
Abstract
While prevailing evidence supports that the amyloid cascade hypothesis is a key component of Alzheimer’s disease (AD) pathology, many recent studies indicate that the vascular system is also a major contributor to disease progression. Vascular dysfunction and reduced cerebral blood flow (CBF) occur prior to the accumulation and aggregation of amyloid-β (Aβ) plaques and hyperphosphorylated tau tangles. Although research has predominantly focused on the cellular processes involved with Aβ-mediated neurodegeneration, effects of Aβ on CBF and neurovascular coupling are becoming more evident. This review will describe AD vascular disturbances as they relate to Aβ, including chronic cerebral hypoperfusion, hypertension, altered neurovascular coupling, and deterioration of the blood-brain barrier. In addition, we will describe recent findings about the relationship between these vascular defects and Aβ accumulation with emphasis on in vivo studies utilizing rodent AD models.
Keywords
INTRODUCTION: ALZHEIMER’S DISEASE AND THE AMYLOID CASCADE HYPOTHESIS
Alzheimer’s disease (AD) was first described by Alois Alzheimer in 1906 after examining the brain of Auguste Deter, a 51-year-old woman with aggressive and progressive memory loss. Alzheimer’s findings eventually became known as the pathological hallmarks of AD [1]. AD is a devastating illness characterized by a progressive decline in cognition, which is currently the 6th leading cause of death in the United States, and the primary cause of dementia [2]. Initially, AD was considered a middle-age disease whereas senile dementia was separately defined as progressive dementia in the elderly. However, since middle-age AD and senile dementia patients share indistinguishable postmortem neuropathology, including the abnormal accumulation of amyloid plaques, both conditions are now identified as AD [3, 4]. The amyloid cascade hypothesis describes the events leading to the development of plaques in AD brains by preferential overproduction of the amyloid-β (Aβ)42 isoform that is prone to aggregation compared to the Aβ40 isoform [5]. This accumulation of toxic Aβ42 isoforms begins when amyloid-β protein precursor (AβPP) is cleaved sequentially by β-secretase BACE1 (β-site APP-cleaving enzyme 1) and γ-secretase [6]. Additional processes that increase the amount of Aβ42 include impaired degradation by neprilysin or insulin degrading enzymes [7, 8], or reduced clearance across the blood-brain barrier (BBB) [9] through low density lipoprotein receptor related protein 1 (LRP1) and receptor for advanced glycation end products (RAGE) found on epithelial cells [10, 11]. Eventually, accumulation reaches a threshold where Aβ42 begins to aggregate into low molecular weight oligomers followed by formation of insoluble plaques that are deposited throughout the brain [12–14]. While femtomolar to picomolar concentrations of Aβ42 play a role in the healthy brain [15, 16], higher nanomolar concentrations of soluble Aβ42 is known to increase neuronal activity that may create an excitotoxic environment [17–20]. Excitotoxicity includes an inflammatory response consisting of microglial and astrocytic activation [21, 22], progressive neuronal/synaptic injury, altered neuronal ionic homeostasis and oxidative injury, and aberrant tau protein hyperphosphorylation and aggregation [23]. The eventual neuronal dysfunction and cell death associated with this cascade of events leads to widespread cerebral atrophy [24] that is prominent throughout the neocortex including the medial temporal lobe, cingulate gyrus, parietal lobe, and frontal lobe [25–27]. Altered neurotransmission and the subsequent neurodegeneration are hypothesized to contribute to the cognitive and behavioral decline observed in AD [28, 29].
The identification of early-onset familial AD caused by genetic mutations of APP, presenilin-1, and presenilin-2 (the catalytic subunit of γ-secretase), strengthened the case for the amyloid cascade hypothesis of AD pathogenesis [30]. These mutations result in a higher ratio of Aβ42/Aβ40 production, increased rates of Aβ oligomerization, and senile plaque formation [28, 31]. Consequently, disease-modifying therapies targeting Aβ production were developed [32], but all clinical trials, to date, targeting abnormal Aβ accumulation have been unsuccessful at slowing or halting AD progression [33, 34]. However, these studies primarily focused on mild-to-moderate AD, presumably when pathological hallmarks and neurodegeneration were already present. It is currently unclear if clinical trials designed to treat AD prior to the development of severe pathological changes might improve patient outcome. However, obstacles exist to implementing this strategy in the human population, such as the lack of a reliable early biomarker for AD.
A surprising discovery came from ‘the Nun Study’ in which cognition in elderly nuns (aged 77–103 years) was assessed with tests including the Mini-Mental State Examination and postmortem neuropathological changes were quantified [35]. A subset of individuals with normal cognition had an abundance of diffuse plaques in the neocortex and hippocampus, indicating the involvement of additional unknown factors in the cognitive symptoms of AD. Findings such as these suggest that the amyloid cascade hypothesis is not the sole contributor to the etiology of AD and overlapping neuropathologies may contribute to disease progression.
CEREBRAL VASCULATURE AND THE TWO-HIT VASCULAR HYPOTHESIS OF ALZHEIMER’S DISEASE
Blood is delivered to the brain by cardiovascular circulation that is regulated by heart rate. The internal carotid artery supplies the frontal, parietal, and lateral temporal lobes, while the vertebral arteries supply the occipital lobe, brainstem, and cerebellum. Under physiological conditions, systemic blood flow delivery into the brain remains relatively constant and entry is regulated by the precise architecture of blood vessels branching into a vascular tree [36]. The large penetrating arteries break off and diverge into arterioles, precapillary arterioles, and finally into capillaries. The regulation of local cerebral blood flow (CBF) requires different cell types distributed along the vascular tree (Fig. 1). While all blood vessels are coated by endothelial cells, the presence of other cell types depends on the region of vascular branching. For example, penetrating arteries have 2–3 layers of vascular smooth muscle cells (VSMCs) whereas arterioles only have a single VSMC layer. Capillaries share a common basement membrane with cells called pericytes, and are covered by astrocytic endfeet that are innervated by local neurons [36]. Neurons, astrocytes, VSMCs, pericytes, and endothelial cells work in concert to form the neurovascular unit (NVU). The NVU regulates local CBF in response to brain activity, a process called neurovascular coupling (NVC) or functional hyperemia [37, 38].
