
Abstract
It is widely recognized that Alzheimer’s disease (AD) has a complicate link to renin-angiotensin system (RAS). It is known that cerebrovascular disease has some connections with AD, but most of the studies are still conducted in parallel or independently. Although previous research came up with large number of hypotheses about the pathogenesis of AD, it does not include the mechanism of RAS-related regulation of AD. It has been found that many components of RAS have been changed in AD. For example, the multifunctional and high-efficiency vasoconstrictor Ang II and Ang III with similar effects are changed under the action of other RAS signal peptides; these signal peptides are believed to help improve nerve injury and cognitive function. These changes may lead to neuropathological changes of AD, and progressive defects of cognitive function, which are association with some hypotheses of AD. The role of RAS in AD gradually attracts our attention, and RAS deserved to be considered carefully in the pathogenesis of AD. This review discusses the mechanisms of RAS participating in the three current hypotheses of AD: neuroinflammation, oxidative stress and amyloid-β protein (Aβ) hypothesis, as well as the drugs that regulate RAS systems already in clinical or in clinical trials. It further demonstrates the importance of RAS in the pathogenesis of AD, not only because of its multiple aspects of participation, which may be accidental, but also because of the availability of RAS drugs, which can be reused as therapies of AD.
INTRODUCTION
Renin is synthesized by proximal glomerulus cells in the afferent arterioles of the kidney. When released into the blood, renin acts directly on angiotensinogen which transforms angiotensinogen into angiotensin I (Ang I). Ang is converted to angiotensin II (Ang II) catalyzed by angiotensin-converting enzyme (ACE) [1]. Ang II has high biological activities and vasoconstriction effect. It induces aldosterone secretion and retains sodium and water by stimulating the adrenal cortical globular zone, increases norepinephrine secretion by stimulating sympathetic nodules and elevates blood pressure by activating sympathetic transmitter and its specific receptors [2]. Besides, Ang II inhibits the renal secretion of renin and stimulates the renal secretion of prostaglandin to maintain normal blood pressure levels. This regulation system from renin to angiotensin is called the renin-angiotensin system (RAS). The physiological functions of RAS, an endocrine system, include maintaining the steady-state of sodium ions in peripheral circulation, regulating body fluid volume, and regulating cardiovascular function. It has been reported that the brain owns an independent brain RAS. The brain RAS is an independent system associated with the development of various physiological functions and diseases of the brain. Many studies have shown that dysregulated brain RAS may be associated with neurodegenerative diseases caused by pathological and physiological changes due to neuroinflammation, oxidative stress, and aging.
Brain RAS is mainly composed of brain renin/pro-renin, cerebral angiotensin conversion enzyme (ACE/ACE2), cerebral angiotensinogen, angiotensin peptide, and its receptors [3]. Moreover, studies have reported a low renin level in the brain. In neuronal cells, renin is secreted as pro-renin in both autocrine and exocrine states. Renin/pro-renin stimulation causes pro-renin to bind to the pre-renin receptor in the brain cleaving angiotensinogen and activating the angiotensin receptor, which may cause cognitive dysfunction [4]. ACE is the main component of brain RAS and is mainly distributed in cerebral vascular endothelium. Several clinical studies revealed that ACE is associated with cognitive dysfunction because ACE inhibitors (ACEIs) can reduce the incidence of Alzheimer’s disease (AD) [5]. Additionally, there exists a correlation between hypertension and central ACE activity and expression. However, underlying mechanism remain unclear. In the early 20th century, Ang I and Ang II were found in the brain. Notably, the Ang II receptor (AT2R) expression is lower in the brain comparing with Ang I receptor 1 (AT1R) expression. AT2R can counteract the neurotoxicity and blood-brain barrier damage caused by the AT1R of Ang II signal transduction. The action of aminopeptidase (AMN) initiates amino-terminal aspartic acid residues conversion into another angiotensin peptide-angiotensin III (Ang III). Studies have shown that Ang III combines with AT1R, AT2R Non-AT receptors [6]. Ang III is converted forms angiotensin IV (Ang IV) catalyzed by AMN. Also, Ang IV can be directly synthesized from Ang II and catalyzed by dipeptide aminopeptidase. Other researchers have reported that Ang IV mediates the protective mechanism against neurodegenerative diseases through Ang IV receptor 4 (AT4R) [7].
AD is a neurodegenerative disease with neurological manifestations and mental symptoms such as progressive memory decline, cognitive dysfunction, language disorders, and even personality changes. By 2019, Alzheimer’s Disease International estimated that more than 50 million people worldwide suffered from AD and this number is estimated to reach 152 million by 2050. AD has been described as one of the most life-threatening diseases globally as several people were diagnosed every three seconds as they feared dementia [8]. In recent decades, research on AD and the exploration of new drugs that inhibits AD the progression has remained unsuccessful. The onset of AD is unknown and its pathogenesis is complex. AD can either be caused by genetic factors or acquired factors. The risk of AD may be related to genetic factors. The mutations of presenilin 1 (PSEN1), presenilin 2 (PSEN2), and amyloid precursor protein (APP) genes are associated with early AD pathogenesis, whereas the late AD is mainly associated with the polymorphism of apolipoprotein E (APOE) gene [9]. Acquired factors such as aging, brain trauma, and aluminum poisoning also increase the risk of AD [10]. In the past, limited reports have linked cardio-cerebrovascular risk factors (CRF) to AD; however, in recent years, considerable research has proved that CRF such as cerebrovascular disease, hypertension, and diabetes play an important role in AD development. CRF increases the susceptibility to AD and accelerates the AD development [11–13]. Although RAS plays an important role in regulating cardiovascular and renal function, fluid-electrolyte balance, and blood pressure, RAS imbalance may also be an important factor causing neurodegenerative diseases. Epidemiological studies have shown that middle-aged hypertension and cerebrovascular deficit may have something to do with dementia risk such as AD [14]. Therefore, it is urgent to find new methods for treating AD to eliminate cardiovascular and cerebrovascular diseases.
The classic renin-angiotensin system (cRAS) is an important signaling system for regulating body fluid volume, sodium homeostasis, and maintaining arterial blood pressure. Through this system, renin splits inactivate angiotensinogen into Ang I. Ang I is thereby converted to Ang II catalyzed by ACE. Ang II affects vasoconstriction and blood pressure regulation through its effect on ATR1. Despite hemodynamic regulation, RAS is involved in many brain activities including memory consolidation and acquisition [15]. A separate brain RAS which contains Ang III, Ang IV, and a new member of RAS containing 7 amino acids-Ang (1–7) and additional brain RAS components play a role in the brain. Studies have shown that separate brain RAS may be associated with cerebral vasodilation and neuroprotection. Moreover, RAS is the main secretory regulator of cardio-cerebrovascular and renal function and plays an important role in regulating the physiological function of the central nervous system (CNS) [16]. Ang IV functions through the ATR4 which is an insulin-regulated peptidase (IRAP). IRAP can regulate the intracellular transport of hippocampal and cerebral cortical pyramidal cells through insulin-responsive glucose transporter 4 (GLUT4) reducing glucose uptake. Of note, Ang IV is a natural inhibitor of IRAP, thereby improving memory by inhibiting IRAP-induced intracellular GLUT4 transport. This increases glucose uptake by neurons. Studies have determined that the activities of aminopeptidase A, B, N, and IRAP are reduced in the brain of AD patients. These enzymes are considered to be associated with learning and memory [17]. Stimulating the Ang IV/AT4 on type 1 tyrosine kinase receptors can slow down the neurodegenerative lesions and increase the hippocampal axon dendrites. The brain damage of Ang II is mainly mediated by the ATR1A subtype [18]. Contrarily, Ang II can repair damaged DNA and promote neuronal differentiation by activating ATR2. Ang (1–7) are mainly secreted by Mas receptor (MASR) with a small part by ATR2. Ang (1–7) interacts with Mas receptors to activate nitric oxide synthase (NOS) in brain neurons producing nitric oxide (NO), which improves learning and memory and promotes neuroprotection [19]. Currently, little evidence has approved that Ang II could inhibit the release of acetylcholine. It is also possible to enhance cognitive benefits through Ang IV-mediated signaling by its receptor of IRAP, which is expressed abundantly in brain regions. Compared with its precursor, Ang IV had been found to be helpful to memory in vivo. Using IRAP inhibition from Ang IV-like compounds as cognitive enhancer required further research. Angiotensin-converting enzyme inhibitors, ARAs, and renin inhibitor, alikram, had effects on formation and action of Ang IV. Some studies have confirmed that local activation of RAS in the brain can damage the brain neurons, while excess level of oxidative stress is an important factor in pathogenesis of AD. Therefore, activating brain RAS may mediate oxidative stress, thereby causing neurological dysfunction [20].
