
Abstract
Background:
Development of Alzheimer’s disease (AD) pathology is associated with impaired blood flow delivery of oxygen and nutrients throughout the brain. Cerebrovascular endothelium regulates vasoreactivity of blood vessel networks for optimal cerebral blood flow.
Objective:
We tested the hypothesis that cerebrovascular endothelial Gq-protein-coupled receptor (GPCR; purinergic and muscarinic) and K+ channel [Ca2+-activated (KCa2.3/SK3 and KCa3.1/IK1) and inward-rectifying (KIR2.x)] function declines during progressive AD pathology.
Methods:
We applied simultaneous measurements of intracellular Ca2+ ([Ca2+]i) and membrane potential (Vm) in freshly isolated endothelium from posterior cerebral arteries of 3×Tg-AD mice [young, no pathology (1– 2 mo), cognitive impairment (CI; 4– 5 mo), extracellular Aβ plaques (Aβ; 6– 8 mo), and Aβ plaques + neurofibrillary tangles (AβT; 12– 15 mo)].
Results:
The coupling of ΔVm-to-Δ[Ca2+]i during AβT pathology was lowest for both sexes but, overall, ATP-induced purinergic receptor function was stable throughout AD pathology. SKCa/IKCa channel function itself was enhanced by ∼20% during AD (Aβ+ AβT) versus pre-AD (Young + CI) in males while steady in females. Accordingly, hyperpolarization-induced [Ca2+]i increases following SKCa/IKCa channel activation and Δ[Ca2+]i-to-ΔVm coupling was enhanced by ≥two-fold during AD pathology in males but not females. Further, KIR channel function decreased by ∼50% during AD conditions versus young regardless of sex. Finally, other than a ∼40% increase in females versus males during Aβ pathology, [Ca2+]i responses to the mitochondrial uncoupler FCCP were similar among AD versus pre-AD conditions.
Conclusion:
Altogether, AD pathology represents a condition of altered KCa and KIR channel function in cerebrovascular endothelium in a sex-dependent and sex-independent manner respectively.
INTRODUCTION
Cerebrovascular endothelium coordinates vasoreactivity of blood vessel networks essential for delivery of oxygen and nutrients throughout brain tissue in accord with metabolic demand [1, 2]. Cerebrovascular “dysfunction”, defined in part by reduced endothelial-mediated vasodilation per actions of nitric oxide (NO) [3, 4], impairs blood flow throughout the brain and thereby contributes to cognitive disorders such as Alzheimer’s disease (AD) [5, 6]. In parallel with NO signaling, another essential pathway for cerebral blood flow regulation entails intracellular Ca2+ ([Ca2+]i) increase and thereby activation of small- and intermediate-Ca2+-activated K+ (SKCa/IKCa) channels to increase the negative membrane potential (Vm) or endothelium-derived “hyperpolarization” (EDH) of the vascular wall [7]. As integral to cerebral blood flow control [8], inward-rectifying K+ (KIR) channels may sustain hyperpolarization following SKCa/IKCa channel activation [9]. Activation of respective K+ channels can also restore or boost actions of NO [10]. Altogether, the endothelial layer of the cerebral resistance arteries commands surrounding smooth muscle to relax and thereby allow vasodilation for necessary blood flow and optimal perfusion of the brain. In such manner, the impact of progressive AD pathology on EDH as a central pathway for matching cerebral blood flow supply to metabolic demand of the brain parenchyma is unknown.
Cerebrovascular endothelial purinergic and muscarinic receptor activation increases [Ca2+]i and subsequently activates SKCa/IKCa channels as the central components of EDH [11, 12]. In such manner, our laboratory [10, 13] and others [12, 15] have demonstrated the regulation of EDH in rodent cerebral arteries via indirect, physiological regulation of endothelial GPCRs in addition to direct, pharmacological activation of SKCa/IKCa channels. It is also worth noting that direct control of endothelial SKCa/IKCa channels in particular has emerged as a potential approach for improving vascular function during aging and the development of chronic disease [16]. In addition, we have recently identified a significant decline in KIR channel function in both males and females during old age (≥24 months) as another potential manifestation of endothelial “dysfunction” [11]. It is not known whether and how GPCRs and K+ channel functions underlying EDH at the endothelial cell level of cerebral arteries are altered with development of AD pathology and, hence, resolving this precise question was the goal of the current study. We tested the hypothesis that cerebrovascular endothelial function, as defined by [Ca2+] i signaling and K+ channel activation in the EDH pathway, is diminished during progression of AD regardless of biological sex. We examined both males and females of a triple mutation mouse model (3×Tg-AD; mutations in the PS1, APP, and tau genes), whereby, off spring progressively develop cognitive impairment (CI; 4– 5 months), extracellular amyloid-β (Aβ; 6– 8 months), and Aβ plaques in combination with neurofibrillary tangles composed of hyperphosphorylated tau (AβT; 12– 15 months) [17, 18].
We measured endothelial purinergic and muscarinic receptor-stimulated increases in [Ca2+]i and subsequent activation of Ca2+-sensitive SKCa/IKCa channels producing EDH during progressive phases of AD. We also determined the impact of the development of AD pathogenesis on direct activation of endothelial SKCa/IKCa and KIR channels. Moreover, as chronic vascular disease can increase mitochondrial [Ca2+]i [19, 20], we tested mitochondrial Ca2+ content/release via dissipation of the inner mitochondrial membrane potential (ΔΨmt) using the mitochondrial uncoupler FCCP [20, 21]. In brief, purinergic receptor activation of EDH was relatively stable throughout progression of AD pathology in both males and females except during late stage AβT pathology where ΔVm-to-Δ[Ca2+]i coupling (i.e., sensitivity of SKCa/IKCa activation to GPCR-induced increases in [Ca2+]i) was lowest. Direct SKCa/IKCa channel activation of EDH with NS309 was relatively stable throughout AD pathology in females while significantly enhanced in males during overall conditions of AD (Aβ+ AβT) versus Pre-AD (Young + CI). In addition, hyperpolarization-induced [Ca2+]i increases following SKCa/IKCa channel activation and Δ[Ca2+]i-to-ΔVm coupling (i.e., sensitivity of Ca2+ influx into cells in response to hyperpolarization) was enhanced by≥two-fold during AD pathology in males but not females. KIR channel function in response to elevated extracellular K+ decreased during progression of Aβ and AβT pathology regardless of biological sex. Finally, other than higher [Ca2+]i responses to FCCP by ∼40% in females versus males during Aβ pathology, mitochondrial Ca2+ release in the cytosol in response to the mitochondrial uncoupler FCCP was stable throughout AD conditions. Altogether, development of AD pathology represents a condition of progressively altered cerebrovascular endothelial ion channel function as described by a sex-based divergence in SKCa/IKCa channel function and a sex-independent decline in KIR channel function.
METHODS
Animal care and use
All animal care use and experimental protocols for this study were approved by the Institutional Animal Care and Use Committee of Loma Linda University and performed in accord with the National Research Council’s “Guide for the Care and Use of Laboratory Animals” (8th Edition, 2011). Experiments were performed using male and female 3×Tg-AD mice inbred using homozygous breeding pairs obtained from the Jackson Laboratories (Wilmington, MA, USA). We chose the 3×Tg-AD study model [(B6;129-Tg (APPSwe,tauP301L) 1Lfa Psen1tm1Mpm/Mmjax); Mutant Mouse Resource and Research Center (MMRRC) stock #034830] as it develops phases of cumulative AD pathology over a reasonable, phased aging process versus aggressive pathology in 5XFAD mice with neuronal and synaptic loss≤12 months [22]. At the same time, the contribution of the aging process alone to pathology in the original background strain (B6129SF2/J; ≤15 months) is relatively minimal to negligible for overall cognitive function represented by synaptic function [23] and learning/memory [18, 24] as examples. In accord with the phases of cumulative AD pathology, we categorized the mice into four groups, as young (1– 2 months, 7 males and 8 females), cognitive impairment (CI; 4– 5 months, 6 males and 6 females), presence of extracellular amyloid-β plaques (Aβ; 6– 8 months, 6 males and 5 females), and presence of Aβ and neurofibrillary tangles composed of tau (AβT; 12– 15 months, 10 males and 8 females) [17, 18]. All animals were housed on a 12 : 12 h light-dark cycle at 22– 24°C with fresh water and food available ad libitum. For individual experiments, animals were selected randomly with respect to AD pathology group and biological sex.