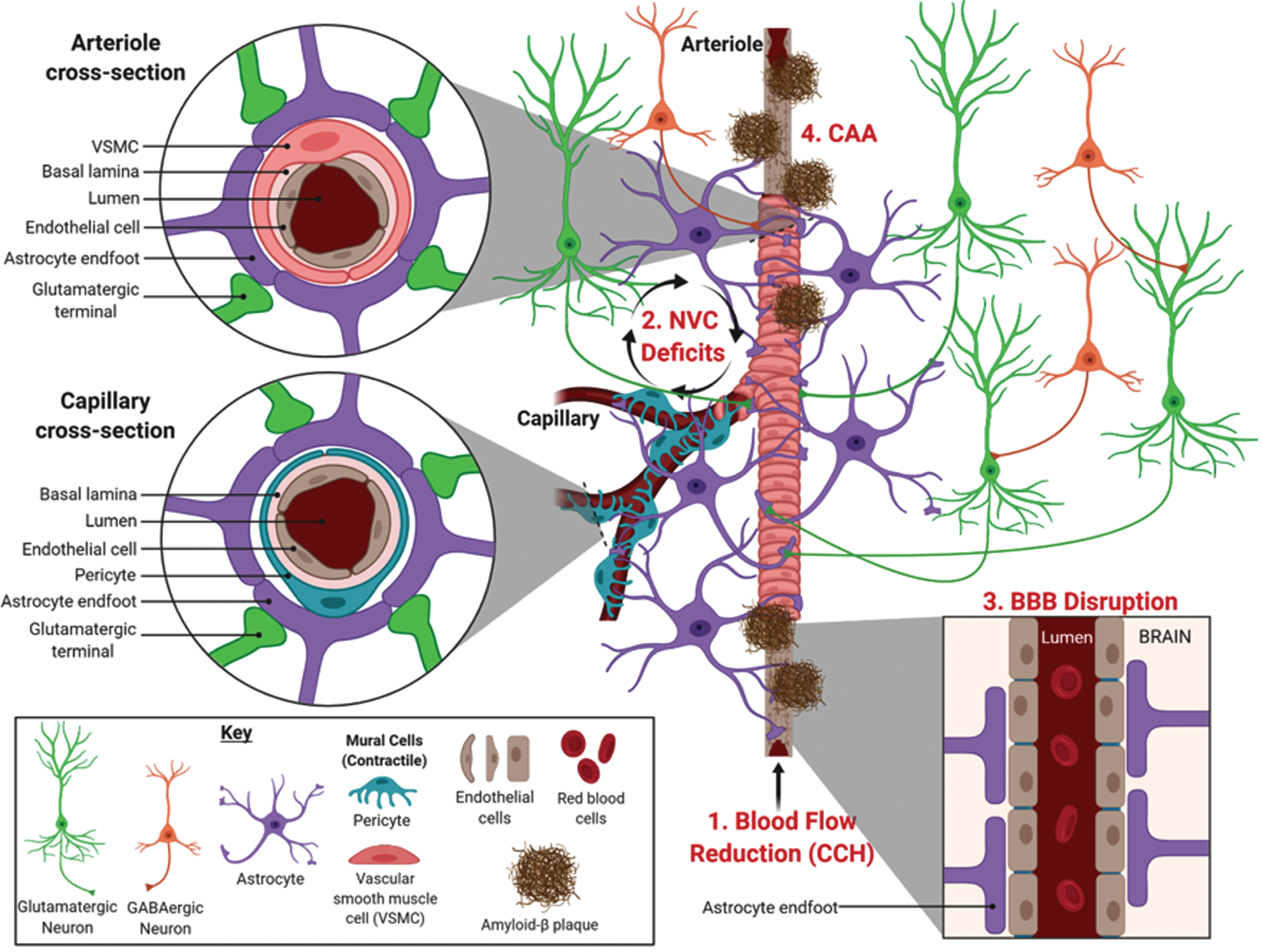
Neurovascular deficits linked to Alzheimer’s disease. The illustration shows the various cell types involved in regulating localized cerebral blood flow along an arteriole bifurcating into capillaries. Neurovascular deficits related to AD include (1) chronic cerebral hypoperfusion (CCH), a reduction in blood flow into the brain that occurs prior to symptomology, (2) neurovascular coupling (NVC) deficits, and (3) blood-brain barrier (BBB) disruption that can be related to (4) cerebral amyloid angiopathy (CAA), characterized by the deposition of Aβ in blood vessels. Image created with BioRender.com.
Historically, it was postulated that the increase in CBF in response to increased neuronal activity was associated with energetic costs and driven by metabolic byproducts, such as carbon dioxide [39], which was termed the ‘negative feedback’ hypothesis. This has been superseded by the ‘feedforward’ CBF hypothesis that involves vasoactive messengers including nitric oxide, which plays a role in neuronal signaling and regulating CBF responses [37, 40–43]. It is now well established that astrocytes also regulate CBF through release of vasoactive substances, such as arachidonic acid and its metabolites, prostaglandins and epoxyeicosatrienoic acids [44, 45]. Thus, the NVU is sometimes more aptly referred to as either the neurogliovascular or gliovascular unit. Astrocytes have perivascular endfeet that wrap around endothelial cells (Fig. 1, cross section) to regulate CBF entry, as well as establish and maintain the BBB integrity [44, 47]. VSMCs and pericytes both regulate CBF by physically controlling vascular reactivity of arterioles and capillaries on which they respectively ensheathe [48]. In particular, pericytes possess the largest vascular resistance in the brain and are estimated to increase CBF by 84% in response to neuronal activity [49]. This makes pericytes promising targets to treat medical conditions involving neurovascular dysfunction [36, 51].