High Ang II levels can increase oxidative stress and promote neuroinflammation, whereas angiotensin receptor antagonists (ARBs) can prevent many AD risk factors and protect neurons [21]. Angiotensin-converting enzyme II activates AT1R by increasing angiotensin-converting enzyme expression and is involved in cerebral vasoconstriction and impaired cognition [22]. The cRAS activity is regulated by the downstream regulatory RAS pathway (rRAS). rRAS is closely associated with pathological features in AD patients and the decline of cognitive function in animal models with CNS diseases [23]. There is strong evidence that excessive brain ACE/Ang II/AT1R axis is involved in oxidative stress, apoptosis, and neuroinflammation causing neurodegenerative changes in some brain diseases [24, 25]. Also, studies have found that AT1R blockade confers neuroprotection. Many studies have reported that the pathophysiological changes of neuroinflammation, oxidative stress, and aging associates brain RAS disorder with neurodegeneration [26]. RAS participates in many AD signaling cascades, for example, regulating cerebral blood flow. This may initiate the formation and degradation of amyloid-β (Aβ) thereby providing a new direction for future AD treatment [27]. In addition, a meta-analysis showed that RAS-targeted antihypertensive drugs achieved better results than other hypertension drugs in reducing the risk of dementia. The results indicated that ACEIs reduced AD incidence by 14%, whereas ARBs decreased the AD risk by 30% [28]. However, there are contradicting reports on ACEIs indicating that they are not optimal drugs for AD treatment. This is because the Aβ clearance process may be interrupted by ACE which is contrary to the pathology of Aβ [29]. Besides, because of improving cerebral blood flow, ACEIs have been shown to reduce AD incidence [30].
ACE2/Ang (1–7)/MASR is a brain RAS axis that counteracts the damage of ACE/Ang II/AT1R axis to brain neurons. Ang (1–7) binding to the MASR in the hippocampal CA1 area stimulates long-term gain effects. This finding indicates the role of the ACE2/Ang (1–7)/Mas receptor axis in learning and memory [31]. Subcutaneous injection of Ang (1–7) was reported to improve the spatial and new object recognition memory in a mice model of myocardial infarction without changing cardiac function [32]. Studies on Mas-KO mice reported that the deletion of MASR gene enhanced contextual fear memory and slowed down their disappearance [33]. Therefore, activating the ARBs and ACE2/Ang (1–7)/MASR axis may be a new strategy against neurodegenerative diseases (Fig. 1).

Alzheimer’s disease versus brain renin angiotensinogen system. Brain RAS is mainly composed of brain renin/pro-renin, cerebral angiotensin conversion enzyme (ACE/ACE2), cerebral angiotensinogen, angiotensin peptide (angiotensin II/angiotensin(1–7)), and its receptors (ATI receptor/MAS receptor)) which are associated with Alzheimer’s disease caused by pathological and physiological changes due to neuroinflammation, oxidative stress, and amyloid-β protein. ACE2/Ang (1–7)/MASR is a brain RAS axis that counteracts the damage of ACE/Ang II/AT1R axis to brain neurons which are synthesized by kidney and liver, respectively.
RENIN-ANGIOTENSIN SYSTEM AXIS AND ITS REGULATORY MECHANISM ON AD
The RAS axis is associated with various disease processes, including hypertension, inflammation, among others. A wealth of studies has investigated the correlation of AD with the RAS (Table 1). This review explores various mechanisms through which the RAS axis improves AD.
Studies on the role of RAS in neurodegenerative diseases
Renin-angiotensin system axis improves AD through inflammatory pathways
Among the two RAS axes, angiotensinogen (AGT) is the most classic axis that generates Ang I catalyzed by renin. Ang I is converted to Ang II via ACE induction. Then, Ang II combines with AT1R to form the ACE/Ang II/AT-1R axis, which mediates common cardiovascular and cerebrovascular diseases and critically function in the progression of neurodegenerative diseases [34–37]. Activating the ACE2/Ang (1–7)/MASR, another RAS axis can reverse damage caused by the ACE/Ang II/AT-1R axis to neurons. Numerous investigations have been geared toward assessing the potential relationship between AD pathogenesis and neuroinflammation. A study using in vitro and in vivo AD models revealed that the expression of p65-nuclear factor Kappa B (NF-κB), tumor necrosis factor-α (TNF-α), and other inflammatory proteins were elevated [38]. Other studies have shown that dysregulated brain RAS has the potential to improve some common neurodegenerative diseases such as AD by mediating inflammatory pathways. Also, the ACE/Ang II/AT-1R axis has been revealed to activate various cell functions and molecular signaling pathways associated with tissue damage and inflammation [39]. The ACE/Ang II/AT1R axis induces microglial activation, upregulates NOX complexes, and increases reactive oxygen species (ROS) production [40, 41]. Except for the activation of the M1 phenotype signal of microglia, activating AT1R may partially inhibit the peroxisome proliferator-activated receptor gamma (PPAR-γ) function, thereby preventing microglia transition to the opposite of the immunomodulatory M2 state. ACE2/Ang (1–7) /MASR axis inversely induces an inflammatory response in the ACE/Ang II/AT-1R axis [42, 43]. Ang II mainly initiates the release of ROS and inflammatory mediators during neurodegenerative disease attacks. The inflammatory cascade induced by Ang II causes cognitive dysfunction and neurodegeneration in the brain [44]. Studies have reported that high Ang II levels mainly induce AT-1R to promote neuroinflammation. Contrarily, ARBs can inhibit AD-associated risk factors, thereby protecting the neurons [45]. On the other hand, the ACE/Ang II/AT1R axis enhances microglia activation and M1 phenotype transformation through NOX, consequently releasing proinflammatory factors including TNF, interleukin IL-1β, NO, and ROS. The neuronal inflammation-mediated AT1R upregulation of M1 microglia in the cortex and hippocampus is associated with cognitive impairment [46]. Notably, since the blood-brain barrier (BBB) can prevent the entry of peripheral toxins into the brain, Ang II, therefore, alters the permeability of BBB through the ACE/Ang II/AT1R axis. As a result, pro-oxidation and proinflammatory effects are exerted, which damage the brain neurons.
