
Abstract
Background:
Recently, many studies have investigated the association between orexin A and Alzheimer’s disease (AD). However, it remains to be determined whether the observed changes in orexin A levels are associated with pathological changes underlying AD, or cognitive function. In particular, a direct association between cerebrospinal fluid (CSF) orexin A levels and cognitive function has not been reported to date.
Objective:
The aim of this study was to identify whether there is a direct association between the orexinergic system and cognitive function in AD.
Methods:
For this study, we included 22 patients with AD and 25 control subjects who underwent general physical, neurological, and psychiatric examinations, neuroimaging, and CSF collection by lumbar puncture were enrolled. Correlations between CSF orexin A levels and CSF AD biomarker levels (i.e., levels of phosphorylated tau [p-tau], Aβ42, and Aβ42/Aβ40) were assessed to confirm the results of previous studies. Moreover, the correlation between CSF orexin A levels and Mini-Mental State Examination (MMSE) and Japanese version of the Montreal Cognitive Assessment (MoCA-J) scores were analyzed.
Results:
There was a significant positive correlation between CSF orexin-A levels and cognitive function (MMSE scores: r = 0.591, p = 0.04, MoCA score: r = 0.571, p = 0.006) in AD patients.
Conclusion:
This is the first study to our knowledge demonstrating an association between cognitive function and CSF orexin A levels in AD. Our results suggest the possibility that orexinergic system overexpression is not always a negative factor for cognitive function In AD.
Keywords
INTRODUCTION
Alzheimer’s disease (AD), which is the most common cause of dementia, is a neurodegenerative disease characterized by progressive cognitive and behavioral impairment [1]. Although the main characteristic of AD is cognitive decline, sleep disturbance is common in AD. Sleep disturbance is a highly disruptive behavioral symptom, and there is association between sleep disturbance and AD pathology. Indeed, epidemiological studies have reported that up to 45% of AD patients have sleep disturbances [2]. In addition, previous studies showed the high risk of development of dementia, particularly AD, in patients with primary insomnia [3, 4]. Tononi et al. suggests that sleep-wake behavior plays an important role in a crucial brain function that is associated with cognition and synaptic plasticity [5]. Insufficient sleep facilitates the accumulation of amyloid-β (Aβ) in the brain, which may potentially trigger earlier neuropathological changes in AD [6]. In fact, previous studies of cerebrospinal fluid (CSF) analysis and Pittsburgh compound B positron emission tomography (PET) showed an association between changes in sleep quantity and quality and Aβ brain burden in cognitively normal older adults [7–9]. Moreover, an association between sleep disturbance and AD pathology has been reported in patients with mild cognitive impairment (MCI) and AD [10–12]. Therefore, an association between sleep disturbance and AD pathology appears to exist.
The hypothalamic neurotransmitter orexin A (hypocretin 1) is considered to be a crucial molecule of the orexinergic system. It is widely known that patients with narcolepsy have a deficiency in orexin A [13, 14] and orexin A contributes to regulation of the sleep-wake cycle by increasing arousal levels and maintaining wakefulness [15]. Through both direct as well as trans-synaptic modulation of various pathways, hippocampal neurotransmission is controlled by orexin A. Therefore, orexin A may play an important role in hippocampal-dependent cognitive tasks. In particular, orexin-mediated modulation of γ-aminobutyric acid (GABA) and glutamate levels in the hippocampus may potentially contribute to disruption of the sleep-wake cycle as well as cognitive function [16]. Recently, the association between orexin A and AD pathology has been investigated [17–26].
However, no studies to our knowledge have found a direct association between cognitive function and orexin A levels in the CSF of patients with AD. Therefore, the aim of this study was to clarify whether there is a correlation between cognitive function (i.e., Mini-Mental State Examination [MMSE] [27] and Japanese version of the Montreal Cognitive Assessment [MoCA-J] [28] scores) and CSF orexin A levels in AD. In addition, we aimed to confirm the results of previous studies investigating the association between the levels of orexin A and AD biomarkers (i.e., levels of phosphorylated tau [p-tau], Aβ42, and Aβ40/Aβ42).