Recent evidence indicates a causal relationship between cerebrovascular dysfunction and the development of AD [52]. Vascular abnormalities are present in at least 50% of AD cases and become more prevalent with increasing age of diagnosis [53]. Furthermore, vascular risk factors associated with AD, such as hypertension and atherosclerosis [54, 55], cause additional damage leading to progressive cerebral hypoperfusion [56]. Over time, homeostatic and hemodynamic disruption damages the cerebral vasculature that perturbs delivery of macromolecules necessary to maintain neuronal activity. The subsequent oxidative stress damages cellular membranes, leads to neuronal and astroglial cell death, and culminates in the cognitive decline observed in AD [57]. Other factors are known to correlate with cognitive deficits including stroke severity, hypertension, atherosclerosis, and arteriosclerosis [58, 59], while macroscopic cerebral infarcts increases the likelihood of AD diagnosis [60]. As such, evidence support that neurovascular dysregulation plays a role in determining the severity of the clinical symptoms of AD.
A neurovascular hypothesis that incorporates a vascular pathogenic component and excessive Aβ accumulation was initially proposed and developed into the two-hit hypothesis of AD [61]. This model hypothesizes that vascular damage, such as BBB disruption that concurrently impairs the machinery for Aβ clearance (hit 1) is followed by the accumulation of Aβ in the brain to vasculotoxic and neurotoxic levels (hit 2) [62, 63]. Supporting this hypothesis are early neuropathology studies that found AD patients had Aβ peptides in [64, 65] and along the walls of cerebral blood vessels [66, 67].
While it is difficult to address the most important question “What causes Alzheimer’s disease?”, and which hypothesis more precisely reflects its pathogenesis, traditional models have focused on the classic AD hallmarks of Aβ accumulation, tau hyperphosphorylation, and neuronal loss [68]. However, retrospective neuroimaging analysis of healthy controls, mild cognitive impairment (MCI), and AD patients indicated that vascular abnormalities occur first, followed by changes in Aβ deposition, metabolic dysregulation, functional impairment, and cerebral atrophy [69]. In addition, studies using arterial spin labeling magnetic resonance imaging (MRI) have detected changes in CBF years before AD symptoms appear [70]. Hence, a growing body of literature support neurovascular dysregulation is a critical factor underlying AD pathogenesis.
Although numerous vascular risk factors and conditions are associated with AD [71], in this review we focus on neurovascular processes observed in AD patients, including (1) chronic CBF reduction, (2) NVC dysfunction, and (3) BBB disruption related to cerebral amyloid angiopathy (CAA). We also discuss findings from recent studies that examine these neurovascular irregularities in AD amyloidogenic animal models. With the advent of transgenic mouse models that develop classical AD pathology, we are able to study how the progression of AD relates to vascular aberrations. While no model adequately recapitulates either familial or sporadic AD, many transgenic amyloidogenic AD mouse models have been developed by incorporating mutations from familial AD. All of the transgenic AD mouse models described in this review display parenchymal Aβ accumulation and some also exhibit Aβ accumulation in blood vessels. These models also vary according to the onset of Aβ accumulation, the rate and spatial distribution of Aβ accumulation, and the extent of cognitive decline. A descriptive table of the models mentioned in this review is presented in Table 1.
Transgenic amyloidogenic AD mouse models and an AD rat model have been developed incorporating mutations from familial AD that display progressive accumulation of Aβ in the brain. All of the transgenic AD mouse models described in this review display parenchymal Aβ accumulation and some exhibit Aβ accumulation in blood vessels
*Transgenic rat model.
CHRONIC CEREBRAL BLOOD FLOW REDUCTION IN ALZHEIMER’S DISEASE
Early ultrastructural studies showed that AD brains with amyloid deposits had abnormally shaped blood vessels [72, 73]. These finding led to the hypothesis that amyloid fibrils caused blood vessel wall deformations and lumen stenosis resulting in reduced blood flow and restricted nutrient entry into the brain [74]. Subsequent studies supported this hypothesis by showing that people with MCI and AD exhibit hypoperfusion in many brain areas, especially those involved in learning and memory [75–79]. The gradual reduction in CBF observed during AD progression is referred to as chronic cerebral hypoperfusion (CCH). It is unclear whether hypoperfusion causes AD or if CBF deficits develop in response to AD neuropathological changes. However, evidence support that hypoperfusion is present at preclinical AD stages [70, 80]. In agreement, patients with chronic vertebra-basilar stenosis, resulting in CCH to the posterior brain regions, display severe cognitive impairment attributed to perfusion deficits [81]. While this study suggests that CCH in itself is detrimental to cognition, the relationship between CBF and cerebrovascular Aβ has been examined using florbetapir positron emission tomography (PET) imaging. The presence of Aβ in cognitively normal, older adults predicted an association between hippocampal hyperperfusion and diminished memory performance, whereas no association was observed in older adults negative for Aβ [82]. However, others have reported that increased Aβ was associated with hypoperfusion that diminished in respect to the stage of cognitive impairment [83]. Although these studies show discordant results regarding the relationship between Aβ and CBF, they suggest that Aβ presence does affect CBF in a spatially dependent manner.