The AT-1R activation induces apoptosis cascade and promotes the expression of inflammatory proteins such as p65-NF-κB and TNF-α. Besides, the Ang II-AT-1R complex induces neuroinflammation, which destroys the remodeling of memory consolidation and tissue repair. Excessive activation of glutamate receptors results in neurotoxicity, elevated expression of neuronal Aβ protein, and excessive tau protein phosphorylation, all of which promote AD progression [47].
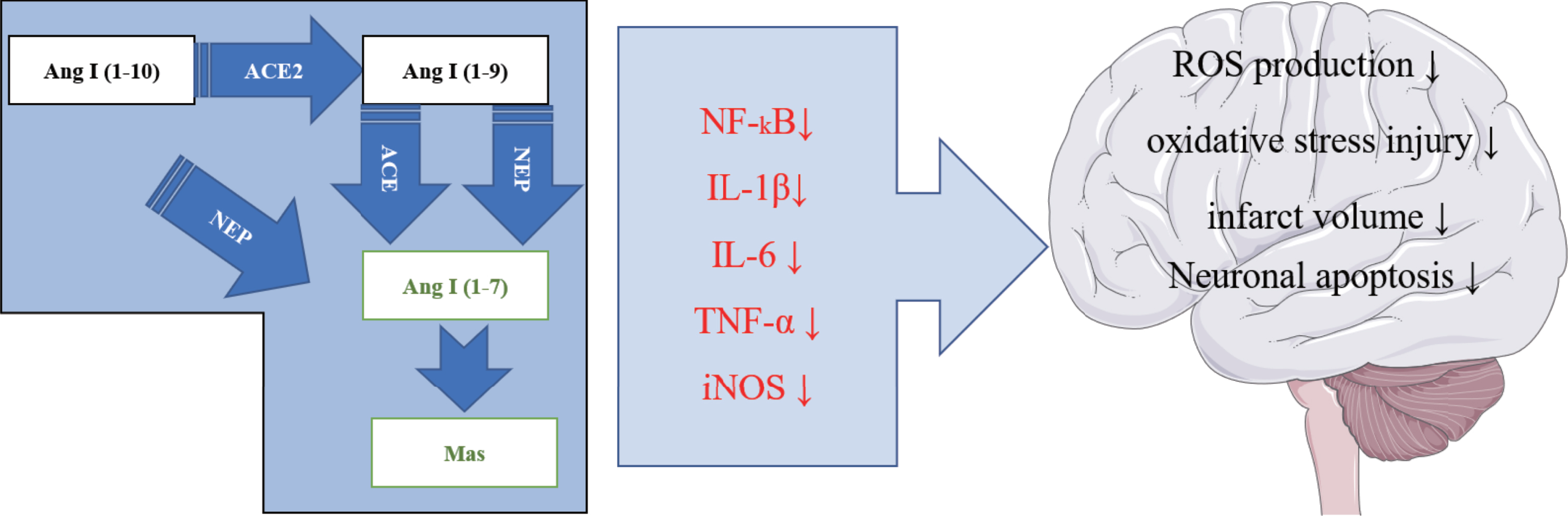
Ang (1–7) is a heptapeptide composed of tyrosine, arginine, aspartic acid, valine, isoleucine, proline, and histidine [48]. It was later identified as a new member of RAS and is mainly produced via Ang II hydrolysis, which is induced by ACE2. Ang (1–7) is generated from three substrates: Ang I (1–10), Ang (1–8), and Ang (1–9) by Ang I or Ang II digestion [49]. Besides, a secondary source of Ang (1–7) is through Ang I hydrolysis to Ang (1–9) catalyzed by ACE2, which then is converted to Ang (1–7) mediated by neutral endopeptidase (NEP). Of note, the mechanism by which ACE2 catalyzes Ang II conversion is more efficient than on Ang I [50]. Ang (1–7) is critical in regulating the inflammatory process; it exerts the anti-inflammatory effects, for example, reduction in leukocyte recruitment, cytokine release, fibrosis, and tissue damage [51–53]. Several studies have revealed that Ang (1–7) participate in the inflammatory response after cerebral ischemia, improve the cascading amplification effect of inflammatory response after cerebral ischemic injury, inhibit the NF-κB-mediated inflammatory response after ischemia, reduce oxidative stress injury and infarct volume, promote nerve function repair, and induce neuroprotection after ischemia [54–56]. Moreover, Ang (1–7) binding to the Mas receptor of G protein-coupled potentially inhibit the MAPK pathway activated by Ang II, exert a protective role in inhibiting cell proliferation, inhibit the inflammatory response, and fibrosis [57]. Of note, the binding regulates inflammation and fibrosis of the peripheral organ and improves neuron glucose utilization.
IL-6, a key cytokine in the AD pathological process, exerts various proinflammatory effects, including the activation of the acute-phase inflammatory response, serve as adhesion molecules for the upregulation endothelial cells, and promote B and T cell differentiation [58]. A study reported that Ang (1–7) reduced IL-6 level in atherosclerotic plaques of apolipoprotein E (ApoE) knockout (KO) based AD-related atherosclerosis mouse models. Also, it reduced the level of monocytes chemotactic protein-1 (MCP-1) and TNF-α. Moreover, it was reported that the Mas receptor antagonists could elevate IL-6 levels [59]. The levels of IL-6, MCP-1m, and TNF-α in ACE2 gene knockout mice were higher than the control group [60]. IL-1 exerts multiple functions in enhancing inflammation. IL-1α and IL-1β have been found to activate ACE2/Ang (1–7)/MASR, thereby lower the IL-1 level. Intraperitoneal injection of lipopolysaccharide (LPS) can cause systemic and brain tissue inflammation in mice. Studies have revealed that LPS-treated MasR knockout mice exhibit significantly high IL-1 plasma levels compared to LPS-treated wild-type (WT) mice. Ang (1–7) was reported to reduce the levels of IL-1 and IL-6 in rat astrocytes after radiation culturing [61–63]. Notably, radiation treatment could cause damage to brain tissue in patients, and its specific mechanism was possibly related to inflammation, thus had the potential to trigger AD [64]. Inflammasome regulates IL-1β production. Therefore, the effect of the ACE2/Ang (1–7)/MASR axis on IL-1β may be mediated by the inflammasome [65]. However, the proinflammatory phenomena caused by ACE2 deficiency are highly attributed to elevated Ang II levels rather than low Ang (1–7) levels; however, the specific mechanism is yet to be fully elucidated.
Following the above findings, it is evident that the RAS function is not limited to cardiovascular and renal physiology. Many researchers have focused on the effect of angiotensin peptide on the inflammatory response. The brain RAS potentially participates in AD progression by regulating the inflammatory response. Besides, studies have reported that the ACE2/Ang (1–7)/MASR axis activation has the potential to reduce the level of inflammatory factors. Therefore, targeting the ACE2/Ang (1–7)/MASR axis activation may provide new insights for future AD treatment (Fig. 2).
Main mechanisms of anti-inflammatory effects of renin angiotensinogen system. IL, interleukin; TNF, tumor necrosis factor; MCP-1, monocyte chemotactic protein 1; NF-κB, nuclear factor kappa B; p-STAT 3, phosphorylated signal transducer and activator of transcription 3; p-ERK1/2, phosphorylated extracellular-signal-regulated kinases 1/2; p-JNK, phosphorylated c-Jun N-terminal kinases; p-p38 MAPK, phosphorylated p38 mitogen-activated protein kinases; VCAM-1, vascular cell adhesion molecule-1; ICAM-1, intracellular cell adhesion molecule-1; MMPs, matrix metalloproteinases; iNOS, inducible nitric oxide synthase.