Solutions and reagents
Preparation and composition of all solutions have been previously described for measurements of [Ca2+]i and/or Vm in intact endothelium freshly isolated from mouse posterior cerebral arteries [13, 25]. Briefly, physiological salt solution (PSS) was prepared for continuous superfusion of cerebral endothelial tubes [(in mmol/L): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 10 Glucose]. Dissection PSS lacking CaCl2 contained 0.1% bovine serum albumin. For isolation of endothelium, dissociation PSS contained reduced Ca2+ (0.1 mmol/L CaCl2), 0.1% bovine serum albumin and an enzyme cocktail (0.31 mg/mL pa pain, 0.5 mg/mL dithioerythritol, 0.75 mg/mL collagenase, and 0.13 mg/mL elastase). For applications utilizing 15 mmol/L KCl, an equimolar decrease of NaCl was applied accordingly in order to maintain osmolarity. Reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) or ThermoFisher Scientific (Pittsburgh, PA, USA) unless otherwise indicated.
Dissection of cerebral arteries and isolation of endothelial tubes
Sequential dissection of mouse posterior cerebral arteries and isolation of arterial endothelium have been previously described and illustrated [11, 25]. Following isoflurane inhalation for anesthesia and decapitation, the brain was removed and arteries were placed in chilled (4°C) dissection PSS. Arterial segments were partially digested in dissociation PSS containing the enzyme cocktail at 34°C for 10 to 12 min. Following digestion, arterial segments were triturated to remove adventitia, smooth muscle cells, and internal elastic lamina.

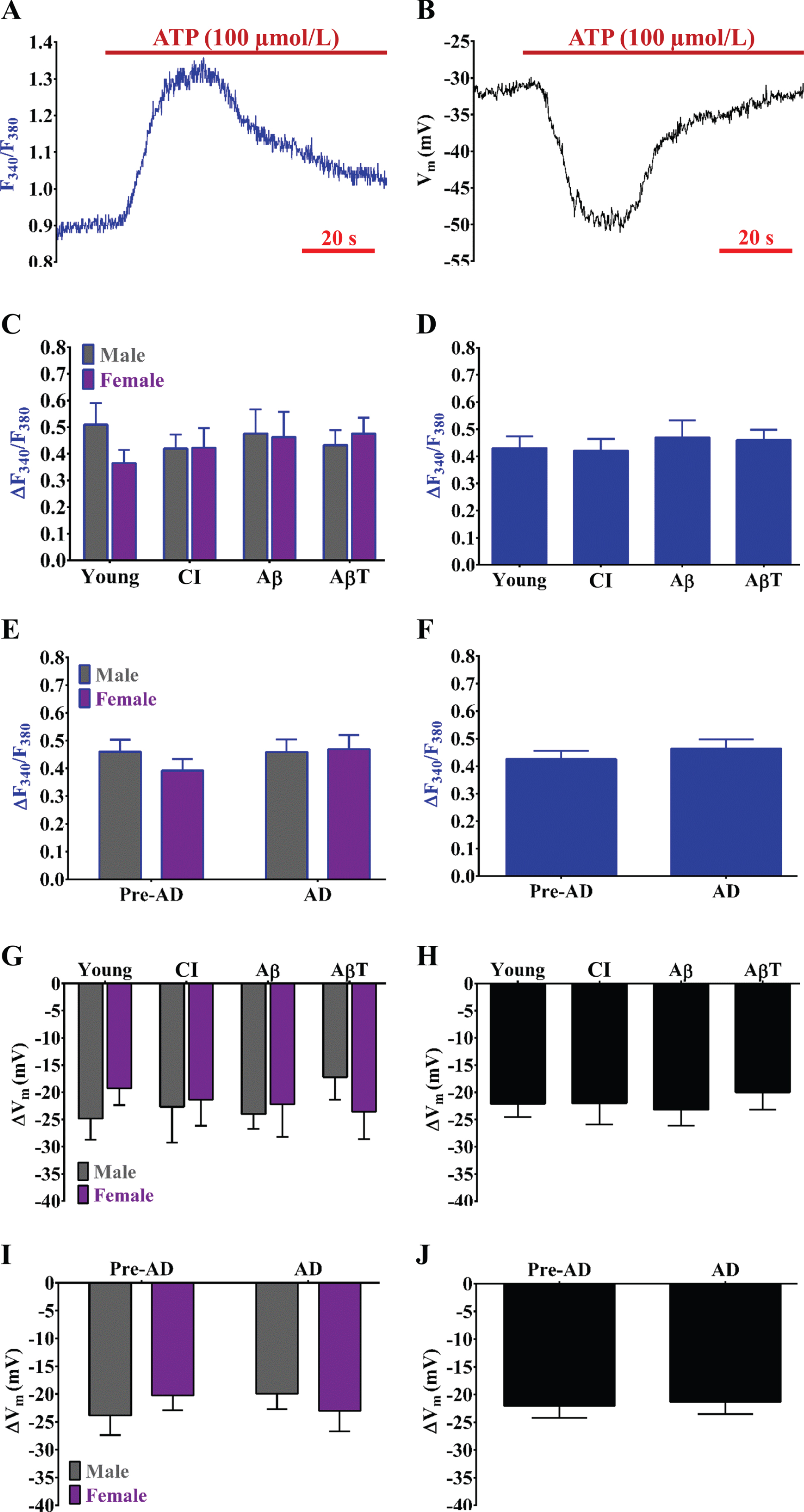
Simultaneous [Ca2+]i and Vm measurements and resting values with progressive AD pathology. A) A schematic of applied simultaneous sharp electrode electrophysiology and Fura-2 Ca2+ photometry measurements in isolated cerebrovascular endothelium (ATP treatment shown for respective traces). B) Summary data for resting F340/F380 as a function of AD group (Young, CI, Aβ, and AβT) and biological sex (Male, Female). C) As shown in (B) as a function of AD pathology only (sexes combined). D) Summary data for resting F340/F380 as a function of pre-AD group (Young + CI) versus AD (Aβ+ AβT) in accord with biological sex. E) As shown in (D) as a function of AD pathology only with sexes combined. F, G, H, I) As shown in (B), (C), (D), and (E), respectively, for Vm. n = number of animals and respective cerebral endothelial tubes (Young, CI, Aβ, AβT: 7, 6, 6, and 10 Males and 8, 6, 5, and 8 Females; combined for AD group only: 15, 12, 11 and 18). CI, cognitive impairment; Aβ, amyloid-β; AβT, amyloid-β+ tau neurofibrillary tangles.
Simultaneous measurement of intracellular Ca2+ and Vm
Our protocol for measurement of [Ca2+]i and Vm in mouse cerebral endothelium has been previously described and illustrated [11, 25] (Fig. 1A). Briefly, [Ca2+]i was measured using Fura-2 photometry with an IonOptix system (Milford, MA, USA) simultaneously with Vm using sharp electrodes and Axoclamp electrometers 2B and/or 900A (Molecular Devices, Sunnyvale, CA, USA) connected to a data acquisition system (Digidata 1550A; Molecular Devices). Respective recordings were temporally synchronized across respective IonOptix and Molecular Devices software suites at a data acquisition frequency of 10 Hz. Using the equation [Ca2+] i = Kd • β • (R - Rmin)/(Rmax - R) [26], [Ca2+]i (in nmol/L) per Fura-2 ratio recordings was approximated using Fura-2 Kd = 282 nmol/L [Ca2+]i [27] and calibration values of Rmin = 0.67, Rmax = 3.25, and β= 3.71 [11]. Temperature was maintained at 37°C during continuous superfusion (∼7 ml/min) throughout experiments.
Pharmacology
Cerebrovascular endothelial GPCR function was assessed using adenosine 5′-triphosphate disodium salt hydrate (ATP, 100 μmol/L) and acetylcholine chloride (ACh, 10 μmol/L) to activate endothelial purinergic and muscarinic GPCRs, respectively [12, 28]. Effective concentrations of SKA-31 (10 μmol/L; Tocris Bioscience, Bristol, UK) and NS309 (1 μmol/L; Tocris) were used to directly evaluate function of SKCa/IKCa channels to produce hyperpolarization [13, 29] and hyperpolarization-dependent [Ca2+]i increases [11, 31]. Elevated level of extracellular KCl ([K+]o: 15 mmol/L) was used to stimulate endothelial KIR channels [13]. Finally, to examine mitochondrial Ca2+ content, a maximal but reversible concentration of FCCP (1 μmol/L) was applied [20, 21]. DMSO solvent for working stocks of SKA-31, NS309, and FCCP was ≤0.1% which we have found to have no effect on endothelial Ca2+ and Vm on its own for ≤5 min applications used for respective drug treatments [32].