Apolipoprotein E (ApoE) is a glycoprotein responsible for triglyceride and cholesterol transport, but the ɛ4 allele is less efficient at this transport and is the second biggest genetic risk factor for AD. Hyperperfusion has been observed in several brain areas, including the medial temporal lobe of cognitively normal or MCI patients that are APOE ɛ4 carriers [84, 85]. This phenomenon has been attributed to a compensatory response in which increased neuronal activation required by APOE ɛ4 carriers during memory formation requires hyperperfusion [86]. However, as the disease progresses in these APOE ɛ4 carriers, reduced CBF is observed [85, 87], which could be the result of vascular damage related to neuropathological effects. Therefore, studies that reported hyperperfusion in the medial temporal lobe in early MCI or in non-amnestic MCI, might have seen skewed results by not controlling for APOE ɛ4 genotype status [88, 89]. The overall trend for the majority of sporadic AD patients is reduced CBF prior to MCI diagnosis that continues to decline with disease progression and is affected by Aβ accumulation.
ANIMAL STUDIES OF CHRONIC CEREBRAL BLOOD FLOW REDUCTION (OR CCH) IN ALZHEIMER’S DISEASE
Although some consider CCH to be ischemia, it is important to note that CCH implies a moderate reduction in CBF over a prolonged period of time. In contrast, ischemic insults completely eliminate CBF and delivery of metabolic substances, such as glucose and oxygen, to specific brain regions resulting in rapid damage to those areas lacking blood supply. Since some animal models that induce CCH result in different levels of brain hypoxia, they are considered to be viable models of ischemia.
The established animal CCH model was first developed in rats by performing bilateral common carotid artery occlusion (BCCAO), also called 2-vessel occlusion (2VO). This was performed by ligating the common carotid arteries (CCAs) while the vertebral arteries remained open resulting in a partial CBF reduction [90, 91]. The BCCAO CCH model reduced cortical and hippocampal CBF leading to impaired spatial learning, deficient memory performance, and damage to neurons in the hippocampal CA1 [92–95]. In addition, the CBF reduction induced by BCCAO is thought to disrupt the NVU leading to increased hippocampal astrocyte reactivity [96, 97]. Since inducing CCH in rats with the BCCAO technique can elicit cognitive deficiencies that recapitulate cognitive symptoms and pathology seen in AD [98], additional studies have examined the relationship between CCH and Aβ in the brain. CCH induced by BCCAO in rats resulted in increased Aβ accumulation in the hippocampus [99], which has been attributed to increased AβPP expression and enhanced β- and γ-secretase activity [100–102]. A recent imaging study in rats confirmed that BCCAO induced higher frontal cortex and hippocampal Aβ levels and a concurrent reduction in glucose metabolism in the hippocampus, entorhinal cortex, and amygdala [103]. Injecting Aβ into the brain of young rats that underwent CCH conferred synergistic spatial memory impairments and increased cortical and striatal AβPP levels compared to CCH or Aβ alone [104, 105], indicating that Aβ can influence CCH-induced changes.
While BCCAO has been employed in numerous studies in rats, ligating arteries is thought to produce a more profound and rapid reduction in CBF than the gradual CCH observed in AD. Moreover, BCCAO is not amenable to study CCH in C57Bl/6 mice because chronically occluding both CCAs results in a high mortality rate [106]. Chronic unilateral right CCA occlusion (rCCAO) was developed to study CCH in mice. This model decreases CBF to ipsilateral brain regions, induces cognitive deficits, and results in ipsilateral white matter lesion (WML) development [107]. In two AD mouse models, rCCAO exacerbates learning and memory deficits compared to genetically- and age-matched controls without rCCAO. In the PS1V97L mouse model, rCCAO exacerbated Aβ expression that was attributed to changes in expression of Aβ degrading enzymes and clearance proteins [108]. In the Tg2576 AD mouse model, learning and memory deficits caused by rCCAO-induced CCH were attributed to hypometabolism [109]. Although these studies suggest that CCH induced by BCCAO in rats or by rCCAO in mice can induce or exacerbate AD-like pathology, the CBF pattern does not fully represent the progressive CCH seen in AD.
Bilateral carotid artery stenosis (BCAS) is an alternative CCH model for mice in which microcoils of different sizes narrow bilateral CCAs, thereby decreasing CBF based on CCA constriction [110]. Effects of BCAS-induced CCH indicate global CBF reduction lasts for several weeks to months [111, 112]. Mice that underwent BCAS have memory impairment, WML formation, and astroglia and microglia activation [110, 113]. CCH induced in mice have decreased hippocampal metabolism and atrophy 6 to 8 months post BCAS, respectively [114], and increased endogenous Aβ accumulation throughout the hippocampus and cortex [115]. Studies using BCAS-induced CCH in amyloidogenic AD mouse models showed a synergistic effect of learning and memory impairment and Aβ accumulation in APPSwInd mice [116, 117], increased parenchymal and cerebrovascular Aβ accumulation in Tg-SwDI mice [118], and increased Aβ aggregation in APP/PS1 mice without increasing total Aβ levels [119]. Like the BCCAO CCH model in rats, the BCAS CCH model in mice is thought to model vascular cognitive impairment because it induces formation of WMLs leading to behavioral/cognitive deficits [120]. Although the BCAS model is popular to study vascular dementia, it provides a useful model to further study CCH in mouse models of AD.
