
Abstract
Numerous experimental and postmortem studies have increasingly reported dystrophic axons and dendrites, and alterations of dendritic spine morphology and density in the hippocampus as prominent changes in the early stages of Alzheimer’s disease (AD). Furthermore, these alterations tend to correlate well with the progressive cognitive decline observed in AD. For these reasons, and because these neurite structures have a capacity to re-grow, re-establish lost connections, and are critical for learning and memory, there is compelling evidence to suggest that therapeutic interventions aimed at preventing their degradation or promoting their regrowth may hold tremendous promise in preventing the progression of AD. In this regard, collapsin response mediator proteins (CRMPs), a family of phosphoproteins playing a major role in axon guidance and dendritic growth, are especially interesting. The roles these proteins play in neurons and immune cells are reviewed here.
INTRODUCTION
The greatest risk factor for developing Alzheimer’s disease (AD) and related dementias (ADRD) is advanced age, and the aging (>65 years) population is currently the largest in history [1, 2]. Thus, ADRD represents one of the greatest public health crises worldwide [1, 3]. Indeed, an estimated 44 million people are currently affected globally, and many more are estimated to develop ADRD in the years to come [1, 3]. AD is characterized by a progressive cognitive impairment beginning as a mild memory loss that gradually, and sometimes precipitously, culminates into deep dementia [4–7]. Indeed, the preclinical and prodromal phases of the disease are thought to last approximately 20 years followed by a clinical duration of approximately 8–10 years [5]. Neuropathologically, the most prominently affected brain structure in AD is the hippocampus and its surrounding entorhinal cortex [8]. Given its many afferent, efferent, and intrahippocampal connections along the hippocampal septotemporal axis facilitating its physiological activity [9–11], the hippocampal complex is widely considered as the center of brain networks implicated in novelty detection, regulation of emotion, fear, stress, and learning and memory [12–14]. Thus, it is not surprising that the most common cognitive disruptions in ADRD include short and long-term episodic memory deficits, spatial memory, navigational impairments, difficulty sustaining attention, mood swings, and loss of motivation [15, 16]. Neuroanatomical and behavioral studies have demonstrated that the hippocampus is involved in multiple learning and memory processes [17, 18], and physiological and neurochemical disciplines have determined the essential role of synaptic plasticity and neuronal signaling systems, respectively [19, 20]. In this review we discuss how alterations in neuronal structures associated with functional connectivity are a main cause of hippocampal-dependent cognitive decline and introduce a family of proteins, CRMPs (collapsin response mediator proteins) that may play important roles for early detection and treatment of AD.
NEUROPATHOLOGICAL CONSIDERATIONS
Imaging, genetic, preclinical, and human postmortem studies have served to identify two cardinal pathological markers of advanced AD: amyloid-β42 (Aβ42)-containing senile plaques and hyperphosphorylated tau-containing neurofibrillary tangles, both of which are believed to lead to neuronal dysfunction and death, and in turn dementia. Indeed, individuals with AD have approximately 30–50% fewer neurons in the entorhinal cortex compared to normal aging. Importantly, synaptic dysfunction is an early and prominent pathological feature of AD that precedes frank neuronal cell death and has been reported to correlate better with cognitive impairments than Aβ or tau pathology [21, 22]. Indeed, gene expression profiling, immunocytochemistry, and postmortem studies with associated learning and memory deficits have demonstrated that abnormalities in axonal connectivity, dystrophic neurites, reduced dendritic arbor, and loss of dendritic spines occur well in advance of neuronal death [23, 24]. It is estimated that cortical synaptic density is reduced by 25–30% and synaptic density per neuron is reduced by 15–35% in the earliest stages of AD [25, 26]. Furthermore, expression levels of pre-synaptic, synaptic, and post-synaptic proteins are reduced in postmortem AD brains compared to controls [27, 28].