Resting [Ca2+]i and Vm were typically allowed ≥2 min to stabilize before application of a pharmacological agent, whereby each application was allowed sufficient time (≥3 min) to record peak [Ca2+]i and Vm responses. In between individual applications, the endothelial tube was washed with PSS to baseline conditions.
Data and statistical analysis
For Ca2+ photometry, data analyses included fluorescence emission collected at 510 nm and expressed as the ratio during excitation at 340 nm and 380 nm (F340/F380) and change in F340/F380 ratio (ΔF340/F380) = peak response F340/F380 – preceding baseline F340/F380. For electrophysiological recordings, data analyses included resting Vm (mV) and change in Vm (ΔVm) = peak response Vm – preceding baseline Vm. For ATP (purinergic receptor activation) experiments, ΔVm-to-Δ[Ca2+]i coupling ratios were calculated among groups as mV of hyperpolarization per nmol/L [Ca2+]i increase. For SKA-31 and NS309 (SKCa/IKCa channel activation) experiments, Δ[Ca2+]i-to-ΔVm coupling ratios were calculated as nmol/L [Ca2+]i increase per mV of hyperpolarization. All statistical analyses were performed using GraphPad Prism (Version 6; GraphPad Software, La Jolla, CA, USA). Unless indicated otherwise, all [Ca2+]i and Vm data represent the number (n) of independent experiments, with each “n” representing one independent experiment using one endothelial tube from one mouse. Not more than one endothelial tube was used for experimentation per animal.
For simultaneous comparison among pathology groups (young versus CI versus Aβ versus AβT or Pre-AD versus AD) and both sexes statistical analysis included a two-way Analysis of Variance (ANOVA) with Tukey’s post hoc correction for multiple comparisons. One-way ANOVA with Tukey’s post hoc was used for data sets with more than two groups for comparison (i.e., among pathology groups with data from sexes combined). The two-tailed unpaired t-test was used for comparisons between two groups as combined pre-AD versus AD data sets. Exceptions to these statistical applications (e.g., significance via two-tailed unpaired t-test comparison between male and female of the same pathology group) are indicated in the Results text and Figure Legends. Note that all statistical analyses were applied per response to each concentration of a pharmacological agent. In addition, per the hypothesis and overall experimental design of the study, none of the data sets were matched, paired, and/or analyzed using repeated measures analysis within and among study groups. Finally, this analytical approach did not consider the role of opposite sexes across dissimilar pathology groups (e.g., AβT male versus CI female).Differences between groups were accepted as statistically significant with p < 0.05. All summary data are presented as the mean±SEM. Note that the absence of statistically significant differences does not necessarily rule out physiological or pathological importance of study group comparisons that indicate p > 0.05.
Purinergic receptor function remains stable during development of pathology. Example simultaneous recordings of (A) Fura-2 for [Ca2+]i and (B) a sharp electrode for Vm before and during ATP (100 μmol/L; purinergic receptor agonist and indirect SKCa/IKCa channel activator) in an isolated endothelial tube of a young male. C) Summary data for ΔF340/F380 during ATP as a function of AD group (Young, CI, Aβ, and AβT) and biological sex (Male, Female). D) As shown in (C) as a function of AD pathology only (sexes combined). E) Summary data for ΔF340/F380 during ATP combined as a function of pre-AD group (Young + CI) versus AD (Aβ+ AβT) in accord with biological sex. F) As shown in (E) as a function of AD pathology only with sexes combined. G, H, I, J) As shown in (C),(D), (E), and (F) respectively for ΔVm. n = number of animals and respective cerebral endothelial tubes (Young, CI, Aβ, AβT: 7, 6, 6, and 9 Males and 7, 6, 5, and 7 Females; combined for AD group only: 14, 12, 11 and 16). CI, cognitive impairment; Aβ, amyloid-β; AβT, amyloid-β+ tau neurofibrillary tangles.
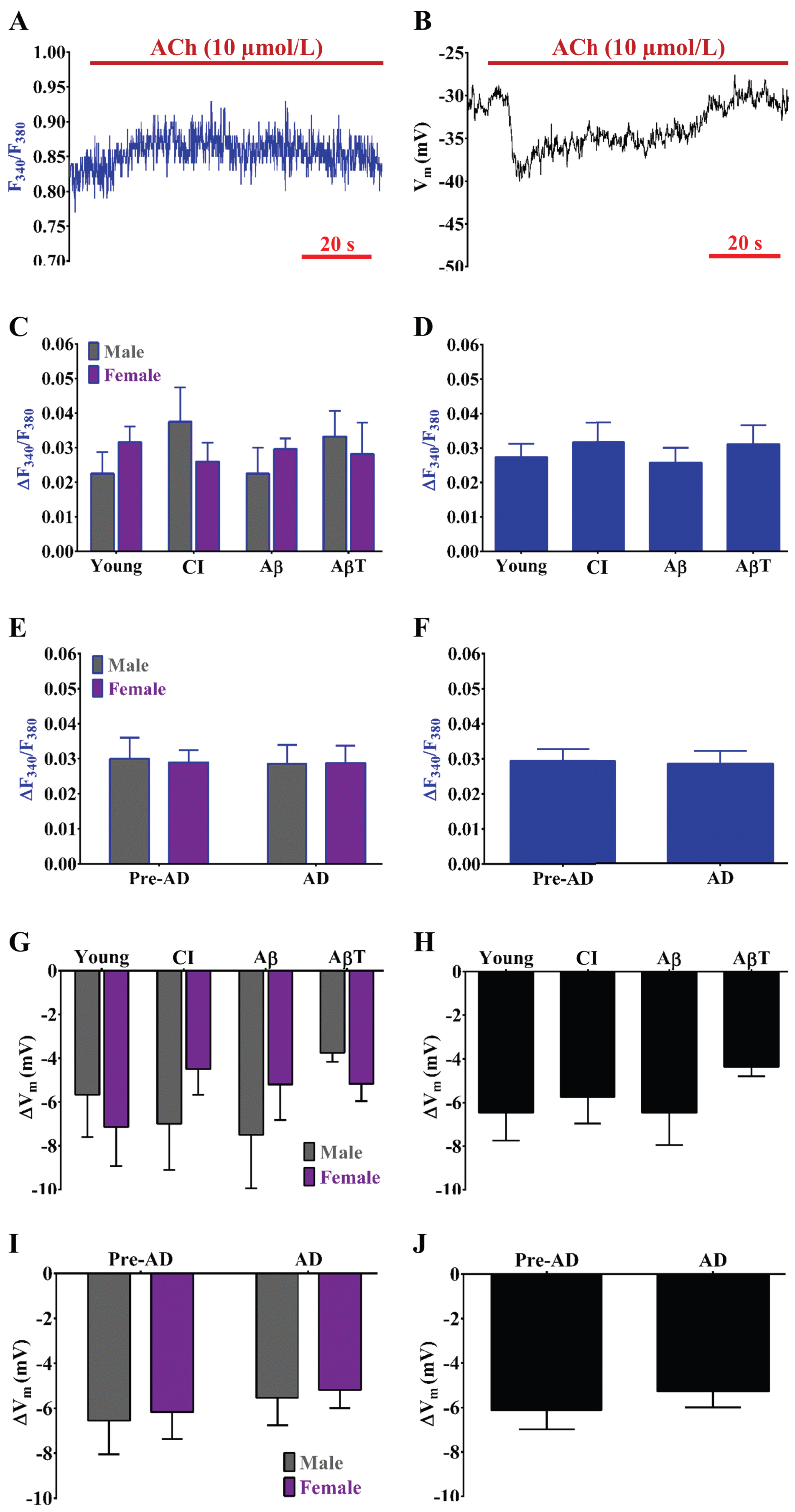
Muscarinic receptor function plays a minor role in cerebrovascular EDH and remains stable during overall AD versus pre-AD pathology. Example simultaneous recordings of (A) Fura-2 for [Ca2+]i and (B) a sharp electrode for Vm before and during ACh (10 μmol/L; muscarinic receptor agonist and indirect SKCa/IKCa channel activator) in an isolated endothelial tube of an Aβ male. C) Summary data for ΔF340/F380 during ACh as a function of AD group (Young, CI, Aβ, and AβT) and sex (Male, Female). D) As shown in (C) as a function of AD pathology only (sexes combined). (E) Summary data for ΔF340/F380 during ACh combined as a function of pre-AD group (Young + CI) versus AD (Aβ+ AβT) in accord with biological sex. F) As shown in (E) as a function of AD pathology only with sexes combined. G, H, I, J) As shown in (C), (D), (E), and (F) respectively for ΔVm. n = number of animals and respective cerebral endothelial tubes (Young, CI, Aβ, AβT: 6, 6, 6, and 8 Males and 7, 6, 5, and 6 Females; combined for AD group only: 13, 12, 11 and 14). CI, cognitive impairment; Aβ, amyloid-β; AβT, amyloid-β+ tau neurofibrillary tangles.
