase. Thus, under specific acute conditions, bilirubin induces tau hyperphosphorylation by activating multiple proteins kinases. However, in 1-month-old and 4-month-old rats, tau-related kinases were not activated possibly due to the disposal of bilirubin, indicating that kinase activation is due to the direct effect of bilirubin. Even though the increased kinase activation disappeared during the 1- and 4-month follow-up, phosphorylated tau proteins persistently existed in rat brain tissues. It has been reported that neurotoxicity could induce nonspecific tau phosphorylation [37]; therefore, we speculate that bilirubin-induced neurotoxicity either by UPS inhibition [15] or unknown factors potentially contributed to tau hyperphosphorylation in rat brain tissues.
Thirdly, another typical pathological parameter Aβ was investigated. It was found that in acute models including
It has been reported that aberrant protein aggregation could further inhibit protein degradation by impairing the UPS [39]. Several groups have shown that full-length AβPP can be ubiquitinated, either under physiological condition [40] or after pharmacologic inhibition of the proteasome [41, 42], supporting the notion that AβPP can undergo ubiquitination-dependent proteasomal degradation [43].As shown in Figure 5, bilirubin treatment in newborn rats and LCV injected rats directly induced decreases in AβPP proteins, which is possibly caused by AβPP cleavage to form Aβ since AβPP mRNA level was stable in the whole process. Secondary to bilirubin treatment, AβPP protein level was slightly decreased in 1-month-old rats but unchanged in 4-month-old rats. This is perhaps because decreased degradation of AβPP due to bilirubin-induced proteasome malfunction and protein aggregation has countered the AβPP protein level decreasing trend resulting from AβPP cleavage.
To summarize, these findings have provided novel evidence that bilirubin potentially has an important role in AD pathogenesis. Even though there is a positive relationship between bilirubin and AD, the plasma bilirubin is generally within the physiological range and there is no evidence for an increased permeability of the BBB in AD patients; therefore, it seems difficult for the physiological level of bilirubin to penetrate BBB, thereby leading to AD [20]. Nevertheless, there are more than one major pathway for the production and increase of bilirubin in neurons. In addition to bilirubin from the peripheral blood, neuronal bilirubin can be from the neuron-autologous intracellular metabolic pathway [44, 45].
From the peripheral pathway, the three most possible causes for increased neuronal bilirubin are 1) low serum albumin concentration, 2) hyperbilirubinemia, and 3) abnormally increased permeability of BBB. At a physiological level, unconjugated (free) bilirubin almost completely binds to serum albumin; once the albumin is dramatically decreased during, for example, cancer, liver disease, and aging, free bilirubin can be released from the albumin [46]. The released free bilirubin is lipophilic and has the potential to get into the peripheral tissues. In hyperbilirubinemia as seen in, for example, pathological jaundice or kernicterus, free bilirubin can surpass the normal binding capacity of albumin and thereby elevates its level in the blood and increases its chance to get into the peripheral tissues [47]. Under these two conditions, BBB abnormal opening is a prerequisite for peripheral free bilirubin to get into the brain tissues. Based on these analyses, only under very specific conditions, i.e., when free bilirubin release is coupled with abnormally high BBB permeability, AD may be induced by bilirubin neurotoxicity. In this study, an abnormal bilirubin increase was exogenously induced through i.p. injection of bilirubin to neonatal rats at 3 days of age when BBB is leaky [48]. The 3 consecutive days of bilirubin exposure coupled with the underdeveloped BBB at the early neonatal stage allowed neuronal entry of bilirubin and induced typical AD-like changes in 1 or 4 months of age. This model more likely mimics kernicterus in newborn babies. Along the same line, we hypothesize that BBB impairing conditions (e.g., acidosis, hypoxia, etc.) likely increase the propensity of hyperbilirubinemia toward inducing AD in adult humans.
The endogenous intracellular bilirubin pathway mainly relies on the heme oxygenase (HO) pathway [49]. HO catalyzes the rate-limiting step of heme degradation in mammals. The products of the reaction are biliverdin, carbon monoxide (CO), and free iron. Biliverdin is then reduced by heme reductase to form bilirubin [50]. It has been suggested that biliverdin and CO have cytoprotective effects against various cellular stresses, but the byproduct of this pathway, bilirubin, is far from being understood [51]. It has been reported that the HO-1 pathway is involved in the occurrence of not only AD but also other neurodegenerative diseases, highly supporting an important role for bilirubin in AD [52–54]. In summary, here we have provided novel evidence that short-term exposure to bilirubin induces encephalopathy similar to AD, and bilirubin encephalopathy maybe an AD-like disease.
In summary, we demonstrated that bilirubin, intraperitoneally injected at newborn or intraventricularly injected at adult, could induce encephalopathy similar to AD in rats. Short-term bilirubin exposure induced bilirubin encephalopathy maybe an AD-like disease. This result could be explained by bilirubin-induced both UPS malfunction and neurotoxicity. These findings provide a new perspective on the pathophysiological role of bilirubin which may be an endogenous pathogenic factor of AD.
Footnotes
ACKNOWLEDGMENTS
We thank Drs. Daolin Tang and Huabo Su for helpful discussions and revision. The study was supported in part by Guangzhou Education System Foundation (1201610014, 1201620494); the National Natural Science Foundation of China (81170608, 81570278, 81602427, 81600089, 81670156); the Science and Technology Program of Guangzhou (201707010046, 201604020001); The National Funds for Developing Local Colleges and Universities (B16056001); Natural Science Foundation research team of Guangdong Province (2018B030312001); Natural Science Foundation of Guangdong Province (2017A030313662).