MATERIALS AND METHODS
Patients
For this study, we included 47 subjects free of psychotropic drugs and who underwent CSF collection by lumbar puncture for diagnosis at the Memory Disorder Clinic in the Department of Geriatric Medicine, Tokyo Medical University: 22 AD and 25 controls.
All subjects underwent general physical, neurological, and psychiatric examinations, extensive laboratory tests, and single photon emission tomography (SPECT) and magnetic resonance imaging (MRI) to establish a clinical diagnosis and to exclude other potential causes of dementia (i.e., stroke, dementia with Lewy bodies, Parkinson’s disease, depression, vitamin B12 deficiency, hypothyroidism, hyperammonemia, intake of drugs acting on the central nervous system or of any type of hypnotic, etc.). After above extensive examination, AD patients were determined by three geriatric neurologists (H.H., K.H., and S.S.). Patients with AD had to meet the National Institute on Aging and Alzheimer’s Association (NIA-AA) criteria for a diagnosis of probable AD [1]. Moreover, to increase the probability that the subjects included in the study actually had AD pathological change, we used the NIA-AA research framework [29]. Biomarkers of Aβ plaques (labeled “A”) are defined as low Aβ42 (<500 pg/mL) or elevation of Aβ42/Aβ40 ratio (0.05>). Biomarkers of tau (labeled “T”) are elevated CSF phosphorylated tau (p-tau) (50 > pg/mL). The cut-off values are determined based on the best value for discriminating AD from other dementias and cognitive unimpaired subjects who expected CSF collection by some complaints in our department (i.e., headache, parkinsonism, and cognitive decline, etc.). Based on above criteria, we defined participants with A+ T+ as “AD” group (n = 22). On the other hand, the control group included subjects with normal neuropsychological measurements (MMSE score ≧25) who underwent clinical neurological investigation, neuroimaging, and lumbar puncture for diagnostic purposes. We defined participants with (A–T–) [29] who had MMSE score ≧25 as control group (n = 25).
This study was approved by the ethics committee of Tokyo Medical University. Written informed consent was obtained from all subjects (either from the patients themselves or their closest relative) before entry, following a detailed explanation of the lumbar puncture and biochemical analysis of the CSF. All procedures were in accordance with the ethical standards on human investigation and with the principles of the declaration of Helsinki.
CSF samples and assays
CSF was collected in polypropylene tubes following standardized conditions and using an atraumatic needle, preferably between 11 AM and 2 PM under fasting conditions to minimize the effects of diurnal variations of CSF biomarkers. Samples were centrifuged (3,000 rpm, 10 min) and preserved at –80°C until analysis.
Levels of p-tau, Aβ40, and Aβ42 were analyzed using a commercially available enzyme-linked immunosorbent assay, as previously described [30]. CSF orexin A (hypocretin 1) was measured at Akita University using an 125I radioimmunoassay kit (Phoenix Pharmaceuticals, Belmont, CA) as previously described [15, 31]. The detection limit was 40 pg/mL. As CSF orexin A levels can be reliably measured in frozen CSF stored for several years [31], CSF samples collected for diagnostic purposes of AD (storage period: 4 days to 4 years in a –80°C freezer) were used.
Statistical analysis
Values were expressed as means±SD and analyzed using Spearman’s rank correlation coefficient and linear regression. A p-value of less than 0.05 was considered to indicate a statistically significant difference between two groups. All analyses were performed using IBM SPSS statistics version 24 software (Chicago, IL).