Axons and dendrites are dynamic structures with critical roles in neural transmission and synaptic plasticity. Proper axonogenesis and dendritogenesis in adulthood is crucial for neuronal network wiring, and essential for normal brain function [29]. Therefore, it is not surprising that abnormalities to these neuronal processes are associated with a large number of neurodegenerative disorders [24, 30–33]. Axon growth and maintenance depends on microtubule-associated proteins, such as tau, that support its cytoskeletal stabilization [34]. It is known that the kinase Cdk5 (activated by p35), present in axon shafts and growth cones, plays a key role in axon growth and maintenance, such that its depletion leads to robust axon guidance defects [35, 36]. Dendritic arborization involves calcium-dependent regulation of the local cytoskeleton via Rho family GTPases, which depending on disease stage and neuronal population, are reduced in various areas of the AD brain including the hippocampus and entorhinal cortex [37]. Dendritic spines are major sites of synaptic contact, receiving and storing input about synaptic strength from axons and transmitting key signals to the cell body. They have a dynamic cytoskeleton enabling them to rapidly change shape in response to stimuli both during development and in the adult brain, and this morphological plasticity is thought to be a structural correlate of synaptic plasticity facilitating long-term memories [38, 39]. Thus, their reduction and/or altered structure found in AD patients and in AD mouse models is clearly associated with alterations in memory formation and storage [39]. Importantly, non-neuronal cells, including glia, have been shown to play a significant role in dendritogenesis [40–42], and in modifying dendritic spine structure and function through various signaling pathways including ephrin-A3 released by astrocytes [43], and CX3CR1 expressed by microglia [44]. This interest in the critical involvement of axonal and dendritic pathology and synapse disassembly has spurred a growing literature in the prevention of synaptic damage and in the capacity of injured neurons to regenerate dendrites and other surrounding structures to ameliorate synaptic function. Among several novel molecules thought to be important in this regard, CRMPs hold significant promise.
CRMPs AS NOVEL TARGETS
CRMPs form a family of five cytosolic phosphoproteins that can assemble in heterotetramers [45]. These proteins regulate axonal growth and guidance through transport of tubulin heterodimers to the distal end of the growing axon and subsequent facilitation of microtubule polymerization [46–53]. CRMP1, CRMP2, CRMP3, and CRMP4 have 75% homology of their amino acid sequence while the phylogenetically divergent CRMP5 shares only 50% homology [54]. CRMPs are highly expressed in the developing brain, and continue to be present in axons and dendrites in several brain regions, into adulthood [54, 55] (Fig. 1). They play important roles in axon growth/guidance and dendrite/spine formation as critical intracellular signaling molecules for semaphorin (Sema3/Plexin/Neuropilin) [56, 57], and various neurotrophic factors, including BDNF and neurotrophin-3 [58].

CRMPs: similarity and expression. Phylogenic conservation (A) and sequence comparison of mouse CRMPs1–5 (B). Summary of relative CRMPs 1–5 expression pattern in mouse CNS (C). ePN, early postnatal; Hippo, hippocampus; DG, dentate gyrus; DRG, dorsal root ganglia; (1): inferior olive complex and reticular formation; (2): pontine nuclei. +++high level; ++moderate level; +weak level; –undetectable signal; ND, not determined.
The role of CRMPs in neurons
CRMP2 is the best-studied among the five CRMP homologs. In neurons, this microtubule-associated protein shares important similarities with tau (reviewed in [59]) including its expression within neuritic tips, in growth cones, and synapses as well as its relevance to AD pathology [60, 61]. Non-phosphorylated CRMP2 binds to tubulin, leading to microtubule formation and stability of actin filaments, thus inducing axon growth [62]. CRMP2 knock-out in the adult hippocampus has been reported to lead to reduced long-term potentiation, abnormal NMDA receptor composition, aberrant dendrite arborization, and defective synapse formation in CA1 neurons, indicating its critical role in neuronal plasticity [63]. Phosphorylation of CRMP2 by Rho kinase (at Threonine 555) or Cdk5 (at Serine 522) and GSK3β (at Threonine 509/Threonine 514/Threonine 518) has, in contrast, been shown to suppress its binding to tubulin consequently impairing tubulin polymerization and neurite outgrowth [46, 64–66] (Fig. 2). Neuronal deletion or inhibition of GSK3β increases microtubule growth speed and enhances axonal and neurite regeneration via CRMP2 [67] even in neurons damaged by Aβ [68]. Thus, the GSK3β-CRMP2 axis appears pivotal for axonal growth and repair.