Renin-angiotensin system axis improves AD through oxidative stress
Reports have indicated that RAS participates in AD pathogenesis; however, most studies have focused on Ang II to elucidate the relationship between RAS and AD. ACE2 exert a protective role in AD progression. A few exogenous ACE2 or Ang (1–7) have been reported to potentially eliminates AD by inhibiting Ang II-induced oxidative stress [66]. Elsewhere, it was confirmed that local activation of RAS in the brain could damage the brain neurons. According to reports, an excess level of oxidative stress is attributed to AD pathogenesis, whereas activation of the brain RAS may mediate oxidative stress causing neurological dysfunction [67, 68]. Other researchers attribute brain damage in AD patients to RAS-activated mitogen-activated protein kinase (MAPK)-mediated apoptosis, NF-κB-mediated inflammation, and oxidative stress responses promoted by redox imbalance [69]. Moreover, free radicals have been found to play an important role in neurodegeneration [70]. Due to high oxygen consumption and lack of antioxidant enzymes, compared with other organs, neurons are more vulnerable to free radical damage. ACE/Ang II/AT-1R axis is involved in oxidative stress causing neurodegenerative diseases in the brain [71]. In another study, Ang II was found to bind to the AT1 receptor and was considered an effector of oxidative stress. Notably, Ang II introduces an active substance, superoxide, in the brain, which is an indicator of oxidative stress [72].
Additional reports indicate that dopamine levels in the striatum can be regulated by AT1R, which is expressed in neurons, microglia, oligodendrocytes, and astrocytes. ROS is a derivative of oxygen, therefore, in AD, excessive ROS causes an imbalanced endogenous antioxidant mechanism in the brain. Such a condition is referred to as oxidative stress in the brain, which damages neuron cells and induces cell apoptosis. Ang II is an active peptide of RAS and stimulates ROS production in blood vessels [73]. Also, it has been demonstrated that mitochondrial dysfunction may be associated with the burden of vascular ROS. Ang II stimulates NAPDH oxidase and may affect mitochondrial ROS production. Excessive NAPDH oxidase is considered to be the main downstream target for brain stem Ang II [74]. Excessive AT1R activation in neurons increases ROS levels via different signaling pathways of the NADPH-oxidase (NOX) complex resulting in oxidative stress, which consequently causes cognitive impairment [75]. Also, Ang II enhances the death of dopaminergic cells, which may be caused by activated oxidative stress from Ang II and AT-1R complex. Reports indicate that AT1R stimulates the NF-kB signaling pathway to mediate oxidative stress, and this may cause the death of AD neurons [75–78]. In addition, Ang II can affect the neuronal N-methyl-D aspartate (NMDA) current, thereby increasing the production of NOX-2 dependent ROS. Moreover, the superoxide produced by NOX activated microglia NF-kB and RhoA/Rho kinase pathways have been revealed to promote NOX stimulation through p38 mitogen-activated protein kinase. Ang II can influence NO production through the AT-2R subtype. These could be regarded as indicators of oxidative stress [79]. Ang II participates in the death of AD neurons when AT-1R activates NADPH oxidase complexes, causing oxidative stress [80].
These studies provide evidence that RAS mediates oxidative stress in neurodegenerative diseases, causing apoptosis and increasing the intracellular ROS levels that result in neuron damage. AT1 receptors in cerebral blood vessels function in cerebral blood flow and automatic regulation of blood vessels, thereby helping to regulate the supply of nutrients and oxygen in the brain. In neurons, excessive activation of AT1 receptors blocks long-term enhancement of the hippocampus, increases oxidative stress, and promotes neuroinflammation. In animal models, the activation of the AT2 receptor plays a key role in the conditional avoidance response. The expression of the ACE2/Ang (1–7)/Mas axis in different regions of the CNS suggests that it may exert neuroprotective effects. Increasing evidence shows that the ACE2/Ang (1–7)/Mas axis protects the brain by: reducing oxidative stress, mediating neuronal autophagy, and other mechanisms, thereby reverse the damage caused by AD to brain neurons. Thus, reducing the levels of ACE2 and Ang (1–7) could damage the cerebral arterial cell and increase the oxidative stress level. On the other hand, activating the ACE2/Ang (1–7)/MasR axis is considered a new neuroprotective method, owing to its potential inhibitory effects on the NF-kB signaling pathway, which results in reduced oxidative stress. Besides, Ang (1–7) plays a multi-target protective role in several pathophysiological mechanisms of brain damage caused by AD. For example, Ang (1–7) reduces the oxidative stress in varying degrees by acting on the Mas receptor on brain injury, reducing neuronal apoptosis, inhibiting cell autophagy, and affects the ACE/Ang II/AT-1R axis metabolism, thereby it contributes to the pathophysiological regulation of AD brain injury [81]. Also, Ang (1–7) decreases the levels of Ang II and AT-1R and reduces oxidative stress and apoptosis of brain neurons, thereby exert an effective inhibitory effect on the ACE/Ang II/AT-1R axis. Activating MasR in neurons increases NO and reduces mitochondrial respiration, thus lowering the oxidative stress.
With an increase in age, MasR expression in neurons gradually decreases [82]. Through an in vivo experiment, it was revealed that Ang (1–7) injection could inhibit oxidative stress and NF-kB activity, thereby reducing the expression levels of tumor necrosis factor IL-1β and COX-2. However, MasR inhibitor A779 reversed the protective effect of Ang (1–7) [83]. Interestingly, from a similar experiment, Ang (1–7) exerted an anti-oxidative stress effect through MasR, however, it was unclear whether MASR was the only functioning receptor on Ang (1–7). Compared with WT mice, ACE2 knockout mice had higher levels of oxidative stress in the brain. Besides, it was found that, in the CNS, Ang (1–7) could reduce the enhancement of oxidative stress-related hormones (such as Ang II, glucocorticoids, norepinephrine, serotonin, and dopamine) in the blood and oxidative stress-related brain regions (such as hippocampus, thalamus, amygdala, and prefrontal cortex). In vitro studies showed that ACE2 deficient macrophages and ACE2 deficient human brain microvascular endothelial cells exhibited enhanced oxidative stress effects; however, overexpression of ACE2 attenuated the oxidative stress induced by Ang II. In another experiment, it was found that the activation of AT2R and ACE2 by drugs could prevent oxidative stress, thus improve AD [84].