RESULTS
Table 1 shows the characteristics of the participants. There were significant differences between two groups in MMSE scores, MoCA-J scores and p-tau. Table 2 shows the correlation between cognitive function and AD biomarker, and CSF orexin A. In whole subjects, there were no significant association between CSF orexin A levels and CSF biomarker, and cognitive function (Table 2). In control group, there was significant correlation between CSF orexin A level and p- tau (r = 0.495, p = 0.026) (Table 2, Fig. 1). On the other hand, there was significant correlation between CSF orexin A level and cognitive function (MMSE and MoCA-J scores) (Table 2, Fig. 2) in AD. MMSE (r = 0.591, p = 0.04; Fig. 2a) and MoCA-J (r = 0.571, p = 0.006; Fig. 2b) in AD.
Characteristics of the participants
MMSE, Mini Mental State Examination; MoCA-J, Japanese version of the Montreal Cognitive Assessment; P-tau, phosphorylated tau; Aβ40, amyloid-β 40; Aβ42, amyloid-β 42.
Correlation between CSF orexin A level and CSF biomarker, and that between CSF orexin level and cognitive function
MMSE, Mini Mental State Examination; MoCA-J, Japanese version of the Montreal Cognitive Assessment; P-tau, phosphorylated tau; Aβ40, amyloid-β 40; Aβ42, amyloid-β 42.
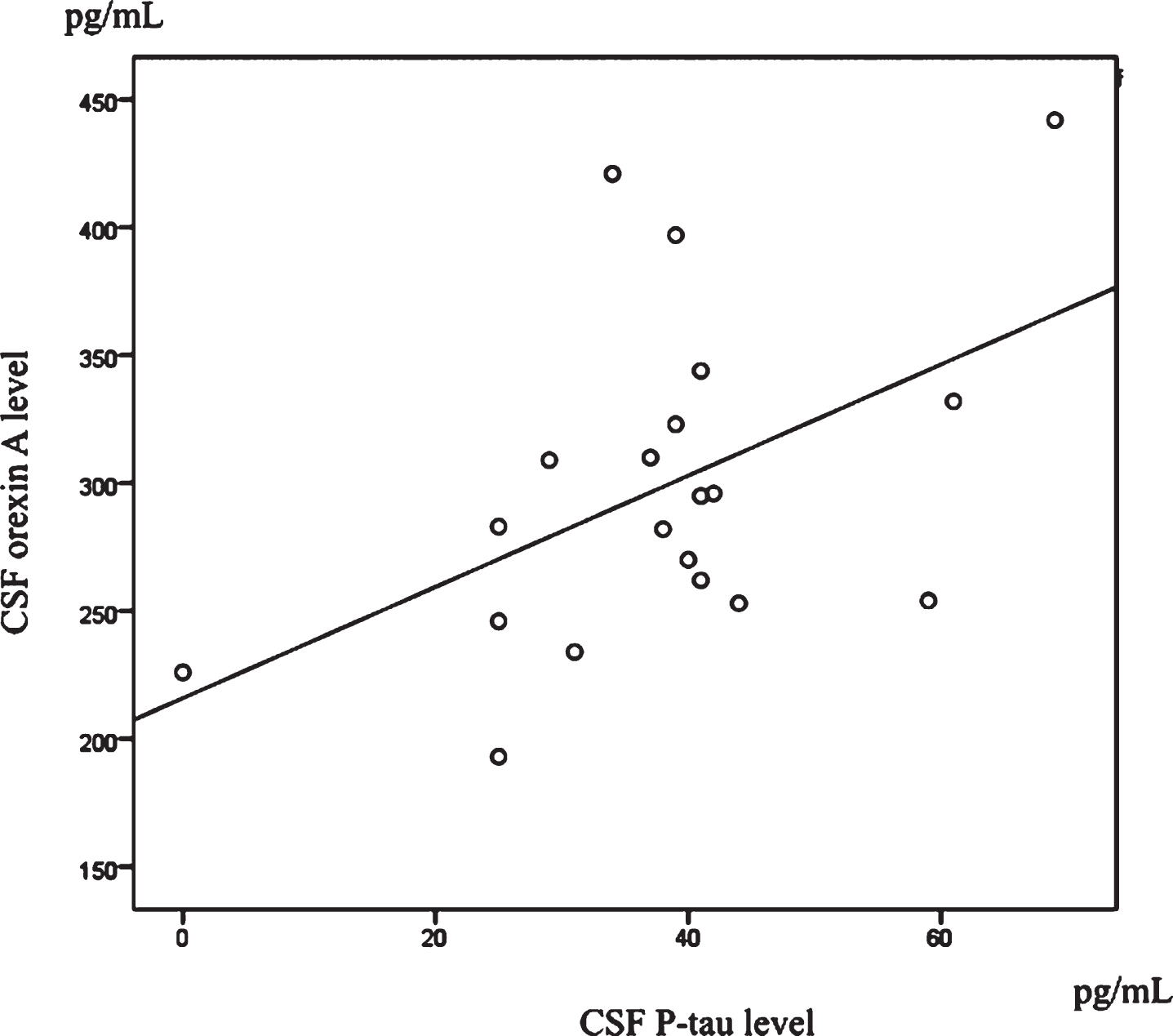
Scatterplots of CSF orexin A and p-tau levels in control group. Significant positive correlations were found between CSF orexin A and p-tau levels in control group.