RESULTS
Our goal was to examine endothelial [Ca2+]i signaling and K+ channel function for hyperpolarization of Vm as components of EDH during development of AD pathology (Fig. 1A). As similar to previous studies examining aging C57BL/6 mice [11, 13], endothelial tubes freshly isolated from 3×Tg-AD mouse posterior cerebral arteries were ∼90 to 100 μm in width and ≥300 μm in length. Per the goal of our study, we simultaneously recorded changes in [Ca2+]i and Vm in response to agonists of endothelial GPCRs and activators of SKCa/IKCa and KIR channels during progressive AD pathology for males and females. Throughout all figures, note that data are presented as independent sexes (summary graphs on left side) in addition to sexes combined (summary graphs on right side). Also, data are presented as individual phases of AD pathology (Young, CI, Aβ, and AβT) in addition to pre-AD (Young + CI) versus AD (Aβ+ AβT) conditions. In brief, statistically significant differences among groups were primarily observed for K+ channel function, whereas GPCR function was relatively stable throughout AD pathology.
Resting cerebrovascular endothelial [Ca2+]i and Vm with progressive AD pathology
Resting F340/F380 was similar among all groups as consistent with an approximate [Ca2+]i range of ∼80 to 120 nmol/L (Fig. 1B– E). In parallel, trends for more hyperpolarized resting Vm (p > 0.05; two-way ANOVA) were observed for both males and females during AβT conditions relative to the other pathology groups of similar sex (Fig. 1F, G). In addition, such observations for resting Vm were not statistically significant (p > 0.05) for overall pre-AD to AD conditions (Fig. 1H, I).
Effect of AD pathology on cerebrovascular endothelial GPCR function in both biological sexes
The impact of progressive AD pathology on cerebrovascular endothelial GPCR function is unknown. Cerebrovascular endothelial purinergic and muscarinic receptor activation increases [Ca2+]i with subsequent activation of activation of SKCa/IKCa channels for hyperpolarization of Vm [11, 12]. As consistent with previous studies for cerebrovascular endothelium [11, 13], activation of purinergic receptors with ATP (ΔF340/F380 ∼0.4 to 0.5, Δ[Ca2+]i ∼300 nmol/L and subsequent ΔVm ∼–20 to –25 mV) is prominent versus muscarinic receptor activation with ACh (ΔF340/F380 < 0.05 and ΔVm≤– 5 mV) (compare Figs. 2 to 3). No trends or statistically significant alterations in ΔF340/F380 and ΔVm responses to ACh were observed for muscarinic receptor function (Fig. 3).
Both ΔF340/F380 (Fig. 2C– F) and ΔVm (Fig. 2G– J) responses to ATP were relatively stable throughout AD pathology with the exception of a trend for a ∼5 mV decrease in hyperpolarization in males during AβT conditions (p > 0.05). In addition, the ΔVm-to-Δ[Ca2+]i coupling ratio during ATP (i.e., sensitivity of SKCa/IKCa activation for hyperpolarization in response to increases in [Ca2+]i following purinergic activation) was lowest during AβT conditions in both sexes (Table 1). However, alterations in the ΔVm-to-Δ[Ca2+]i coupling ratio (mV hyperpolarization produced per nmol/L increase in [Ca2+]i) during ATP were not statistically significant throughout development of AD pathology (Table 1).
Effect of AD pathology on cerebrovascular endothelial SKCa/IKCa channel activation for hyperpolarization and subsequent increases in intracellular Ca2+ in both biological sexes
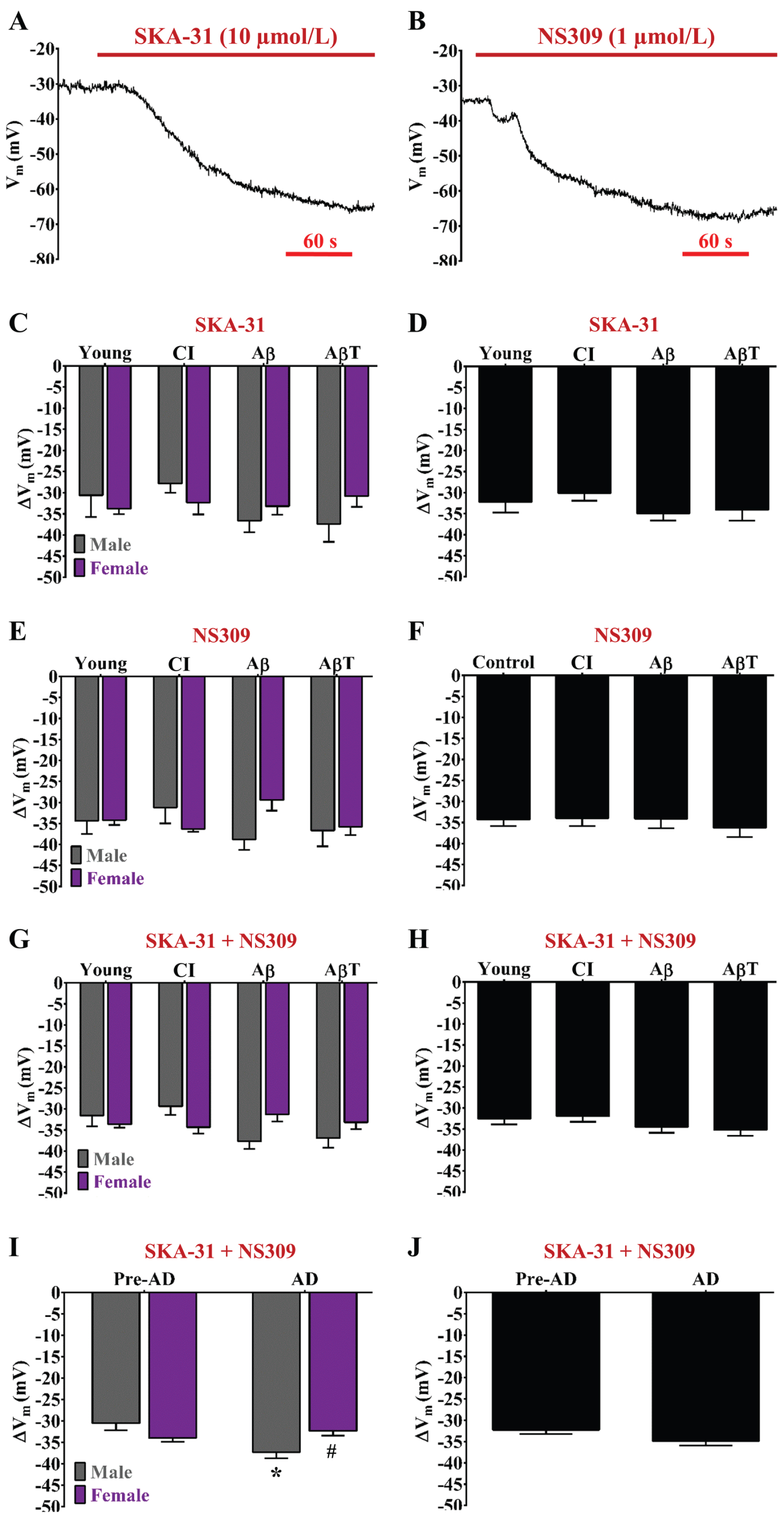
AD pathology enhances SKCa/IKCa channel function for Vm hyperpolarization in males but not females. Example recordings of Vm before and during (A) SKA-31 (10 μmol/L; mild SKCa/IKCa channel activator) and (B) NS309 (1 μmol/L; potent SKCa/IKCa channel activator) in an isolated endothelial tube of a young female. C) Summary data for ΔVm during SKA-31 as a function of AD group (Young, CI, Aβ, and AβT) and biological sex (Male, Female). D) As shown in (C) as a function of AD pathology only (sexes combined). E, F) As shown in (C) and (D) respectively for NS309. G) Summary data for ΔVmduring SKA-31 and NS309 combined as a function of AD group and biological sex [data from (C) and (E) combined]. H) As shown in (G) as a function of AD pathology only with sexes combined [data from (D) and (F) combined]. I) Summary data for ΔVm during SKA-31 and NS309 combined as a function of pre-AD group (Young + CI) versus AD (Aβ+ AβT) in accord with biological sex. J) As shown in (I) as a function of AD pathology only with sexes combined. n = number of animals and respective cerebral endothelial tubes (Young, CI, Aβ, AβT: 5 to 7, 5 to 6, 5, and 5 to 8 Males & 5 to 6, 6, 5, and 5 to 6 Females; combined for AD group only: 10 to 13, 11 to 12, 10, and 10 to 14). See Table 2 for corresponding Δ[Ca2+]i/ΔVm ratios during SKCa/IKCa channel activation.*p<0.05 (two-way ANOVA), AD Males versus Pre-AD Male;#p < 0.05 (two-way ANOVA), AD Female versus AD Male. CI, cognitive impairment; Aβ, amyloid-β; AβT, amyloid-β+ tau neurofibrillary tangles; AD, Alzheimer’s disease.