The above research findings proved that RAS critically influence the level of oxidative stress in the brain, either as an enhancer or inhibitor. However, brain damage in AD patients is most partly due to the development of severe oxidative stress, leading to a series of complications such as cognitive dysfunction caused by severe cell apoptosis. Studies have found that the activation of the ACE2/Ang (1–7)/Mas axis can lower the oxidative stress response. Drugs that reduce the level of oxidative stress in the brain by activating the ACE2/Ang (1–7)/Mas axis may offer a reliable direction for AD treatment in the future (Fig. 3)

Renin-angiotensin system axis improves AD through oxidative stress. Proposed Ang-(1–7)/Mas signaling pathways counter-regulating the AngII/A T1R axis. AngII is produced by ACE from AngI and cleaved by ACE2 and other peptidases to form Ang-(1–7). Binding of AngII to AT1R activates p38 MAPK, Erk1/2, PLC, as well as PI3K, and this effect can be attenuated by Ang-(1–7) activation of Mas. Following activation by Mas, phosphatases, like DUSP-1 and PTP1B, inhibit kinase activity. AT1R-mediated activation of oxidases leads to the formation of ROS. In contrast, NO release can result from the activation of Mas via Akt phosphorylation. Several agonists (green arrows) and antagonists (red lines) have been developed for the various components of this system, like ACE inhibitors (ACEI) and AT1R blockers (AT1RB). AngII/AT1R as well as Ang-(1–7)/Mas signaling pathways regulate gene expression in the nucleus via different transcription factors (NF-κB, STAT1/3, FOXO1).
Renin-angiotensin system axis improves AD by influencing the Aβ deposition pathway
In recent years, a large part of research related to AD has focused on the amyloid cascade hypothesis and cholinergic hypothesis. The amyloid cascade hypothesis described the significant pathogenic effect of Aβ peptides cleaved from amyloid-β precursor protein (AβPP) and accumulation in the brain due to the imbalance between its production and clearance [85–88]. Histopathological features of AD in the brain are senile plaques (neuritis plaques) formed by Aβ and NFTs generated from tau protein aggregates. The two main Aβ-proteins, Aβ42 and Aβ40, are easily aggregated to form oligomers and fibrils. In normal conditions, the brain eliminates toxic peptides through enzyme degradation, receptor-mediated efflux transport, and perivascular drainage. The damage of these two clearance mechanisms may lead to the accumulation of Aβ peptide, consequently damaging the neuronal membrane, increasing oxidative stress, changing receptor-mediated signal transduction, changing membrane pores, increasing intracellular calcium ions, and damaging the mitochondria. These changes could also lead to a continuous loss of cortical cholinergic projections [89]. The deposition of Aβ also promotes the formation of pathological phosphorylated of tau protein. Abnormal aggregation and deposition of tau protein lead to form nerve fiber tangles and progressive lose neurons [90]. However, so far, the failure of various amyloid-targeted drug development challenges the amyloid hypothesis in AD, an increasing number of people prefer the link between RAS and AD [91]. Notably, it has been proved that RAS also exerts a significant effect on the formation of amyloid plaques [92].
RAS participates in many aspects of the AD cascade, for example, in the regulation of cerebral blood flow, which is critical in the formation and degradation of Aβ, thus may provide a new direction for the therapeutic schemes of AD in the future. Among patients diagnosed with AD, a large proportion of them show different severity of cerebrovascular disease, age-related plaques as well as cerebral amyloid angiopathy with Aβ characteristics associated with AD [93]. RAS also participate in the clearance of Aβ deposition. Ang II interferes with BBB by activating AT1 receptors on brain endothelial cells. These mechanisms increase the amount of Aβ in the brain by enhancing inflow from the peripheral circulation. It is generally believed that Aβ results from neuritis plaques formed by oligomerization and fibrosis of AβPP in AD. Some researchers argue that AβPP mediates the activation of Ras-ERK and supports the assumption that AβPP acts on upstream of the RAS signaling cascade, affirming the role of AβPP in the deregulation of AD cell cycle and neurodegeneration [94]. Studies have shown that neuronal exposure to oligomer Aβ42 and AβPP expression promoted the Ras/ERK signaling cascade and activated glycogen synthase kinase 3 (GSK-3) [95–98]. According to the analysis of the human brain, which showed significantly increased RAS expression, GSK-3 was activated, while AβPP and tau were phosphorylated. Whether ACE could degrade Aβ remains controversial, and if so, to what extent can it be degraded? Notably, in vitro experiments showed that ACE could cleave the aggregation-prone peptide Aβ40, release Aβ1–7 and Aβ8–40, which do not form plaques with less cytotoxicity [94]. In human neuroblastoma cells, overexpression of ACE could inhibit the expression of Aβ40 and Aβ42 receptors. ACE could also promote the conversion of Aβ42 to Aβ40 in Tg2576 mice [99]. Elsewhere, a study found that in mouse brain tissue homogenates, ACE and ACE2 could convert the more soluble and more toxic Aβ43 (a high amyloid form of Aβ that could cause plaque formation) to Aβ40 [100]. Another study evaluated the effect of Aβ deposition by hybridizing the AD model (AβPP mice) with a mouse line characterized by overexpression of ACE in bone marrow monocytes. Notably, the levels of soluble and insoluble brain Aβ42 decreased in its offspring mice, but Aβ42 levels were partially increased after ACEIs treatment [101]. ACE could degrade Aβ peptides to inhibit the development of amyloid plaques. Other studies have confirmed the effect of ACE enzymatic action in the degradation of Aβ peptides [102, 103]. Intraventricular injection of Ang II can lead to increased Aβ and tau lesions in elderly normal rats, causing cognitive dysfunction. ACE can hydrolyze the most neurotoxic peptides of Aβ42 and Aβ43 to generate amyloid peptides, which are less sensitive to aggregation and senile plaque formation [104]. Also, ACE can metabolize the most abundant of Aβ40, potentially reducing the deposition and aggregation of Aβ42. A study reported that the serum ACE-2 activity of patients with AD was lower than the control group. These studies have also confirmed that ACE could reduce Aβ, the main risk factor for the occurrence and development of AD [105]. Of note, the specific process could be summarized as follows: Aβ43 is converted to Aβ42 mediated by ACE-2, then Aβ42 is cleaved by ACE-1 into less toxic Aβ40 and Aβ41. RAS enzymes metabolize amyloid plaques by reducing the most toxic amyloid peptide consisting of 40–43 amino acid sequences [106, 107]. It is worth mentioning that ACE could degrade Aβ peptide directly, and the higher activity of ACE was detected in the brain of patients with AD. The level of angiotensinogen in the cerebrospinal fluid of patients with AD was high [108]. Thus, the elevated activity of ACE could be utilized as a new strategy for AD. However, it was hypothesized that because ACE can reduce Aβ while Ang II can mediate an effective anticholinergic effect, the observation of angiotensin receptor antagonist (Aras) on the pathogenesis and progression of AD exert a specific effect. It is unreasonable how ACE inhibitors exert a similar effect on the incidence of AD and the progression in some cases, and whether these drugs would also enhance the deposition of Aβ. ACE inhibitor Captopril is used to treat AD transgenic model Tg 2576 mice; ACE transformed Aβ43 and Aβ42 into less toxic of Aβ40. Ang II also participates in the clearance of Aβ by increasing the BBB permeability of AT1R and activating the unproven advanced glycosylation end-product receptor (RAGE) in the brain. Ang II induces inflammation, oxidative stress, and vascular disease through AT1R, which induces the accumulation of Aβ, causing AD [92, 110]. However, AT2R signaling is beneficial in learning and memory. ARBs were found to inhibit AT1R signaling, which transferred the role of Ang II to a beneficial pathway (AT2R signaling). Patients and mice with AD have in most cases been found to be characterized by excessive activation of RAS [111]. Based on previous experiments, the brains of AD patients after death are characterized by elevated ACE levels in the frontal cortex, hippocampus, and caudate nucleus regardless of whether patients had hypertension or other cardiovascular diseases [112]. These levels were related to the pathology of AD. Besides, the level of Ang (1–7) was reduced in the sporadic AD mouse model, which could be associated with tau hyperphosphorylation [113]. In another study of the brain tissue of AD patients after death, ACE2 activity was reduced, and ACE2 was negatively correlated with ACE1 activity, which were related increased Aβ and tau pathology [114]. This study also found that the ratio of Ang II to Ang (1–7) (a proxy indicator of ACE2 activity) increased, an indication that the conversion rate of Ang II to Ang (1–7) is decreased, and the activity of ACE2 and β-secretase has a strong negative correlation with ACE2. Based on the above observation, it was suggested that ACE2 may partly be contribute to the regulation of AβPP amyloidosis. This concurred with findings from a recent study that showed that the level of peripheral serum ACE2 in AD patients was in low-degree, and the activity of ACE2 was negatively correlated with parenchymal Aβ load [115]. However, some studies have reported no difference in the level of ACE activity in the brain microvessels between AD patients and normal controls [116]. Investigation of ACE levels and activity in cerebrospinal fluid in several brain regions has yielded more contradicting results, including reports of reduced ACE levels [117]. Besides, the activity of ACE has no significant difference.