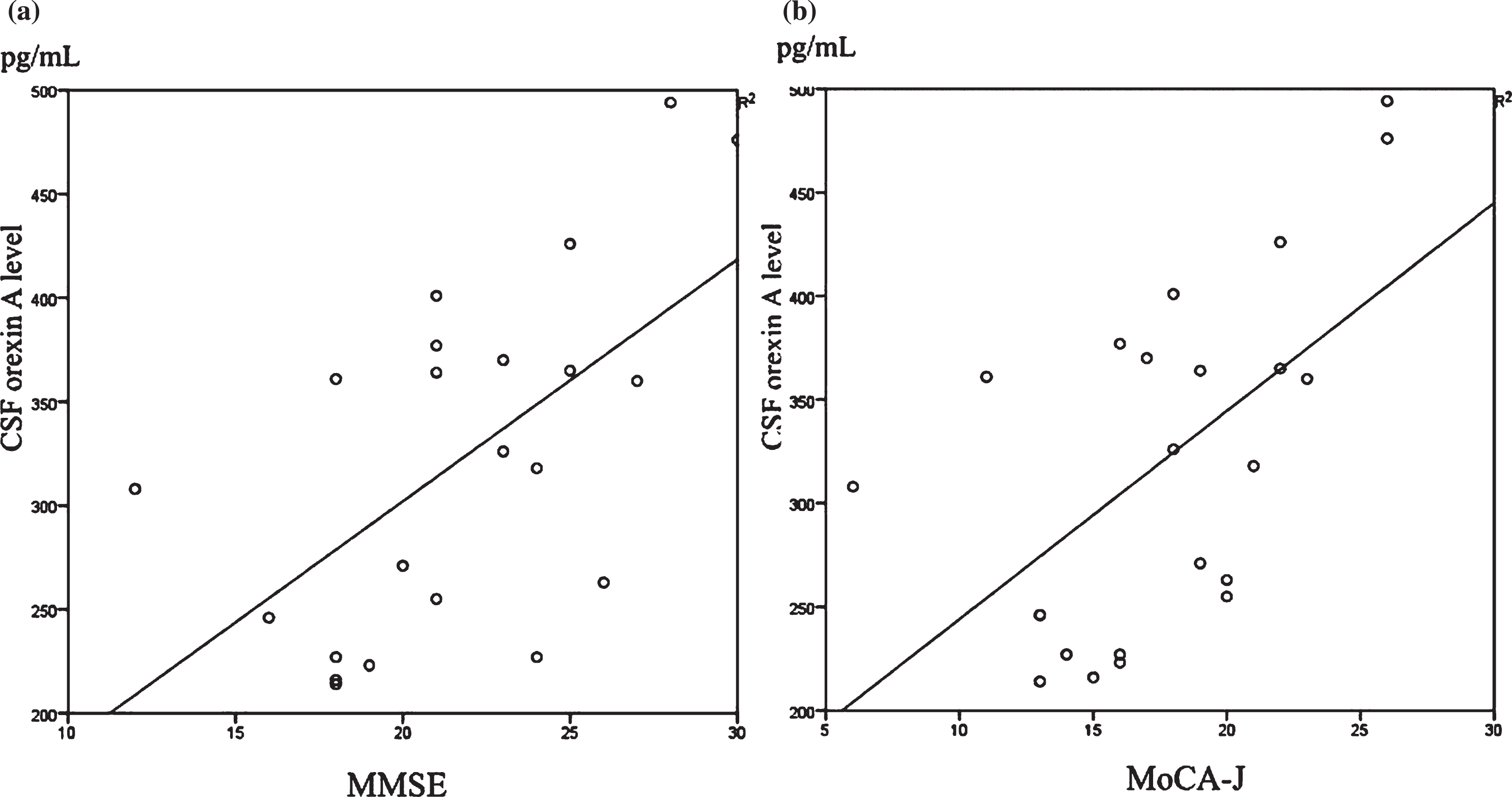
Scatterplots of CSF orexin A levels and MMSE scores (a) and MoCA-J scores (b) in AD group. Significant positive correlations were found between CSF orexin A levels and both cognitive function scale scores.
DISCUSSION
We found a significant positive correlation between CSF orexin A levels and cognitive function. This is the first study demonstrating an association between cognitive function and CSF orexin A levels in AD.
Recently, associations between orexin A and AD pathology have been investigated, and previous studies suggest that overexpression of the orexinergic system might be associated with AD pathology [10–12, 17–26]. However, it remained to be determined whether the changes observed in orexin A levels are associated with pathological changes underlying AD, or occur secondary to changes in the sleep-wake cycle or cognitive function. Previous studies showed, in non-demented people, there is an association between CSF Aβ levels and poor sleep quality [9, 33]. On the other hand, there is no study that showed the association between CSF Aβ levels and sleep quality in AD. Moreover, previous studies [11, 12] and our study were unable to find correlation between CSF orexin levels and CSF Aβ42 levels as an AD biomarker. Concentrations of Aβ42 in the CSF were found to decrease as a function of estimated years from expected symptom onset, reaching significantly low levels 10 years before expected symptom onset [34, 35]. Therefore, the lack of a correlation between Aβ42 levels and orexin A levels in our present study as well as previous studies might be owing to the possible effects of low CSF Aβ42 levels reaching a plateau in both MCI and AD patients.
In fact, one recent study results could not support the hypothesis that sleep disturbance predict subsequent cognitive decline [36], and the significance of CSF orexin A levels in patients with AD remain to be clarified [11, 33]. Moreover, in patients with MCI and AD, CSF orexin A levels were only found to directly correlate with CSF tau levels [11, 23]. Previous studies showing the association between levels of CSF orexin A and tau in patients with AD [11, 23] suggest that dysregulation of the orexinergic system, found as increased CSF orexin A levels in AD patients, is associated with tau-mediated neurodegeneration. In contrast with these previous studies, Gabelle et al. could not find association between CSF orexin A and tau [12] similar to our study. Unfortunately, we could find an association between CSF orexin A and p-tau levels in only control subjects but not in AD patients. Heterogeneities in sample size, populations, CSF sample collection time, methods for CSF orexin-A measurement, and associated comorbidities, such as sleep disturbances, may explain these discordant results.