The ΔVm/Δ[Ca2+]i ratio during purinergic receptor activation via ATP throughout AD pathology across sexes (see Fig. 2)
A relatively higher ΔVm/Δ[Ca2+]i ratio supports a higher sensitivity of KCa activation for hyperpolarization in response to increases in [Ca2+]i.ΔVm/Δ[Ca2+]i units = [(mV hyperpolarization)/(nmol\bulletL –1)]; all data are presented as the mean±SEM; n = one endothelial tube from one animal.
We have previously found that endothelial SKCa/IKCa channel function is reduced in cerebral endothelium of old female C57BL6/N mice (versus young female and old male) in response to an effective concentration (∼EC50; 1 μmol/L) of NS309 [11]. However, the effect of AD pathology on cerebrovascular endothelial SKCa/IKCa channel function is unknown in accord with biological sex. Hyperpolarization responses to EC50 concentrations of SKA-31 (10 μmol/L; Fig. 4A) versus NS309 (1 μmol/L; Fig. 4B) were not significant throughout all study groups (compare Fig. 4C to 4E) demonstrating that either SKCa/IKCa channel opener and its respective EC50 concentration activates SKCa/IKCa channels to the same extent regardless of AD pathology group and biological sex. Whether data from respective SKCa/IKCa channel openers are separate (Fig. 4C, E) or combined (Fig. 4G), SKCa/IKCa channel function increased in males while having remained relatively stable in females throughout development of AD pathology. We observed similarity among respective pre-AD conditions (young, CI) and AD conditions (Aβ, AβT) within ∼30 to 40 mV of hyperpolarization (Fig. 4G, I). However, a significant ∼20% increase (p < 0.05; two-way ANOVA) in SKCa/IKCa channel function was observed in males when examining overall AD (ΔVm; Aβ+ AβT: – 37±2 mV, n = 23 independent measurements of SKA– 31 or NS309 from 13 endothelial tubes from 13 animals) versus pre-AD conditions (young + CI: – 31±2 mV, n = 23 from 13 endothelial tubes) (Fig. 4I). In contrast, for females, AD conditions (ΔVm; Aβ+ AβT: – 32±1 mV, n = 21 from 11 endothelial tubes) did not significantly alter hyperpolarization responses relative to pre-AD conditions (young + CI: – 34±1 mV, n = 23 from 12 endothelial tubes) (Fig. 4I). Note that progressive AD pathology does not have an apparent effect on ΔVm responses to SKA-31 and/or NS309 when data from sexes are combined (Fig. 4D, F, H, J).
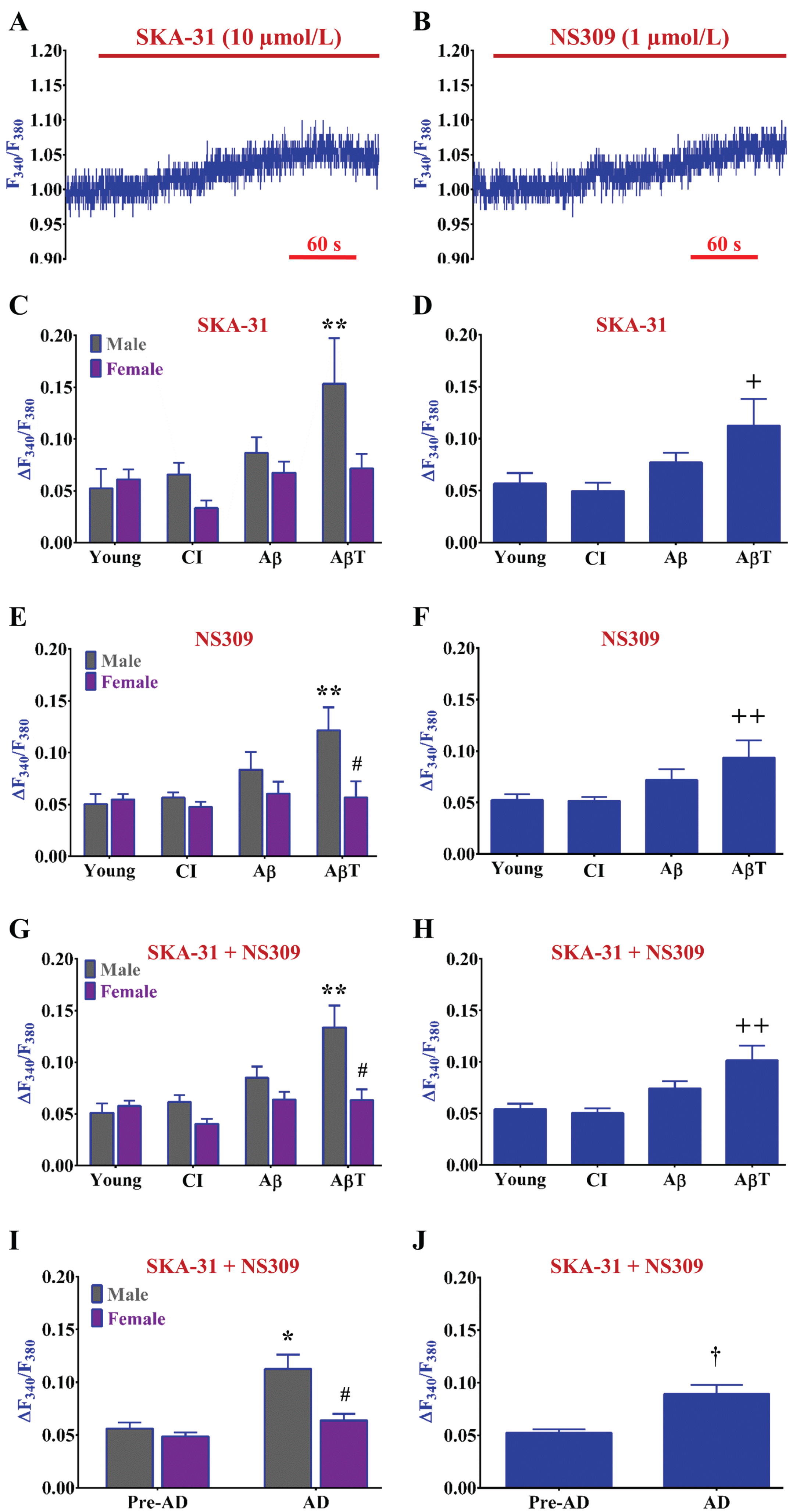
Development of AD pathology progressively enhances [Ca2+] i responses following SKCa/IKCa channel activation and hyperpolarization in males but not females. Example recordings of F340/F380 before and during (A) SKA-31 (10 μmol/L; mild SKCa/IKCa channel activator) and (B) NS309 (1 μmol/L; potent SKCa/IKCa channel activator) in an isolated endothelial tube of a young female. (A) and (B) are simultaneous F340/F380 recordings with V m in Fig. 4A and 4B respectively. C) Summary data for ΔF340/F380 during SKA-31 as a function of AD group (Young, CI, Aβ, and AβT) and biological sex (Male, Female). D) As shown in (C) as a function of AD pathology only (sexes combined). E, F) As shown in (C) and (D) respectively for NS309. (G) Summary data for ΔF340/F380 during SKA-31 and NS309 combined as a function of AD group and biological sex [data from (C) and (E) combined]. H) As shown in (G) as a function of AD pathology only with sexes combined [data from (D) and (F) combined]. I) Summary data for ΔF340/F380 during SKA-31 and NS309 combined as a function of pre-AD group (young + CI) versus AD (Aβ+ AβT) in accord with biological sex. J) As shown in (I) as a function of AD pathology only with sexes combined. n = number of animals and respective cerebral endothelial tubes (Young, CI, Aβ, AβT: 5 to 7, 5 to 6, 5, and 5 to 8 Males & 5 to 6, 6, 5, and 5 to 6 Females; combined for AD group only: 10 to 13, 11 to 12, 10, and 10 to 14). See Table 2 for corresponding data on Δ[Ca2+]i/ΔVm ratio during SKCa/IKCa channel activation. *p < 0.05 (two-way ANOVA), AD Males versus Pre-AD Males; **p < 0.05 (two-way ANOVA), AβT Males versus Young and CI Males; #p < 0.05 (two-way ANOVA), Female versus Male of same AD pathology group; +p < 0.05 (one-way ANOVA), AβT pathology group versus CI; ++p < 0.05 (one-way ANOVA), AβT pathology group versus Young and CI. CI, cognitive impairment; Aβ, amyloid-β; AβT, amyloid β+ tau neurofibrillary tangles; AD, Alzheimer’s disease.