Another approach to recapitulate CCH seen in AD is the 2-vessel/bilateral gradual CCA occlusion/stenosis (2VGO or BCCS or GCAS) model. Instead of ligating CCAs, ameroid constrictors are used to partially constrict the CCAs bilaterally that narrow over time leading to progressive cerebral hypoperfusion. In rats, compared to the BCCAO model, using 2VGO improved survival while eliciting an attenuated CBF decrease and neuroinflammatory response. However, both CCH models showed comparable impaired working memory [121]. In mice, 2VGO elicits a distinct progressive CBF reduction compared to the rapid and profound decrease observed after BCAS [122, 123]. But, 2VGO does not consistently induce hippocampal neuronal loss. Although 2VGO in these mice impaired working memory, they were comparable to sham controls in regard to hippocampal-dependent reference learning [122]. The 2VGO model has been used extensively in the Koji Abe laboratory to examine the effect of CCH in APP23 mice. Overall, compared to age- and genotype-matched controls, CCH in APP23 mice enhances motor and cognitive deficits while increasing hippocampal cell death. [124–127]. CCH in APP23 mice also decreases LRP1 and increases RAGE expression in vascular endothelial cells, which would perturb Aβ clearance resulting in parenchymal and cerebrovascular Aβ deposition [125]. CCH also induced NVU dissociation as measured by decreased astrocyte to blood vessel immunofluorescent staining overlap that could be a consequence of Aβ accumulation [124].
Since 2VGO results in increased mortality after 28 days when ameroid constrictors close, another CCH model developed in mice is the asymmetrical gradual carotid artery occlusion. This method consists of one CCA progressively occluded with an ameroid constrictor while a microcoil is placed on the other CCA resulting in decreased CBF and hippocampal neuronal loss [128, 129]. However, despite the promising translational implications of this model, it has not been tested in AD mouse models yet.
The majority of studies using AD mouse models are in agreement that CCH exacerbates Aβ accumulation and increases neuronal death. When interpreting results from these studies, it is important to discern the differences in CCH and AD models employed. As mentioned, BCAS creates a prolonged CCH followed by CBF recovery making this suitable to model vascular dementia [114]. Still, since BCAS is a long-lasting CCH model, it might be the best model currently available to study CCH in mice. More studies using the BCAS CCH in AD mouse models are warranted to further understand the impact of CCH on Aβ accumulation and to identify potential therapeutic interventions to counteract the negative CCH effects. As stated, a recurring theme is that mice tend to be more vulnerable to CCH than rats. It is plausible that 2VGO in TgF344-AD rats might be a viable model to study the relationship between CCH and Aβ accumulation.
BLOOD PRESSURE AND ALZHEIMER’S DISEASE
Cerebral autoregulation refers to myogenic, autonomic, and metabolic mechanisms that maintain adequate blood perfusion to the brain despite changes in blood pressure [130, 131]. Although a single study of sporadic AD patients showed impaired cerebrovascular autoregulation [132], most studies have shown that cerebrovascular autoregulation is unaffected in AD patients [133–135]. However, it is established that having either low blood pressure (hypotension) or high blood pressure (hypertension) increases the likelihood of developing dementia and AD [136–138]. In particular, elevated systolic blood pressure (BP) is a major vascular risk factor for developing AD and is also associated with cerebrovascular disease, including stroke and cerebral infarcts [54, 140]. Accordingly, hypertension-induced vascular changes, such as small vascular lesions and BBB damage [141], possibly contribute to the development of CCH and cognitive deficits [142]. A neuroimaging study showed AD patients with hypertension had worse cognitive function and reduced hippocampal metabolism compared to normotensive AD patients. Interestingly, no differences in Aβ were observed between these AD patients [143]. Another study showed hypertension in AD patients accelerated the rate of cognitive decline only in AD patients under the age of 65 [144]. A study in older patients (aged 71–85) with moderate AD found decreases in BP as the disease and cognition worsen [145], whereas others showed a decrease in BP years before cognitive decline [146]. Although a comprehensive report concluded that reducing BP in hypertensive people does not help prevent cognitive decline or the development of dementia, methodological variation of these studies resulted in self-reported problems with the analyses performed [147].
The discordant results of these studies may be attributed to the differential effect that CCH or BBB deterioration would have on BP in subjects, thereby skewing the selection criteria. For example, someone who was hypertensive prior to AD diagnosis could have developed a cardiovascular abnormality, such as CCH, which would result in lower BP readings and a subsequent “normotensive” categorization during the study design. Therefore, prospective, longitudinal studies are warranted to examine the effect of lowering BP on the development of dementia or AD that carefully factor in differences in BP and presence/absence of vascular irregularities, such as CCH. One study following MCI patients for 6 years found that higher plasma levels of atrial natriuretic peptide, involved in diuresis and lowering BP, were associated with conversion from MCI to dementia or probable AD. However, MCI patients with increased atrial natriuretic peptide levels that received antihypertensive treatment had a lower likelihood of converting to probable AD [148]. It is thought that elevated atrial natriuretic peptide is indicative of disturbed vascular function, which could be used as an early biomarker to determine the optimal therapeutic treatment window in MCI patients at risk for converting to AD. Overall, we know that hypertension is implicated with the development of dementia and AD, but the effect of using antihypertensive medications to halt the progression from early cognitive symptoms into AD is still inconclusive. In addition to continuing studies to test the effectiveness of antihypertensive treatments, we must identify and study alternative targets related to hypertension-induced effects, such as damaged blood vessels from microinfarcts.