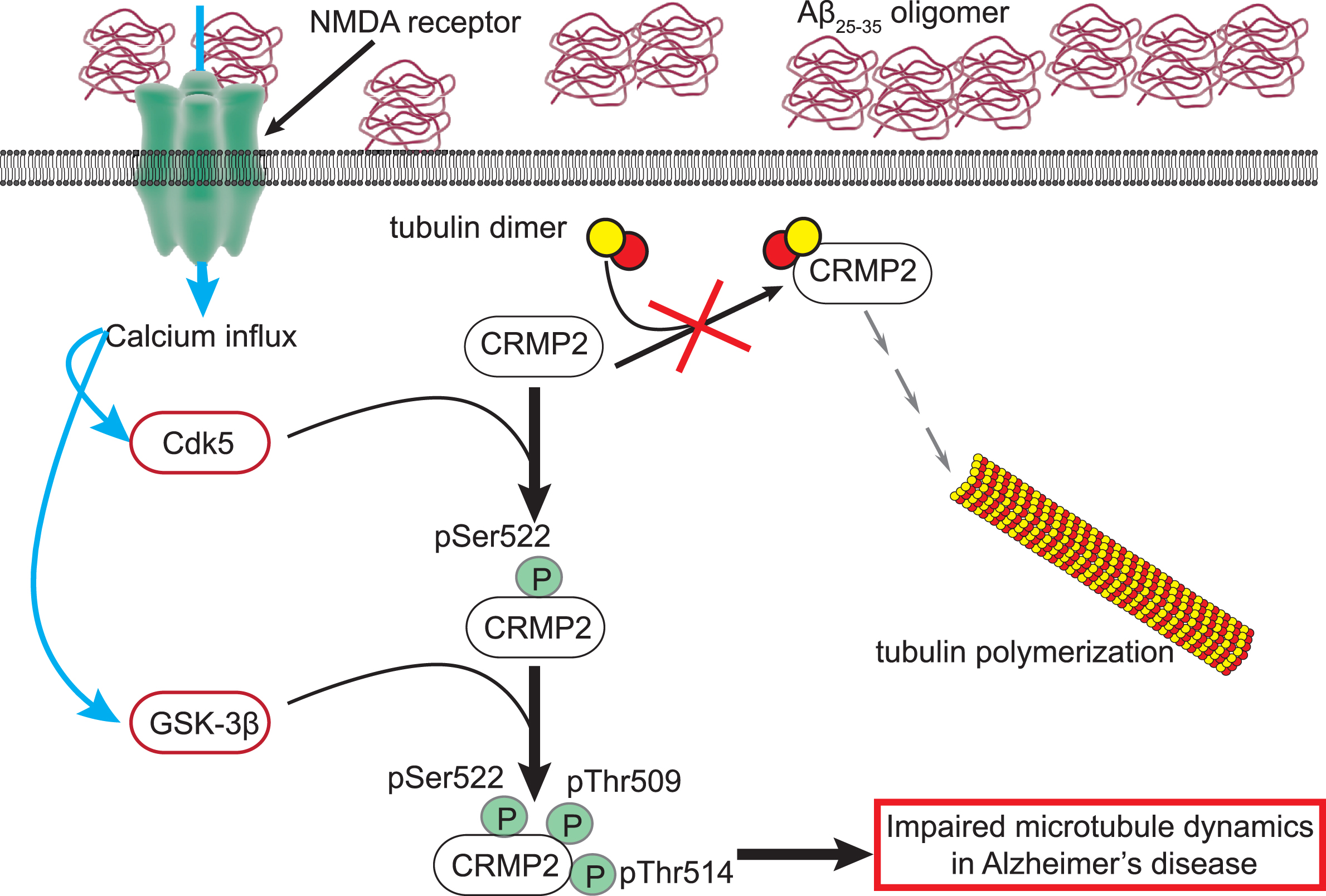
Impairment of CRMP2 function in AD. The physiological function of CRMP2 is to bind to tubulin heterodimers and promote their polymerization to assemble microtubules. In Alzheimer’s disease (AD), extracellular Aβ25–35 oligomers can directly bind and activate the N-methyl-D-aspartate (NMDA) receptor at the surface of neurons. The resulting calcium influx leads to activation of the kinases Cdk5 and GSK3β. Cdk5 will first phosphorylate CRMP2 on the residue serine 522 (pSer522). This renders CRMP2 amenable for phosphorylation by GSK3β on the threonines 509 and 514 (pThr509, pThr514). This hyperphosphorylated CRMP2 loses its ability to bind tubulin and facilitate microtubule assembly. This mechanism may participate in the impaired microtubule dynamics in AD.