In conclusion, these studies strongly demonstrate that reduced activity of ACE2 in the brain may induce the onset of AD and is related to the activation of the central classic RAS axis that deposits harmful Aβ in the brain as they cannot be eliminated in time. Several possible mechanisms link the pathogenesis of AD to the reduction of ACE2 activity. It can be inferred that under normal circumstances there may be a delicate balance between positive and negative effects on the processes affecting cognitive function and which in turn are probably affected by the mechanism of action of ACE inhibitors (Fig. 4). Firstly, the decrease in ACE2 activity would be caused by the low conversion of Ang II to Ang (1–7), increasing the Ang II level. An increased ratio of Ang II/Ang (1–7) has, in most cases, been reported in other chronic diseases related to central axis hyperactivity. Secondly, ACE2 is mainly responsible for the generation of Ang (1–7) from Ang II. It then activates MasR to regulate the brain damage caused by the ACE1/Ang II/AT1R axis while enhances learning and memory processing capabilities. Studies have shown that ACE2 converts highly toxic Aβ43 to Aβ42, then Aβ42 is either cleaved by ACE1 to Aβ40 or transformed into Aβ41 to a lesser toxic form. Therefore, ACE2 activity in AD can enhance the deposition of Aβ43 in the early stage and prevent the cleavage in downstream of Aβ42 by ACE1, indicating that the ACE2/Ang (1–7)/Mas pathway potentially function in reducing the deposition of Aβ to exert neuroprotective effects and improve cognition. However, more investigations are needed to explore the molecular mechanisms of ARAs or ACE in enhancing cognitive function. Obviously, improving the effect of hypertension and contributing to the degradation of Aβ is the predisposing factor.

Proposed interaction between the renin–angiotensin system and Aβ metabolism. The putative interaction between the ACE pathway and the AβPP metabolism pathway that leads to the formation of Aβ is illustrated. Under normal (i.e., drug-free conditions) ACE can have the positive potential to degrade Aβ that is formed from AβPP processing. The amount of ACE available to do this is partly genetically linked such that some genotypes that are found to have increased prevalence in Alzheimer’s disease are also associated with lower levels of ACE. Under this model it is also clear that following the action of ACE on Ang I there may be a negative effect on cognitive performance through Ang II-mediated depression of acetylcholine (ACh) release and also negative effects with respect to increased probability of hypertension. With the introduction of an ACE inhibitor, which will negatively affect ACE activity, the positive benefits to Aβ degradation may be reduced or abolished, but equally the negative effects resulting from Ang II action on either ACh release or hypertension are reduced. With the introduction of an ARB, the successful management of Ang II-mediated hypertension and reduced ACh release are maintained while still allowing ACE activity to go unaltered and potentially act to positively reduce Aβ concentrations.
APPLICATION OF TARGET ON RENIN-ANGIOTENSIN DRUGS IN TREATMENT OF AD
In recent years, AD, as a progressive neurodegenerative disease, has made great progress in its research field. Preclinical AD, AD induced mild recognition, and dementia of AD are the three stages of progressive neurodegenerative disease. Although current pharmacological methods only improve symptoms rather than prevent or delay cognitive decline, many common drugs are being re-evaluated to assess their potential value in AD. Currently, there is an urgent need to improve the treatment methods of AD. The existing drugs are limited to the fact that they target specific downstream neurological dysfunction rather than intervening in the underlying pathological mechanism of upstream. Compared with monotherapies, combined therapies could act on more biological processes in AD, including Aβ accumulation, tau phosphorylation, chronic inflammation, oxidative inflammation, and may confer more beneficial effects. With the discovery of the ACE2/Ang (1–7)/Mas axis and local RAS in different tissues, including the brain, the role of RAS in the pathophysiology of many neurological and psychiatric diseases has been gradually described. Although AD is the most common cause of dementia across the globe, its pathophysiological mechanism remains elusive, thus a few possible mechanisms should further be validated. So far, no treatment has been described to prevent the pathogenesis of AD. Of note, RAS is thought to be involved in AD neurogenesis through vascular and amyloid pathways. The clinical failure of amyloid targeting drugs has led to doubts about the amyloid hypothesis. Based on recent research, RAS was found to play a crucial role in the pathogenesis of AD. Thus, scholars turned their attention to the research on drugs that intervene in the RAS system to treat neurodegenerative diseases. By 2010, except for 10 compounds (exclude licensed drugs), 77 compounds had been described in preclinical development or Phase III clinical trials that could target amyloid Aβ protein or neurofibrosis and neurochemical defects. Besides, the remaining 10 compounds were revealed to target other important components involved in AD pathogenesis, including oxidative stress targeting, mitochondrial dysfunction, serotonin imbalance, neuronal preservation, fatty acid maintenance, cGMP signaling conduction, and regulation of cholesterol reduction. Notably, the pathways targeted by these 10 compounds are barely related to cardiovascular-related mechanisms.
As mentioned before, RAS molecules influence neuroinflammation, oxidative stress, Aβ deposition, and lesions. Animal experiments have shown that RAS-targeted antihypertensive drugs exert cognitive protective effects on AD models through various pathways, for example, by delaying the formation and accumulation of Aβ, anti-oxidative stress mechanism, and reducing the inflammatory response. Ang II can promote brain tissue damage through angiotensin type 1 receptors, whereas Ang (1–7) exert potential neuroprotective effects. Local brain RAS and systemic RAS have been shown to play a role in the pathogenesis of AD, therefore, are considered potential drug targets [118]. Currently, common drugs that regulate RAS include CCBs, ARBs, ACEIs thiazide drugs, and β receptor blockers and central sympathetic antihypertensive drugs (CASBs). Studies have found that CCBs, AT1RBs, and ACEIs may have a more significant protective mechanism for cognitive decline than thiazide drugs, β receptor blockers, and CASBs. In the following section, we reviewed the three mechanisms application of RAS-related potential treatments in AD.