ANIMAL STUDIES OF HYPERTENSION AND ALZHEIMER’S DISEASE
While many animal studies described above pertain to CCH, very few animal studies have explored the effects on Aβ and hypertension. It has been shown that intra-arterial infusion of Aβ40 into the right common carotid artery increased mean arterial blood pressure (MAP) in hypotensive rats (MAP < 100 mm Hg), but had no effect in normotensive (MAP = 100–129 mm Hg) or hypertensive rats (MAP > 139 mm Hg) [149–151]. The Arendash et al. study also showed that the Aβ42 isoform did not increase BP in hypotensive rats. However, after decreasing BP with the vasodilator sodium nitroprusside, both Aβ40 and Aβ42 infusion induced a return to baseline BP, supporting that Aβ can contribute to hypertension by constricting blood vessels. These studies support that increased soluble Aβ can exacerbate cerebrovascular dysfunction by inducing cerebral and peripheral vasoconstriction contributing to hypertension during the early disease stages. However, as amyloid aggregates in the brain, cerebrospinal fluid levels drop potentially resulting in a shift from a hyper to hypotensive state that reduces amyloid clearance through the BBB. As such, a feedforward mechanism results contributing to reduced CBF, further Aβ accumulation, and the eventual cognitive and functional decline contributing to MCI and AD progression.
Inducing hypertension in wildtype mice increased Aβ levels in the cortex and hippocampus as well as surrounding blood vessels that led to BBB deterioration [152, 153]. An increased RAGE expression in capillaries was also observed, indicating hypertension might result in upregulated RAGE-mediated Aβ influx into the brain [153]. Inducing hypertension in APP/PS1 and TgSwDI mice led to increased brain Aβ accumulation, damage to blood vessels, and accelerated cognitive deficits compared to genotype-matched normotensive controls [154, 155]. Compared to wildtype mice, APP/PS1 mice were more responsive to hypertensive treatment, but less responsive to antihypertensive treatment. Reduced hippocampal CBF was also observed in these mice, signifying that hypertension coupled with hippocampal Aβ accumulation combines to disrupt cerebral hemodynamics [156]. In addition, inducing hypertension in APP/PS1 mice exacerbates cognitive performance compared to normotensive APP/PS1 mice, which was attributed to decreased functional connectivity in the brain measured by resting state blood oxygen-level dependent (BOLD) fMRI [157]. This study employed useful methodology in rodents that yielded deficits that are comparable to those found in people with cognitive decline in resting state fMRI studies. When examining age on vascular parameters, 16–18-month-old APP/PS1 mice had higher systolic BP, decreased cortical and thalamic CBF, and altered hippocampal vasoreactivity that contributed to their behavioral and cognitive impairments compared to age-matched control mice [158]. These studies support that hypertension has deleterious effects on AD-like neuropathology, and the potential benefits of treating hypertension to quell AD progression should be explored further.
NEUROVASCULAR COUPLING DYSFUNCTION IN ALZHEIMER’S DISEASE
Changes in CBF in humans are typically measured with imaging techniques, including BOLD fMRI and FDG-PET imaging. BOLD fMRI relies on the observation that CBF increases to areas with neuronal activation. In FDG-PET imaging, radiolabeled fluorodeoxyglucose accumulation in tissue reflects metabolic changes in response to neuronal activity [159, 160]. Recently, a noninvasive MRI alternative using arterial spin labeling perfusion to monitor CBF has been developed that matches metabolic patterns observed with FDG-PET and reflects CBF more precisely than BOLD fMRI [161]. These neuroimaging techniques that measure CBF changes are considered to reflect NVC. By measuring brain activity with BOLD fMRI, patients with MCI, mild AD, or AD exhibit disruptions in functional connectivity in a resting state default mode network (DMN). The DMN is comprised of widespread cortical regions, including the prefrontal, parietal, and temporal cortices and the hippocampal formation [162–164] that are commonly affected in AD [165]. A study combining arterial spin labeling perfusion and resting-state fMRI in AD patients confirmed functional connectivity abnormalities in DMN regions, and also showed regional CBF disruptions in brain areas affected in AD [166]. By measuring Aβ levels with PET using Pittsburgh Compound-B and resting-state DMN with BOLD fMRI it was shown that disrupted functional connectivity in DMN regions is associated with increased Aβ deposition in AD patients even though deposition was also evident in cognitively normal subjects [167]. To measure changes in neuronal activity, electroencephalogram recordings have shown weaker alpha and beta rhythms, but enhanced delta and theta rhythms in the DMN of AD patients [168, 169]. Reduced associations between BOLD signaling and alpha band power was also observed in the DMN of probable AD patients [170]. While deficits in DMN connectivity have been linked to AD, these studies assess brain activity in a specific network during rest, which reflect global NVC anomalies. Incorporation of memory tasks or other sensory stimuli during BOLD fMRI measurements have found region-specific NVC deficiencies in AD patients [171–174]. For example, memory encoding tasks show decreased activation of the medial temporal lobe [172], while exposure to visual stimuli indicates reduced CBF in numerous brain regions of AD patients [175] compared to healthy elderly controls. Recent studies have verified these results in mild AD patients by showing normal CBF in the visual cortex at rest, but upon presentation of visual stimuli, hypoperfusion becomes evident [176]. Retinal NVC measurements are a newer, less invasive method for determining pathophysiological brain processes while providing information regarding the NVU integrity [177]. Retinal vessel reaction to a non-invasive flicker stimulation found that AD patients displayed more pronounced and delayed reactive dilation compared to MCI and healthy controls, which was attributed to a compromised NVU [178].
Many of these imaging studies have examined MCI or early-stage AD to understand how NVU disruption changes during the course of AD progression. With this information, a predictive biomarker can be developed for earlier probable dementia diagnosis allowing for improved therapies and patient outcome. Overall, these studies support a reduction in NVC of AD patients, which may hinder amyloid clearance leading to its accumulation and deposition particularly within the DMN. Additional prospective, longitudinal studies are needed to gain a better understanding of the role of NVC in AD progression.