In AD, CRMP2 is hyperphosphorylated by Cdk5, Rho kinase, and GSK3β in mouse models as well as in human samples [66, 69–72]. CRMP2 phosphorylation sites by Cdk5 and GSK3β cluster on an “Alzheimer epitope” identified in the late 1990s [64, 73]. Phosphorylation of CRMP2 at these sites can be directly induced by amyloid Aβ25–35 oligomers [70]. Aβ25–35 oligomers can directly trigger NMDA receptor function [74] which in turn facilitates Cdk5 [75, 76] and GSK3β activation [77, 78]. The Aβ25–35 oligomers-dependent increase of CRMP2 phosphorylation could be prevented by suppressing the interaction between CRMP2 and the NMDA receptor [79]. This inactivated the channel by diminishing its localization at the plasma membrane [80–83], thus indicating that increased CRMP2 phosphorylation in AD is through Aβ25–35 activation of NMDA receptors. Deleting the Cdk5 phosphorylation site of CRMP2 prevented impairments in LTP and memory formation induced by Aβ25–35 oligomers [70]. CRMP2 phosphorylation by Cdk5 and GSK3β dissociates its interaction with tubulin oligomers consequently impairing tubulin polymerization and neurite outgrowth [46, 65], two cellular mechanisms relevant in AD [84, 85] (Fig. 3). These observations have defined CRMP2 hyperphosphorylation as an early event [69] that is necessary for AD-related neuronal dysfunctions [66, 70].

Schematic of the role of CRMPs in synaptic structure growth and maintenance. The left panel in the diagram depicts the wide array of physiological functions CRMPs play in the healthy neuron. These functions include tubulin polymerization, microtubule elongation, regulation of growth cones related to axonogenesis and dendritogenesis, dendritic spine formation, and important functions related to vesicle transport and autophagy. The right panel reflects how dysregulation of CRMPs perturbs their physiological functions and contributes to the cellular deterioration present in AD. Figure created using Biorender.
Phosphorylated CRMP2 also interacts with Numb, an adaptor protein which regulates the transport and processing of amyloid-β protein precursor and can increase amyloidogenesis [86]. Indeed, phosphorylated CRMP2 has been detected in postmortem AD- and Lewy body dementia-affected brain tissues [65, 72]. And in AD transgenic mouse models, it is significantly increased in hippocampus well before plaque and tangle formation [69]. Recently, CRMP2 was found to be modified at more than 50 different sites by advanced glycation end products (AGE) [87]. Gradual accumulation of AGE-modified adducts is recognized as a factor promoting protein aggregation and amyloidosis in AD [88, 89]. Consistent with this, glycated CRMP2 was irreversibly locked into a multimeric conformation because of di-Lys bonds within and between tetrameric complexes [87]. Thus, the aggregation of CRMP2 in AD secondary to glycation may be a factor contributing to the formation of amyloid plaques.
In AD, a recent theory proposes that trans-synaptic transfer of pathological Tau and other prion-like proteins participates in the propagation of the disease [90]. Although CRMP2 does not possess a prion-like domain, typified by a region of low complexity or prone to aggregation, there are several lines of evidence that CRMP2 could be a nucleation factor. First, CRMP2 can form higher level aggregates modified by AGE products [87]. Second, CRMP2 can be secreted by neurons and have extracellular action on NMDA receptors [91, 92]. Third, CRMP2 can be found in both neurofibrillary tangles [71] and in amyloid plaques [93]. Altogether, this indicates that CRMP2 can aggregate both intra- and extracellularly, can be secreted by neurons and have post-synaptic functional consequences. Thus, pathological manifestations of AD may involve trans-synaptic transfer of phosphorylated/glycated CRMP2.