Anti-inflammatory effect of the renin-angiotensin system
Both central and peripheral systems may be associated with neuroinflammation and cerebrovascular regulation. ACE2/Ang (1–7)/Mas axis is linked to the anti-inflammatory effects in the RAS system. Increasing evidence showed that ACE2/Ang (1–7)/Mas axis targeting compounds could reduce CNS inflammation and exert neuroprotective roles. A wealth of studies have proved that ARBs directly inhibits the pathological and neurocognitive manifestations of AD [119–121]. Intravenous infusion of ICV-Ang IV can inhibit the expression of inflammatory factors in the brain of rats with insufficient cerebral blood perfusion. For instance, in rat microglia cells, Ang (1–7) could work on MasR, therefore, directly lower the transcription of proinflammatory factors IL-1β and TNF and increase the level of anti-inflammatory factors IL-10. The AVE0091 analog of the MasR conjugate Ang (1–7) reduces the intracerebral neuroinflammation in the premature animal model SAMP8 mice by inducing microglia M2 translocation and improving cognition. ARBs can counteract the proinflammatory effects of Ang II/AT1R on microglia. Also, ARBs may directly activate PPAR-γ to transform microglia to the M1 phenotype without any alteration by AT1R antagonism. The adipose factor is a biologically active substance secreted by adipose tissue, such as monocyte chemoattractant factor 1 and tumor necrosis factor α. Adiponectin also secretes anti-inflammatory adipokines, such as adiponectin, which can promote inhibitory effects against neuroinflammation in the brain [122]. In a retrospective study of patients with hypertension and those suspected to have AD, RAS blockers (RASB) could reduce neurological inflammation and oxidative stress in the brain by regulating adipokine and glucose homeostasis to slow the decline of cognitive function in AD [123]. A wealth of reports have revealed that the incidence of AD in women is higher than that in men, suggesting the relationship between estrogen deficiency and AD. For instance, a study found that cognitively deprived ovarian AD rats showed cognitive impairment in the Morris water maze and new object recognition test, increasing the level of inflammatory biomarkers (TNF-α and IL-1β), and ACE1/Ang II/AT1 [124]. The combined effect of the two drugs on reducing neuroinflammation was more significant, similarly, the feedback of RAS regulation was more significant. Olmesartan’s treatment of AD transgenic model AβPP23 mice was found to 1) improve their cognitive ability, independent of the antihypertensive effect; 2) reduced the expression of inflammatory factors; and 3) did not influence the level of Aβ protein in the brain [125]. In non-AD patients, ARBs have unique cognitive protective effects as opposed to ACE inhibitors such as hydrochlorothiazide [126]. In the cognitive and prognostic trials of the elderly, the candesartan treatment group showed a lower cognitive decline rate than the placebo group. In contrast, the placebo group showed a minimum mental state examination (MMSE) score of 24–28 times, but not the entire sample. Epidemiological and clinical studies have shown that the prevention of Ang II signal transduction may provide a wide range of benefits in AD [127]. In addition to improving comorbid hypertension, these drugs may reduce the common cerebral blood flow in AD and Aβ deposition. ACE inhibitors or ARB may inhibit cognitive function by inhibiting Ang II-mediated release of acetylcholine and blocking the activation of harmful inflammation pathways [128]. This is considered one of the most significant controllable factors of AD. By comparing the effect of antihypertensive drugs on the scores of micropsychological examinations in 617 AD hypertensive patients, the antihypertensive drugs and cholinesterase inhibitors exerted a synergistic effect [129]. In addition, some phosphodiesterase subtypes (PDE4B, PDE4D, PDE5A, and PDE7A) have been predicted as targets for aHTN drugs, and the inhibitory effects of losartan and HCTZ on different PDE subtypes have been experimentally confirmed [130]. Combination of diuretics, RAAs blockers, and calcium channel blockers slow cognition decline significantly. Furthermore, systematic pharmacology of chemical genomics-identified molecular targets provides a systematic pharmacological explanation for the synergy of clinical drugs. Improving the level of inflammation in the brain is one of the primary goals of AD treatment [131]. The diuretic 1-CCB-1-RAAS combination can act on several AD-related protein targets directly, including key proteins from different pathological pathways of AD, such as neurotransmitter clearance enzymes (acetylcholinesterase and butyryl cholinesterase), oxidative stress enzymes (MAOB, NOS1, and NOS2), cell signaling pathways enzymes (PDE4B, PDE4D, PDE5A, PDE7A, MAPK, and COX-2), microtubule-associated protein (TAU), neurotransmitter receptors (Cholinergic receptor m cholinergic 2 and b-2 adrenergic receptor), and neuroinflammation of related nuclear receptor (PPAR-γ) [132]. p38 MAPK, another cell signal regulator, has been proposed for the treatment of AD. Activation of p38 MAPK induces phosphorylation of threonine and serine residues in various transcription factors and kinases, as well as the upregulation of cellular stress inflammation [133]. Additionally, p38MAPK has been proved to exert a direct effect on the hyperphosphorylation of tau protein, one predictive factor for the pathology of AD. Therefore, the interaction between amlodipine, MAPK, and HCTZ may be associated with the role of neuroinflammation as a protective mechanism of diuretic, RAAS, and CCB combination therapy. However, there may exist an interaction between HCTZ and COX-2 enzymes. The COX enzyme (prostaglandin H synthase) catalyzes the synthesis of prostaglandins such as prostaglandins, prostacyclins, and thromboxanes, all of which play important roles in the inflammatory pathway. COX enzymes are considered the main target for nonsteroidal anti-inflammatory medicine [134]. Notably, the selective COX-2 inhibitors, rofecoxib and naproxen, have been used to suppress cognitive decline in mild or moderate AD patients in phase III clinical trials, but have not shown significant efficacy. Considering the importance of neuroinflammation in AD pathology, nonsteroidal anti-inflammatory drugs might still hold great potential as new anti-AD drugs [135]. In a Dock study, both losartan and amlodipine were predicted to target PPAR-γ. Besides, losartan metabolite EXP3179 was found to have partial agonist activity against PPAR-γ because of its significance in AD. PPAR-γ comprise three nuclear receptors (α, γ, and δ), regulating a group of genes involved in lipid and energy metabolism. Considering its role in lipid and carbohydrate metabolism, PPAR-γ agonists (rosiglitazone and pioglitazone) have been developed treatment options for type II diabetes [136]. In addition, PPAR-γ agonists have the potential to treat AD in multiple aspects of AD pathology, especially in combating neuroinflammation. Currently, the PPAR-γ agonist pioglitazone has transition to phase III clinical trials, which can slow brain inflammation in AD patients and improve cognitive decline [137].
Application of renin-angiotensin system against oxidative stress
Compared with other tissues, brain is vulnerable to oxidative imbalance because of high oxygen consumption, abundant easily oxidized polyunsaturated fatty acids, and weak antioxidant defense system. Therefore, either enhanced active oxygen production or damaged brain antioxidant system will affect oxidative imbalance of redox balance, resulting in excessive ROS. Oxidative imbalance and subsequent damage by oxidative stress are widely reported in AD. Ischemic neuronal damage and excessive activation of glutamate receptors lead to calcium influx and excessive oxidative stress. In AD, especially those related to presenilin-1 mutations, calcium homeostasis disorders may lead to mitochondrial dysfunction and higher oxidative stress. In neuronal culture, the toxicity of Aβ makes these cells more susceptible to excitotoxicity which involves calcium influx, leading to changes in the cytoskeleton, which is similar to AD nerve fiber tangles [138]. Interestingly, this neurotoxicity is reduced when neurons are cultured in calcium-deficient media. Nilvadipine can prevent spatial memory impairment in rats caused by cerebral ischemia combined with Aβ overload, which is related to preventing neuronal apoptosis and reducing the level of oxidative stress. However, amlodipine does not confer this protective effect, which raises the molecular specificity of thiazide drugs in preventing cognitive decline [139].