Even if there is an association between the orexinergic system and AD pathology, the direct association between the orexinergic system and cognitive function in AD remains unclear. Previous studies were unable to find an association between CSF orexin A levels and cognitive function. Moreover, in our study, there was an association between CSF orexin A and p-tau levels in only control subjects but not in AD patients. On the other hand, we could find an association between CSF orexin A and cognitive function in only AD patients but not in control subjects. Hence, these results are lines of evidence that there is a yet unclear mutual association among orexinergic system dysfunction, AD pathology, and cognitive function. As previous studies [11, 23] showed that CSF orexin A levels increased in association with the progression of AD severity, we expected CSF orexin A levels and cognitive function to be negatively correlated in AD patients. However, surprisingly, we found that CSF orexin A levels and cognitive function were positively correlated. Therefore, we hypothesized the possibility that orexinergic overexpression, which corresponds to a progression of AD pathology, is not always a negative factor for cognitive function. Liguori et al. [11] documented that cognitive impairment is associated with sleep structure deterioration. The association between sleep alteration and dementia is very likely to result from pathological changes in AD, particularly upon dysregulation of the cholinergic system, which is typical of the disease. Therefore, based on previous studies [37, 38], the authors concluded that sleep impairment may be caused by an imbalance between cholinergic and orexinergic systems, with an overexpression of the latter because of the absence of the cholinergic feedback owing to its damage. Gabelle et al. hypothesized that high CSF orexin A levels in AD patients may be a result of increased orexin release, as a compensatory mechanism involving the lateral hypothalamus in the neurodegenerative process of AD [12]. We agree with this proposal of a compensatory mechanism by Gabelle et al. Indeed, basal forebrain cholinergic neurons, which are wakefulness-promoting neurons [39], die during the neurodegeneration in AD. This may lead to the upregulation of other arousal systems, including orexin-producing neurons.
Therefore, we hypothesized that orexinergic overexpression, which occurs as a compensatory mechanism for cholinergic dysfunction, may have positive effects on cognitive function. Based on this hypothesis, there are two possibilities to explain the association between CSF orexin A levels and cognitive function. First, there is the possibility that patients with AD, whose orexinergic system is overexpressed to compensate for cholinergic dysfunction, are able to maintain cognitive function to a certain extent. The second possibility is that wakefulness, which is controlled by orexin overexpression, may affect cognitive function.
On the other hand, a possible explanation for the lack of an association between CSF orexin A levels and cognitive function in previous studies may be that the effects of the orexinergic system on cognitive function are not that strong. To explain these hypotheses, we will need to consider differences between studies regarding the participants’ AD stage, sample size, CSF collection time, associated comorbidities, such as sleep disturbances, as well as the method used for orexin A quantification (radioimmunoassay versus fluorescence immunoassays or enzyme immunoassays). Therefore, to better understand the role of the orexinergic system in AD, further longitudinal studies investigating wake-promoting and AD biomarkers in the CSF, sleep-wake profiles, and Aβ burden quantified by amyloid PET are required.
This study has several crucial limitations. Firstly, this study was carried out in a single memory disorder clinic; therefore, the number of patients enrolled was relatively small. Secondly, sleep-wake profiles were not assessed by clinical interviews or objective measurements, such as actigraphy or polysomnography. However, the main aim of this study was to identify the role of the orexinergic system in cognitive function. Thirdly, cognitive function was evaluated only by MMSE and MoCA-J. Finally, in our study, the absence of healthy controls is not a trivial limitation. However, there is a fundamental ethical problem in collecting CSF from normal subjects by lumbar puncture, for purposes other than diagnosis. Therefore, further studies including larger subject numbers, assessment of sleep-wake profiles, more detailed neuropsychological assessments, normal control subjects, and pathological analyses are needed to confirm our results.
In conclusion, this study is the first study to our knowledge that found a direct positive association between CSF orexin A levels and cognitive function. Our results suggest the possibility that orexinergic system overexpression is not always a negative factor for cognitive function.