Although CRMP3 has not been studied extensively, what is known suggests this protein may be a promising candidate for the protection or rescuing of dysfunctional dendrites. Similar to its CRMP2 homolog, CRMP3 expression has been identified in mammalian growth cones in the hippocampus, suggesting its involvement in neurite growth [94]. Indeed, CRMP3-deficient mice (CRMP3–/–) were shown to exhibit dystrophic dendrites in CA1 and DG regions of the murine hippocampus [53, 95]. In contrast, its over-expression in cultured hippocampal pyramidal neurons induced lamellipodia genesis, neurite initiation, spine-like formation, dendritic outgrowth, and activation of voltage-gated Ca2 + channels [96] (Fig. 3). Furthermore, CRMP3 overexpression protected dendrites against dystrophy induced by prion peptide PrP106–126 [97]. Interestingly, a 2.5-fold reduction in CRMP3 protein expression has been demonstrated in the entorhinal cortex of AD patients compared to cognitively-intact controls [98–100]. These findings collectively suggest that CRMP3 may be a promising target for preventing or reversing dendritic dysfunction.
CRMP5 was originally identified as a novel CRMP3-interacting partner [101], and in parallel, as the target of auto-antibodies present in neurological paraneoplastic syndromes [102–105]. CRMP5 gene expression has been detected in the cerebral cortex, cerebellum, dentate gyrus, and other regions of the hippocampus. Its expression in neurons is developmentally regulated by the transcription factor Sox5 [106], with a reduction in expression as the neurons mature [102, 107]. In adult brain, CRMP5 is highly expressed in the neurogenic areas like the dentate gyrus where it negatively regulates neurogenesis [108]. Within the hippocampus and dentate gyrus, CRMP5 staining is present in CA1–CA3, granular cell bodies and dendrites but not in the axonal mossy fibers projections to CA3 [109]. Its localization in the filopodia of growth cones converges with its role in filopodial dynamics and growth cone development [110]. CRMP5 interacts with other CRMPs and with cytoskeleton proteins to regulate neurogenesis, neuritogenesis, dendrite/axon outgrowth, and guidance [108]. In mouse hippocampal neurons, CRMP5 interacts with tubulin in the growing axon thus halting its growth by displacing CRMP2 [48]. This interaction is regulated by CRMP5 phosphorylation by GSK3β at T509, T514, and T516 (Fig. 3). The increased function of GSK3β in AD suggests that CRMP5 may be highly phosphorylated which would promote its interaction with tubulin. In cultured hippocampal neurons of CRMP5–/– mice, BDNF-induced dendritic branching was inhibited, suggesting a role of CRMP5 in neurotrophin activity [111]. Adding to the complexity, CRMP5 functions in neurite outgrowth and is also involved in the regulation of mitophagy (removal of unhealthy mitochondria), a step necessary for proper establishment of neuronal polarity [48, 113].
These conflicting observations suggest that CRMP5 activity may be development/species/cell type-specific, likely due to its interaction with different proteins and post-translational modifications. Given its important role in the generation and maintenance of the proper dendritic structures and the possible contribution of dendrite dystrophy in the impaired memory function associated with paraneoplastic limbic encephalitis in patients with CRMP5 auto-antibodies [114, 115], it has been hypothesized that CRMP5 might play a role in AD. A recent study [116] examined the role of CRMP5 in the dendritic dystrophy of the 3xTg-AD transgenic mice model. These mice exhibited higher levels of CRMP5 gene and protein expression in the hippocampus as compared to controls. In addition, the social behavior and memory impairments observed in these mice were rescued by knockdown of CRMP5 expression while they were exacerbated by its overexpression, through decreasing AMPA receptor surface expression [116]. The increased CRMP5 expression in AD [116] combined with its role in promoting mitophagy [112] echoes to the known dysfunction of mitochondria in the AD brain [117]. CRMP5 expression can increase mitochondrial fragmentation [112], a feature of AD [117]. Thus, the increased CRMP5 levels may push mitochondria into autolysosomes thereby contributing to the impaired mitochondrial homeostasis and bioenergetic deficit in AD. These observations support an important role of CRMP5 in AD by 1) halting the remodeling of microtubules, 2) decreasing excitatory transmission through inhibition of GluA1 surface expression, and 3) by impairing mitochondrial function. Of course, it should be noted that these neuropathological features are not exclusive to AD, as they are common to several other neurodegenerative diseases such as Parkinson’s disease, Huntington’s disease, and amyotrophic lateral sclerosis [118–121]. While more advanced studies are needed to explore these points further, we propose that CRMP5 may be a viable therapeutic target in ADRD and other neurodegenerative diseases.