ANIMAL STUDIES OF NEUROVASCULAR COUPLING DYSFUNCTION IN ALZHEIMER’S DISEASE
Measuring NVC functionally requires simultaneous measurements of neuronal/astrocytic activity and vascular changes. Since this is technically challenging, there is a scarcity of research examining NVC in rodent models of AD, but most studies indicate NVU dysfunction. The TgF344-AD rat model has diminished cerebrovascular reactivity that correlated with increasing Aβ load in vessel walls as measured by changes in dilatory capacity induced by hypercapnia [179]. At 9 months TgF344-AD rats also have neuronal network dysfunction between the hippocampus and medial prefrontal cortex that is observed after onset of Aβ plaque deposition but prior to cognitive deficits [180]. Two-photon imaging of 3xTg-AD, TgSwDI, and Tg2576 mice that overexpress Aβ in cerebral blood vessels showed abnormalities in both astrocytic and vascular activity in response to whisker stimulation [181]. Moreover, 3xTg-AD mice display impaired neurovascular coupling induced by neuronal-derived nitric oxide signaling [182]. In agreement, hAPPJ20 mice showed NVU disruption characterized by astrocytic endfoot separation from blood vessels as a result of Aβ deposition [183]. Others have shown neutrophils cause CBF deficits in APP/PS1 and 5xFAD mice [184].
A recent study in human tissue and in rat brain slices showed that Aβ oligomers act specifically on pericytes to constrict capillaries [185]. Thus, pericytes have become a potential target to alleviate conditions characterized by blood flow deficiencies [186]. Pericyte deficient mice are a useful model for studying their contribution to NVC. These mice develop neurovascular uncoupling, diminished cerebral oxygen supply, and metabolic stress [187, 188]. Crosses of pericyte-deficient mice with amyloid mouse models of AD may elucidate the role of specific BBB cell types in AD progression.
BLOOD-BRAIN BARRIER, CEREBRAL AMYLOID ANGIOPATHY, AND ALZHEIMER’S DISEASE
The brain microvasculature supports the exchange of critical metabolic substrates between the brain and circulating blood [189]. Importantly, the BBB protects the brain from entry of toxic compounds and eliminates waste products, including Aβ, via LRP1 and RAGE [10, 190]. It is thought that impaired Aβ clearance across the BBB is involved in the pathogenesis of AD [191–193], and deficient Aβ clearance, rather than Aβ overproduction, results in aggregation and plaque pathology [9, 69]. AD brains consistently display CAA, characterized by Aβ deposition along the walls of cerebral blood vessels and leptomeningeal blood vessels that are located between the subarachnoid and pia mater [67]. Although CAA is considered a clinically distinct phenomenon than AD, it shares common cerebrovascular and neurodegenerative properties suggesting a mechanistic link [194]. Originally, sporadic CAA was characterized according to the absence or presence of Aβ deposition in capillaries, the latter referred to as capillary cerebral amyloid angiopathy [195]. More recently, this has been reclassified to detail the cortical penetrance of accumulation along capillaries [196], which has been reviewed elsewhere [197]. Examination of autopsy-confirmed AD cases reported an overlap with CAA in which 82.9% of AD patients exhibited at least mild CAA in parenchymal or leptomeningeal vessels and 25.6% with moderate to severe CAA throughout various brain regions [66]. CAA is prevalent in postmortem AD brains and correlates with both Aβ deposition and dementia severity as assessed by the clinical dementia rating scale [198]. In agreement, CAA severity is associated with lower cognition proximal to death of the patient [199].
A recent study showed that microvessels from the parietal cortex of AD patients or those with advanced CAA contained higher concentrations of vascular Aβ40 and Aβ42 [200]. Vascular levels of P-glycoprotein and neprilysin, which are involved in Aβ degradation, were lower in persons with AD, positively correlated with cognitive function, and inversely correlated with vascular Aβ40 levels. In contrast, BACE1, a protein required to produce Aβ, was increased in AD, negatively correlated with cognitive function, and positively correlated with Aβ40 in microvessel extracts. These studies support the idea that CAA in AD reflects failed cerebral Aβ clearance along perivascular lymphatic drainage pathways and could be a factor in the etiology of AD [201].
It is important to know that CAA is the most common form of cerebral small vessel disease, which encompasses diseases affecting small arteries, arterioles, venules, and capillaries. Initial studies indicated no differences in BBB permeability between early AD and non-demented individuals [202]. However, advances in neuroimaging have allowed the assessment of BBB deterioration in patients with cerebral small vessel disease [203], but only a few studies have examined BBB integrity in AD patients. These studies indicate increased BBB permeability in early AD patients and a correlation between reduced CBF and increased BBB leakage rate [204, 205].
ANIMAL STUDIES OF CAA AND BBB DYSFUNCTION IN ALZHEIMER’S DISEASE
Several different animal models exhibit vascular Aβ deposition similar to observations in human CAA. Compared to control mice, 6–12-month-old TgCRND8 have cortical arterioles with increased tortuosity as well as decreased size and CBF [206]. These structural and functional deficiencies observed in TgCRND8 mice were rescued by inhibiting Aβ oligomerization. Similar to TgCRND8 mice, Tg-SwDI mice develop cerebral microvascular amyloid pathology with reduced CBF, vasoreactivity, and cognitive deficits that can all be prevented by pharmacologically blocking Aβ oligomerization [207]. Transplanting epithelial progenitor cells into the hippocampus of APP/PS1 mice repaired BBB damage and led to improved cognitive performance [208]. Removing Aβ from leptomeningeal and cerebral vessels with immunotherapy in transgenic CAA mouse models improved responsivity to vasodilators [209]. These studies strongly suggest that Aβ accumulation in cerebral blood vessels plays a critical role in cognitive decline, which highlights the importance of additional research further investigating CAA models. For example, the stroke-prone spontaneously hypertensive rats were crossed with TgF344-AD rats to develop a novel mixed vascular dementia and AD model. These rats have robust Aβ accumulation, gliosis, and behavioral alterations [210], which would be useful to study the relationship between Aβ accumulation and damage to cerebral blood vessels.