The Δ[Ca2+]i/ΔVm ratio during SKCa/IKCa channel activation via SKA-31 and NS309 throughout AD pathology across sexes (see Figs. 4 and 5). A relatively higher Δ[Ca2+]i/ΔVm ratio supports a higher sensitivity of Ca2+ influx through non-selective cation channels (or plasma membrane perforations) in response to hyperpolarization produced by KCa activation. Δ[Ca2+]i/ΔVm units = [(nmol\bulletL –1)/(mV hyperpolarization)]; all data are presented as the mean±SEM; n = one endothelial tube from one animal, n′= individual measurements per NS309 or SKA-31, number of endothelial tubes/animals (e.g., n′= 12,7 represents 12 measurements during NS309 or SKA-31 from 7 different endothelial tubes from 7 different animals)
†p < 0.05 (two-tailed unpaired t-test), AD versus Pre-AD. *p < 0.05 (two-way ANOVA), AβT/AD Male versus Young/Pre-AD Male; **p < 0.05 (one-way ANOVA), AβT versus Young & CI; #p < 0.05 (two-way ANOVA), Female versus Male of same AD group.
The hyperpolarization of Vmvia K+ channel activation can enhance Ca2+ entry into endothelial cells via an enhanced electrical driving force [11, 30]. Thus, we also simultaneously determined whether SKA-31 or NS309 indirectly influenced [Ca2+]i signaling following SKCa/IKCa activation. Following hyperpolarization of Vm in response to SKA-31 or NS309 (Fig. 4), F340/F380 increased by ∼0.05 to 0.10 accordingly (Δ[Ca2+]i ∼25 to 50 nmol/L) (Fig. 5A, B). As with ΔVm, ΔF340/F380 responses were not significant (p > 0.05) to EC50 concentrations of SKA-31 (Fig. 5A) versus NS309 (Fig. 5B) throughout all study groups (compare Fig. 5C to 5E). Whether data from respective SKCa/IKCa channel openers are separate (Fig. 5C, E) or combined (Fig. 5G), ΔF340/F380 responses following SKCa/IKCa channel activation progressively increased with advancement of AD pathology in males by approximately three-fold in AβT animals versus young. In contrast, ΔF340/F380 responses remained stable throughout AD pathology in females (Fig. 5G). With some exception (p < 0.05, CI Female versus CI Male; two-tailed unpaired-test), we observed similarity for ΔF340/F380 responses among respective pre-AD conditions for both sexes (Fig. 5G, I). Altogether, AD conditions approximately double ΔF340/F380 responses in males versus pre-AD conditions following EC50 activation of SKCa/IKCa channels (Pre-AD: 0.06±0.01; AD: 0.11±0.01) (Fig. 5I). In comparison for females, ΔF340/F380 responses were relatively similar throughout development of AD (0.06±0.01 versus Pre-AD, 0.05±0.01) (Fig.5I). Note that stable ΔF340/F380 responses in females are not apparent when only AD pathology is considered with data combined from both sexes (Fig. 5D, F, H, J).
With reference to Figs. 4G and 5G, the Δ[Ca2+]i-to-ΔVm coupling ratio (nmol/L increase in [Ca2+]i per mV hyperpolarization) following SKCa/IKCa activation significantly increased in males during AD pathology (1.83±0.31 versus Pre-AD, 1.03±0.14; p < 0.05; two-way ANOVA) (Table 2). In contrast, the Δ[Ca2+]i-to-ΔVm coupling was relatively stable throughout overall pre-AD (0.75±0.06) and AD (1.09±0.13) conditions in females.
Effect of AD pathology on cerebrovascular endothelial KIR channel function in both biological sexes
KIR channels govern cerebral vasodilation and blood flow [33, 34] but the potential impact of progressive AD pathology on cerebrovascular endothelial KIR function remains unknown. Stimulation of cerebrovascular endothelial KIR channels with 15 mM KCl elicits a modest Vm hyperpolarization response [ΔVm ∼– 5 to – 10 mV; [11, 13]; Fig. 6A, B). The hyperpolarizing ΔVm responses to 15 mM KCl significantly decreased by ∼50% during AD conditions (Aβ and AβT: – 4±1 mV, n = 22) relative to the young group (– 8±1 mV, n = 11) regardless of sex (Fig. 6C– F). Corresponding changes in ΔF340/F380 (≤0.03) to 15 mM KCl were inconsistent or altogether absent for detection.
Effect of AD pathology on cerebrovascular endothelial [Ca2+]i responses to mitochondrial uncoupling
The development of chronic disease generally involves increased mitochondrial matrix Ca2+ [19, 20]. Thus, we examined whether endothelial mitochondrial Ca2+ content and thereby its capacity for Ca2+ release into the cytosol, alters throughout development of AD pathology via mitochondrial uncoupling using FCCP [20, 21]. FCCP is a proton ionophore that collapses the ΔΨmt (typically – 180 mV in reference to the cytoplasm) which reduces the affinity for Ca2+ ions within the mitochondrial matrix [21]. As a result, a slow increase in [Ca2+]i to FCCP ensues versus a rapid [Ca2+]i increase following purinergic receptor stimulation (compare Fig. 7A or 7B to Fig. 2A). Other than higher [Ca2+]i responses to FCCP by ∼40% in females (0.48±0.02, n = 5) versus males (0.33±0.02, n = 5) during Aβ conditions (p < 0.05; two-tailed unpaired t-test; Fig. 7A– C), there was no significant difference in general among AD versus pre-AD conditions (Fig. 7E). Any visible sex differences for ΔF340/F380 responses during FCCP are masked when only AD pathology is considered with data from sexes combined (Fig. 7D, F).

AD pathology impairs KIR channel function for Vm hyperpolarization regardless of sex. Example recordings of Vm before and during 15 mmol/L KCl (KIR channel activator) in an isolated endothelial tube of a (A) Young female and (B) Aβ female, respectively. C) Summary data for ΔVm during elevated KCl as a function of AD pathology (Young, CI, Aβ, AβT) and sex (Male, Female). D) As shown in (C) as a function of AD pathology only (sexes combined). E) Summary data for ΔVm during elevated KCl combined as a function of pre-AD group (young + CI) versus AD (Aβ+ AβT) in accord with biological sex. F) As shown in (E) as a function of AD pathology only with sexes combined. n = number of animals and respective cerebral endothelial tubes (Young, CI, Aβ, AβT: 5, 5, 6 and 6 Males and 6, 6, 5, and 5 Females; combined for AD pathology only: 11 per group). *p < 0.05 (two-way ANOVA), Aβ/AβT/AD group versus Young/Pre-AD group of like sex; +p < 0.05 (one-way ANOVA), AβT group versus Young group; ++p < 0.05 (one-way ANOVA), Aβ group versus Young & CI groups; †p < 0.05 (two-tailed unpaired t-test), AD versus Pre-AD. Note in Panel C that p < 0.05 for AβT Females versus AβT Males via two-tailed unpaired t-test but not two-way ANOVA. CI, cognitive impairment; Aβ, amyloid-β; AβT, amyloid-β+ tau neurofibrillary tangles; AD, Alzheimer’s disease.