The role of CRMPs in immune cell functions
Although cells differ in their architecture and function, all cells rely on a dynamic cytoskeleton that can rapidly reorganize and adaptively respond to signals in their environment. While the ability for CRMPs to remodel the cytoskeleton has been best characterized in neurons, immune cells also rely on CRMPs to mediate many of their physiological functions—from phagocytosis to cellular motility—all of which relies on extensive changes in morphology. And importantly, these functions are known to facilitate synaptic plasticity [122]. To date, there is a paucity of research examining the pathophysiological functions of CRMPs in immune cells in the context of AD. However, several studies have examined the function of CRMPs in immune cells using various neuroinflammatory insults. While neuroinflammation is a characteristic hallmark of many neurodegenerative diseases, including AD, the pathologies are nuanced and disease-specific [123]. Nonetheless, what is known may be useful for extrapolating general mechanisms by which CRMPs might contribute to immune cell-mediated pathology in AD.
CRMP2 was the first member of the CRMP family to be studied in immune cells, and as such, has received the most attention. The most notable research has focused on its role in T-lymphocyte motility and migration during the course of human T-leukemia virus-1 (HTLV-1) infection. Importantly, HTLV-1 and other viruses such as Herpes Simplex Virus-1 (HSV-1) can usher in an oxidative and inflammatory environment in the brain which mirrors the pathophysiology of AD and may even engender or exacerbate AD pathology [124]. In regard to HTLV-1, CRMP2 is highly expressed in primary T-lymphocytes isolated from HTLV-1 infected donors compared to those from healthy donors. In addition, these T-lymphocytes also exhibit heightened migratory rates as compared to healthy donors; an effect that can be blocked by administration of a CRMP2-blocking antibody [125]. Interestingly, the virus effectively hijacks the locomotive machinery through modulating the phosphorylation status of CRMP2, increasing its recruitment to the cytoskeleton, and enhancing the proteasomal degradation of the short isoform of CRMP2 which, under non-pathological circumstances, serves as an antagonist to the full-length isoform of CRMP2 [126]. Similarly, other neurovirulent viruses, such as rabies or Canine distemper virus, induce expression of CRMP2 in T-lymphocytes which is associated with enhanced migration, disease severity, and recruitment to the CNS [127]. Importantly, immune cell recruitment to the CNS is also thought to be a feature of AD [128, 129]. This recruitment is dependent on chemokine signaling, and given that CRMPs, specifically CRMP2, transduce chemokine signaling in T-lymphocytes [130], their role warrants further investigation.
While CRMP2 expression is decreased after treatment with the endotoxin, lipopolysaccharide (LPS) [131], CRMP4 is markedly increased following LPS challenge [132]. In addition, LPS-treated BV-2 microglial cells also revealed an increase in CRMP4 expression and protein concentration that was commensurate with increased nuclear translocation of NF-κB and enhanced expression of proinflammatory cytokines. Knocking down CRMP4 expression abrogated the immune response, interfered with the nuclear translocation of NF-κB, and prevented activated BV-2 migration and phagocytosis through dysregulated F-actin dynamics [132]. Similarly, genetic deletion of CRMP4 has been shown to reduce glial scarring from reactive astrocytes and microglia in response to spinal cord injury [133] as well as ameliorate microgliosis in a model of Parkinson’s disease [134]. Microglial dysfunction contributes significantly to chronic neuroinflammation in AD as well as impaired clearance of Aβ oligomers [135]. Research elucidating the specific mechanisms in which CRMPs release cytokines or control phagocytosis would be critical in diverting microglia from a pathological response.
Together, these studies highlight that CRMPs play a role in immune cells, and while we are beginning to reveal the basic mechanisms, more work is needed to elucidate the role of CRMPs in immune cells specifically in the context of AD. A better understanding of CRMP expression and function in immune cells, as well as the post-translational events that shift their normal function, would be of immense importance in identifying therapeutic targets aimed at taming dysregulated immune cells during AD.
CONCLUSION
Dystrophic axons and dendrites and dysfunctional synapses are at the heart of the neurodegeneration-causing memory deficits in AD. Therefore, novel molecules, such as CRMPs, known to play key roles in the formation, reshaping, or reparation of synaptic structures, and in important immune cell functions have the potential to make an impact in the early detection and treatment of AD.