REVISITING THE VASCULAR HYPOTHESIS FOR SPORADIC ALZHEIMER’S DISEASE
Vascular risk factors for AD are known to alter CBF although the mechanisms associated with these changes have not been fully elucidated. As the vascular and two-hit hypothesis for AD stipulate, aging, genetic, and environmental factors compromise the cerebral vasculature and BBB integrity [62, 71]. Microinfarcts or microhemorrhages may be early contributors to BBB deterioration leading to CBF deficits (Fig. 2). Subsequently, BBB deterioration, NVC dysfunction, and reduced CBF impede physiological clearance of Aβ causing its accumulation and aggregation in the CNS and cerebral vasculature. A second alternative is that pathological Aβ accumulation occurs independently of neurovascular dysfunction. Since Aβ constricts the cerebral vasculature, its accumulation results in further CBF reductions [149, 211]. The scarcity of metabolic substrates needed by astrocytes and neurons causes oxidative stress and cell death that starts in regions requiring higher energy demands, such as the cortex and hippocampus. Over time, this extends to other cortical areas causing widespread neurodegeneration. The alarming part about the neurovascular complications associated with AD development, is that they affect similar cell populations within the NVU creating a vicious cycle of BBB deterioration. This could explain the rapid progression from MCI or early AD to late stage AD. Perhaps, the association between small vascular lesions and neurodegenerative diseases has been lacking due to visualization methods. Initially, these vascular lesions were only observed during postmortem analysis; however, recent advances in human neuroimaging methods are now able to detect cerebral microinfarcts [212]. By employing advanced imaging methods, we can identify ischemic small vessel disease on a macro and microstructural scale [213], allowing us to design prospective, longitudinal studies to better understand the role of small vascular lesions in AD pathogenesis and other neurodegenerative disorders.

Vascular Theory of Alzheimer’s disease. Hypertension and other vascular risk factors contribute to the likelihood of cerebral blood vessels damage and microinfarcts. This causes blood-brain barrier (BBB) deterioration and diminishes blood supply to the affected cerebral regions. As the BBB breaks down, cells involved in neurovascular coupling are affected and consequently, are not able to properly regulate and supply local cerebral blood flow (CBF). Concurrently, the abnormal accumulation of amyloid-β (Aβ) in the brain parenchyma and in cerebral blood vessels contributes to the local CBF deficits. Whereas soluble Aβ can decrease CBF by directly constricting blood vessels, Aβ deposition is known to contribute to CBF reduction or chronic cerebral hypoperfusion. Over time, essential macromolecules to support neuronal network activity becomes limited leading to oxidative, neuronal and glial cell damage, and eventual neurodegeneration. Image created with BioRender.com.
SUMMARY: VASCULAR DYSREGULATION IN ALZHEIMER’S DISEASE
For nearly 30 years the majority of AD research has been conducted under the assumption that the cause of the disease is dependent upon accumulation of Aβ plaques and hyperphosphorylated tau tangles, which leads to a series of cellular events ultimately causing neurodegeneration and cognitive decline [214, 215]. Although the familial form of AD is caused by mutations in genes resulting in the abnormal accumulation and perturbed clearance of Aβ and hyperphosphorylated tau, the cause of sporadic AD that represents over 95% of all cases is still unclear [216–218]. While evidence supports that Aβ accumulation contributes to or exacerbates sporadic AD, many studies indicate neurovascular dysfunction occurs prior to Aβ accumulation and contributes to the development and/or progression of AD. Regardless of which hypothesis stands the test of time, a better understanding of the relationship between neurovascular dysfunction and AD is warranted. In addition to focusing on abnormal Aβ accumulation as the key event in the pathogenesis for AD, we need to identify alternative mechanisms involved in AD pathogenesis. However, since many rodent models of AD have been developed that overexpress Aβ and tau pathology, much research has been devoted to understanding mechanisms related to these proteinopathies. Fortunately, over the past decade, there has been an increased interest in understanding how neurovascular dysfunction relates to AD and the relationship between Aβ accumulation and the NVU. The focus of this review was to describe neurovascular dysfunction linked to AD as a contributor to AD pathogenesis. The hope is that a deeper understanding of how the cellular components of the NVU are affected in early AD will lead to the identification of novel targets to prevent or repair neurovascular dysfunction thereby slowing or stopping AD progression. Understanding CBF dysregulation during the development and progression of AD could help identify novel targets for disease modifying therapies. The interrelatedness of CBF dysfunctions indicates the complexity of fully understanding individual contributions to dementia. But, it also suggests that multiple factors (including age, sex, environment, and genetic predilection) may initiate the neurodegeneration and cognitive decline observed in AD. As such, individualized treatment therapies meant to repair the NVU while clearing accumulated AD proteinopathies may prove useful in a subset of patients.
Footnotes
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health [NIA R01AG057767 and NIA R01AG061937], from the SIU Foundation at the School of Medicine [Harriss and Fannie Belle Roe Malan Research Endowment and the Illinois Health Improvement Association Research Endowment], the Center for Alzheimer’s Disease and Related Disorders, and the Kenneth Stark Endowment.
We are grateful to the SIU SOM Medical Library staff, including Sheri Daniels, Ann Gonterman, Lydia Howes, Jaedyn Maltimore, and Mackenzie Sanner for their help with the literature search and locating articles.