Overall AD versus pre-AD pathology does not significantly alter [Ca2+]i responses to mitochondrial uncoupling. Example recordings of [Ca2+]i during FCCP (1 μmol/L; proton ionophore that dissipates the inner mitochondria membrane potential and releases mitochondrial Ca2+ into the cytosol) in an isolated endothelial tube of a (A) Aβ male and (B) Aβ female mouse respectively. C) Summary data for ΔF340/F380 during FCCP as a function of AD pathology group [Young, cognitive impairment (CI), Amyloid-β (Aβ), and Amyloid-β+ tau neurofibrillary tangles (AβT)] and sex (Male, Female). D) As shown in C as a function of AD pathology only (sexes combined). E) Summary data for ΔF340/F380 during FCCP combined as a function of pre-AD group (young + CI) versus AD (Aβ+ AβT) in accord with biological sex. F) As shown in (E) as a function of AD pathology only with sexes combined. n = number of animals and respective cerebral endothelial tubes (Young, CI, Aβ, AβT: 5, 5, 5 and 6 Males and 6, 6, 5, and 5 Females; combined for AD groups only: 11, 11, 10, and 11). Note in Panel C that p < 0.05 for Aβ Females versus Aβ Males via two-tailed unpaired t-test but not two-way ANOVA. CI, cognitive impairment; Aβ, amyloid-β; AβT, amyloid-β+ tau neurofibrillary tangles.
DISCUSSION
Optimized blood flow in accord with metabolic demand to and throughout the brain maintains healthy cognitive function throughout the human lifespan [35]. Chronic alterations in cardio- and cerebrovascular function may trigger the earliest stages of dementia known generally as cognitive impairment. During advanced stages of neurodegenerative disease with significantly diminished energetic resources, brain parenchyma may accumulate toxic levels of peptides such as amyloid-β and hyperphosphorylated tau. Concomitant with the general production and accumulation of toxic peptides and metabolites, diminished vascular clearance of toxic peptides via venular and meningeal lymphatic networks may occur as a well, perhaps playing a more prominent role during the terminal stages of dementia [36]. As an organ in and of itself, vascular endothelium is critical for the perfusion of all other organs, and thereby a central player in the co-development of multiple chronic morbidities such as neurodegeneration, diabetes, hypertension, kidney disease, heart failure, and cancer [10]. While gradually deteriorating to conditions of advanced AD, it is unknown how development of AD in particular influences signaling inputs that modulate the interface between [Ca2+]i and electrical signaling underlying cerebrovascular endothelial function to maintain normal cerebral blood flow. Key components of such modulation of cerebral blood flow include endothelial GPCRs and K+ channels. In order to better understand how cerebrovascular endothelial function interfaces with AD etiology, we employed a study model (3×Tg-AD mice [17, 18]) that fully develops cumulative phases of AD pathology over ∼12 months (middle age of normal, wild-type C57BL/6 mice [37]). Although the sensitivity of SKCa/IKCa activation to ATP-induced intracellular Ca2+ increases was lowest during AβT pathology as similar to old (≥24 months) male C57BL/6 mice [11], purinergic receptor function was not significantly affected by overall AD pathology versus pre-AD conditions. However, SKCa/IKCa channel function itself and the sensitivity of Ca2+ influx to hyperpolarization of membrane potential was enhanced in males during AD (Aβ+ AβT) versus pre-AD (young, CI) conditions while stable throughout in females. When data for both sexes are combined to examine overall progression of AD pathology, disparate effects of male versus females are not apparent. As with old C57BL/6 animals [11], KIR channel function declined by >40% during AD conditions regardless of biological sex. In addition, with the exception of relatively prominent [Ca2+]i responses in Aβ females to the mitochondrial uncoupler FCCP, there were no statistically significant differences in mitochondrial Ca2+ release among overall AD versus pre-AD pathology. In the context of the recognized literature thus far, efforts for illuminating the scientific and clinical implications of these findings have been included below.
Cerebrovascular endothelial GPCR activity to produce EDH is stable during overall AD pathology
Purinergic receptors play a major role in cerebrovascular endothelial cells in addition to neuronal-glial interactions for the control of cerebral blood flow and brain parenchymal function and metabolism [38]. Our previous work [11, 13] and the current study (Fig. 2) illustrate significant ATP activation of EDH (ΔVm: ∼– 20 mV) in mouse posterior cerebral arteries. As similar to old C57BL/6N mice [11], ATP-evoked EDH decreased during AβT in males but was relatively stable or enhanced throughout in females compared to young animals with absence of pathology (Fig. 2). Although not reflective of purinergic receptor function alone, we also observed ΔVm-to-Δ[Ca2+]i coupling to be at its lowest during late stage AD (AβT) following purinergic receptor stimulation (Table 1). However, our data illustrate overall stability of purinergic receptor function during AD pathology as a whole versus pre-AD conditions. Thus, although cerebrovascular purinergic receptor function may decrease with conditions of old age itself [11, 39], it has not been as straightforward for interpretation during development of AD. We suspect that, for aging studies, loss of respective sex hormone signaling may partially explain the divergence in purinergic receptor function in males versus females in animals that are ≥24 months of age [11]. Indeed, for reference, estrogen can blunt endothelial purinergic receptor relaxation of smooth muscle cells in cerebral arteries of young ovariectomized females supplemented with exogenous estrogen [40]. In the current study, the animal study model was 3×Tg-AD animals <16 months (middle age and younger), whereby significant alterations in sex hormone receptor function may have not been a contributing factor. Although, lower levels of androgens and estrogens have been found in ≥80-year-old AD human subjects [41], and as shown in carefully applied interventions in gonadectomized experimental animals, they may limit development of AD pathology [42].
Especially in contrast to skeletal muscle endothelium, ACh activation of cerebrovascular endothelial muscarinic receptors produces a small (ΔVm ∼– 5 mV) and transient (≤30 s) EDH response [13] (Fig. 3). We have previously found that this EDH response significantly declined by ∼– 3 mV in both male and female mice during old age as ≥24 months [11]. In the current study, we observed a ∼– 1 mV reduction in ACh-evoked EDH in both males and females during AD versus pre-AD that was not statistically significant (Fig. 3). At least with respect to vascular GPCR control of cerebral blood flow versus direct modulation of neuronal function, the muscarinic receptors may play an adjunct role to purinergic receptors. In addition, these receptors may also produce NO instead as alternative to EDH for dilating cerebral blood vessels and thereby promoting blood flow to and throughout the brain [15].
Cerebrovascular endothelial K+ channels during AD pathology: Sex-dependent divergence in SKCa/IKCa channel function but sex-independent reduction in KIR channel function for direct activation of EDH
The idea of using K+ channel openers for protection of cerebrovascular endothelium against the effects of amyloid-β has been around for over 20 years [43]. Most recently, selective pharmacology of endothelial SKCa/IKCa channels has been utilized as a promising therapy to maintain overall cardiovascular and cognitive health with aging with little to no overt systemic toxicity or pathology [16]. Vascular NO bioavailability decreases with old age [44, 45] and AD [3, 4].While hyperpolarization through SKCa/IKCa channel activation has the capability to restore NO [10], it is conceivable that endothelial K+ channel modulators can counteract cerebrovascular impairment preceding and accompanying cognitive impairment and AD. We have previously found that EC50 activation of cerebrovascular endothelial SKCa/IKCa channels was slightly enhanced with male old age but significantly declined with female old age [11]. In the current study using 3×Tg-AD animals where old age was not a contributing factor, a similar pattern of pre-AD to AD transitioning (versus young to old age) was observed as enhancement in males (ΔVm; pre-AD and AD: – 31±2 and – 37±2 mV) versus slight decline in females (pre-AD and AD: – 34±1 and – 32±1 mV) (Fig. 4).
The coupling of [Ca2+]i with hyperpolarization of Vm following activation of SKCa/IKCa channels was also progressively enhanced with AD pathology in males while relatively stable throughout groups in females (Fig. 5 and Table 2). Thus, this “hyperpolarization-induced Ca2+ entry” mechanism likely via transient receptor potential channels [10, 30] may serve as a more effective feed-forward mechanism of K+ channel activation and/or production of NO [31] in males versus females during development of AD (Fig. 5 and Table 2). Although, as maximum vasodilation can be achieved with only 10 to 15 mV of EDH [46] in comparison to this mechanism effectively operating at ∼30 mV of hyperpolarization needed to detect significant changes in [Ca2+]i, the physiological relevance or therapeutic utility of this mechanism may be marginal unless submaximal GPCR stimulation is concomitantly at play as well [30].
On the other hand, the presence of the “hyperpolarization-induced Ca2+ entry” mechanism may be a key feature of pathology as Aβ oligomers associated with AD symptoms have been found to mediate Ca2+ influx through perforations in neuronal membranes [47]. Thus, it may not be surprising to observe progressively increasing [Ca2+]i responses due to Ca2+ entry following direct membrane hyperpolarization with advancing AD pathology in vascular cells as well. Although, females were observed to be resistant to [Ca2+]i increases following SKCa/IKCa activation versus males during AD (Fig. 5 and Table 2). With estrogenic infrastructure and signaling as a potential candidate [48], the basis for this sex difference in Δ[Ca2+]i-to-ΔVm coupling in cerebrovascular endothelial cells during the development of AD pathology needs further investigation.
As with any studies of chronic disease during old age [11], the basis of a sex difference in SKCa/IKCa channel function and whether it significantly impacts physiological regulation of cerebral blood flow in AD female animals is unknown and a subject for future study. Indeed, as shown in males, a modest upregulation of K+ channel function with age may maintain local vasodilation underlying at least a minimal level of tissue perfusion to sustain life [10]. It should be noted that females in the general population have a higher incidence of AD even after correcting for age [49, 50]. The apparent diminishment of cerebrovascular EDH as defined by SKCa/IKCa channel function in females versus males [51] and alterations with age [11] may help to explain and treat the chronic cerebral hypoperfusion that precedes and accompanies AD pathology [52].
As channels that serve as extracellular K+ sensors to modulate K+ efflux underlying Vm hyperpolarization, cerebrovascular endothelial KIR channels also modulate cerebral blood flow and perfusion [33, 34]. KIR channels may be integral to the control of cerebral blood flow during resting conditions on their own in addition to being interdependent with activation of SKCa/IKCa channels for EDH [33, 53]. Our previous findings indicate a significant decrease in KIR channel responses to 15 mM KCl by ∼40% in old versus young C57BL/6 animals regardless of biological sex [11]. Overall, we observed a similar pattern in the current study when transitioning from pre-AD to AD conditions with a ∼50% decrease in KIR channel responses in both males and females (Fig. 6). A likely mechanism for this general decrease in KIR channel function entails altered membrane lipid and cholesterol content that accompanies old age and/or development of AD pathology [33, 54].
Altered vascular ion channel function with age and chronic pathology can be due to increased mitochondrial Ca2+ buffering and release [20]. Also, AD pathology in particular may be associated with mitochondrial toxicity [50] typically described by mitochondria pathologically overloaded with Ca2+[20]. Depletion of mitochondrial Ca2+ using FCCP [20, 21] evoked higher [Ca2+]i responses by ∼40% in females versus males during Aβ pathology but there was no significant difference in general among AD versus pre-AD conditions (Fig. 7). Since estrogen receptor signaling protects against mitochondrial toxicity of amyloid-β [50], it is possible that this effect of mitochondrial Ca2+ content is more profound in old (∼24 months) female 3×Tg-AD animals where estrogen receptor-dependent regulation is likely at its lowest in the mouse lifespan preceding death.
Experimental considerations
The current study reflects an unprecedented characterization of cerebrovascular endothelial GPCR and K+ channel function per developmental phases of AD. To an extent reasonably possible, we endeavored to model physiological conditions with regard to temperature (37°C) and continuous laminar flow over freshly isolated and intact endothelium [25]. Using this study tool, we have found that endothelial physiology is retained with regard to Ca2+ homeostasis and Vm regulation [10, 11]. Our simultaneous measurements of endothelial [Ca2+]i and Vm are technically challenging while particularly exhaustive in the current study across 8 study groups with inclusion of sex as a biological variable. In such manner, we discuss the limitations of our approach in the context of future directions next.
The current study did not utilize age-matched B6129SF2/J mice (hybrid of C57BL/6J and 129S1/SvImJ strains) as the original background strain suitable for generating 3×Tg-AD mice. Thus, our present comparisons with wild-type conditions are limited to our previous work using another non-transgenic, “normal” strain as C57BL/6N mice provided by the National Institute on Aging [11]. The circumstantial reasons for such a decision entail use of homozygous 3×Tg-AD offspring (i.e., ideal littermate controls not available during breeding schema) and the potential for a massive logistical undertaking involving 16 study groups total for one straightforward study. A scientific reason includes evidence of negligible contribution of the aging process alone to the neuronal and cognitive health of B6129SF2/J mice relative to progressive neurodegeneration clearly observed for 3×Tg-AD mice up to middle age (∼15 months) [18, 24]. Regardless, differences in the maintenance of neurovascular function among various inbred (e.g., C57BL/6J & N) and outbred (e.g., CD-1) strains up throughout old age (≥24 months) is an outstanding question that remains to be addressed for future experiments.
Mechanistic resolution for various GPCR and ion channels was limited in the current study as we did not test for precise roles among cholinergic receptor (nicotinic versus muscarinic) and purinergic receptor subtypes (X versus Y) throughout conditions. Although, evidence supports the presence of endothelial cholinergic (not nicotinic) muscarinic receptors in the mouse cerebral circulation [55, 56]. Also, our current knowledge supports the presence of P2X receptors on smooth muscle cells and P2Y receptors on rodent cerebrovascular endothelial cells, whereby ATP activates respective GPCRs for vasoconstriction and vasodilation respectively [12, 57]. Exceptions to this vascular architecture have been reported for the endothelium of peripheral mesenteric arteries containing functional P2X1 receptors [58]. Further, as Aβ increases the expression of P2X receptors in PC12 cells and hippocampal neurons [59], a similar shift in purinergic receptor expression may occur in in vascular cells during AD. The potential emergence and diminution of various vascular GPCR and/or ion channel isoforms throughout the development of AD pathology is also a crucial direction for future research.
Finally, we did not track hormonal cycles or measure estrogen levels in this study. However, as a hormone with chronic actions throughout young to middle age life, we suspect that, for the most part only long-term depletion (weeks to months versus ∼5 days) of estrogen during conditions of old age would significantly alter its otherwise genomic and corresponding physiological imprint on female endothelial cell function [60]. In support of this notion, past studies of rodent endothelial function of mesenteric and cerebral arteries have typically taken place ∼1 month (2 weeks minimum) following an extreme surgical intervention to deplete estrogen (ovariectomy) with and without long-term estrogen replacement in order to observe significant alterations in nitric oxide and/or EDH signaling [61, 62]. Further, it is also important to note that the current study utilized a cellular study tool that did not include the presence of blood flow and circulating hormones during data collection. Regardless, we indeed understand the remaining importance of resolving the context and corresponding mechanisms by which the phase of the estrus cycle does [63] or does not [64] significantly impact moment-to-moment cerebral blood flow regulation.
Summary and conclusions
AD is currently the 6th leading cause of death in the United States [65]. Approximately 5.8 million Americans are living with AD, whereby more than 95% of patients are over the age of 65 [65] as a demographic which is projected to rise to ∼21% by 2040 [66]. In the general population, cerebral hypo perfusion is associated with accelerated cognitive decline and an increased risk of dementia [52]. Here, we examined components of endothelium-derived hyperpolarization as a moment-to-moment regulator of cerebral blood flow in a model of AD that progressively develops cognitive impairment and AD pathology as extracellular Aβ plaque burden and neurofibrillary tangles [17, 18]. We found that GPCR activity as purinergic and muscarinic receptor function did not significantly alter among AD versus pre-AD conditions but K+ channel function indeed changed, whereby enhancement of SKCa/IKCa channel function was unique to males and KIR channel function decreased regardless of sex. These observed alterations in K+ channels as a function of AD pathology may be due to an accumulation of pathological contributors such as mitochondrial Ca2+ burden and excess lipid and cholesterol content in plasma membranes [33, 54]. Altogether, we reinforce that AD is also a condition of cerebrovascular pathology, whereby we have now identified sex-dependent and sex-independent roles for endothelial KCa and KIR channels respectively. Especially in light of recent developments for therapy [16], we are confident that selective endothelial K+ channel pharmacology may offer promising therapeutic strategies to maintain overall cardio/cerebrovascular and cognitive health.
Footnotes
ACKNOWLEDGMENTS
This research has been supported by National Institutes of Health grants R00AG047198 and R56AG062169 (to E.J.B.). The content of this original article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.