Studies have shown that ACEIs and Sartans can reduce hereditary hypertension, high renin goldblat hypertension, diabetes induced by streptozotocin, and long-term cognitive decline of scopolamine poisoning rats. Losartan may prevent and restore AD markers by reducing AT1-mediated oxidative stress and “normalizing” memory-related AT4 receptors [140]. ACEIs or Sartans inhibit the activation of AT1 receptors by reducing nicotinamide adenine dinucleotide phosphate oxidase, promoting nitric oxide production, activating guanylate cyclase and cGMP formation, which in turn lead to the release of glutamic acid, and improving cognitive function [141], which ultimately enhance neurovascular coupling and induce synaptic plasticity, both of them are recognized mechanisms to enhance learning and memory processes [142]. Bradykinin join in the neurotoxic effects of Aβ-induced oxidative stress synaptic dysfunction in mice [143]. As mentioned above, Ang II binds to two major receptors in the brain, AT1 and AT2, which have opposite effects. AT1 activation causes vasoconstriction, smooth muscle hypertrophy, endothelial dysfunction, oxidative stress, and chronic inflammation, while AT2 activation reduces the infarct size after ischemic injury by increasing cerebral perfusion in ischemic penumbra, reduces production of superoxide, and activates neuronal repair system by promoting neuronal cell differentiation and axon growth. ARBs block AT1 instead of AT2, and may also increase Ang II and upregulate AT2. This selective blocking of AT1 may excessive stimulate AT2, which indicating that ARBs may have potential therapy on cognitive dysfunction. AT2 hypothesis comes from a recently specific AT2 receptor agonist experiments. In this experiment, C21, a direct agonist of the AT2 receptor, increase nerve growth and differentiation, and improve cognitive ability. Diuretics, especially potassium-sparing drugs, have achieved unexpected results in treatment of AD. Because low potassium concentration is associated with increased oxidative stress, potassium-sparing diuretics can regulate RAS, reduce oxidative stress levels, accelerate cellular metabolic waste, and reduce neuronal damage [144]. Statins is a class of heterogeneous drugs that are widely used in the primary and secondary prevention of atherosclerotic diseases. The ability of statins to penetrate brain is related to their own lipophilicity. In addition, different transport proteins and BBB integrity may play a role in the entry of statins into the brain. In different studies, there are conflicting findings regarding the integrity of the BBB in AD patients [145–147]. One recent study showed that changes in the serum/cerebrospinal fluid albumin ratio reflect human BBB dysfunction, which is a specific change in dementia and is the most obvious change in vascular dementia. These different additional factors may explain the results of animal models that observed which have hydrophilic statins in brain tissue. Different studies explore the problem of entering the brain by analyzing fat-soluble and hydrophilic statins separately [148]. Epidemiological investigations and meta-analysis found that when comparing the lipophilic and hydrophilic groups, there are no difference effects on cognition [149]. But some people think that fat-soluble statins have harmful effects. Studies have shown that using fat-soluble statins is associated with more favorable structural integrity of white matter in elderly people with low cognitive ability but not diagnosed with AD. Animal models reported the beneficial effects of lipophilic atorvastatin, which prevents excessive oxidative stress in the step brain, reverses hippocampal cell damage, and improves learning memory and spatial cognition [150]. ARBs, such as valsartan, is generally considered as safe and better tolerance than other drugs. Captopril and Valsartan have protective effect on memory function and neuronal damage of AD. It was found that captopril and valsartan can significantly increase the activity of superoxide dismutase and catalase in brain tissue, reduce the content of malonaldehyde and oxynitride (NOX), therefore, significantly reduce histopathological damage [151, 152]. The inhibitory effect of captopril and valsartan on RAS enhances the antioxidant function of brain, reduces oxidative stress in AD, and saves neuronal damage. Captopril reducing brain damage caused by ischemia suggests that continuous activation of RAS under neuropathological and physiological conditions impairs cognitive function by stimulating AT1 receptors, accompanied by reduction in cerebral blood flow and increase in ROS production [153]. Telmisartan can prevent AD in nonhypertensive dose, therefore AT1 receptor antagonists or ACE inhibitors as a specific AT1 inhibitor, have a certain inhibitory effect on recognition of AD mice, and prevents neurodegenerative diseases. A study found that STZ-induced dementia significantly increased the levels of malondialdehyde (as a marker of ROS) and nitrogen oxides (as a marker of RNS) in the rat brain. This enhancement of oxidative stress markers may be because of increasing ACE activity and the formation of Ang II, which stimulate NADPH oxidase and have a key effect on the development of oxidative stress [154]. In this study, the use of captopril and valsartan during AD can reduce the generation of free radicals of ROS and reactive nitrogen species (RNS). Previous studies have demonstrated that under different brain pathophysiological states, the activation of AT1 receptor is produced by ROS/RNS. Therefore, inhibiting brain RAS may be a possible way for RAS inhibitors to inhibit ROS or RNS production in rats [137]. According to previous findings, ACE inhibitors like Captopril can be used directly as a free radical scavenger [155]. In addition, inhibiting RAS may reduce the activity of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, which is a key enzyme that produces oxygen free radicals.
The role of the renin-angiotensin system to improving Aβ pathology
In recent years, research progress on the pathophysiology of AD and the renin-angiotensin system pathway has revealed that ARBs are ideal treatments for AD. There is evidence that both RAS axes (ACE1/Ang II/AT1 and ACE2/Ang (1–7)/MasR) are involved in the amyloid hypothesis (Aβ cascade) and the vascular mechanism of AD. However, the existing link between hypertension and AD is unclear. The potential neuroprotective role of ARBs is not in any way associated with the reduction of blood pressure. Of note, animal studies have shown that the cognitive protection is related to reduce production and oligomerization, increase Aβ degradation, and its vascular effects (improve BBB, increase cerebral blood flow, restore endothelial function, and reduce inflammation) [156]. Besides, clinical studies have found that the use of ARBs in AD patients can reduce Aβ deposition in the brain and protect patients from mild cognitive impairment, prodromal of AD, and dementia. However, no clinical trial for ARBs has been conducted in AD. ARBs in people at risk of cognitive impairment may have surprising effects [157]. Although the mechanism of ACE1/Ang II/AT1 and ACE2/Ang (1–7)/MasR axis of RAS has been described, ARBs may be explored ideal drugs in the treatment of AD. However, future research on ARBs should focus on combining experimental ARBs or ACE inhibitors with standard treatment, especially because prescriptions of these readily available drug categories are for persons with different cardiovascular diseases. Many related patents already or soon to expire makes these drugs very attractive if they found that be beneficial on publicly funded healthcare systems. In addition, these drugs have been proven safe for years, thus have passed standard clinical trials in large populations. Once a drug is found to be beneficial, it can be quickly availed to patients. ARBs inhibit damage caused by ACE1/Ang II/AT1 to neurons and activate ACE2/Ang (1–7)/MasR, this promotes their protective mechanisms in preventing cognitive impairment and strengthening neuroprotection. Some antihypertensive drugs have been shown to improve AD incidence and cognitive decline in AD patients, and their mechanism may be related to the clearance of Aβ in the brain. For example, dimethylnaphthalene acetoacetate (DIZE) is currently recognized as an ACE2 activator. A study found that intraperitoneal injection of DIZE could enhance the activity of ACE2 (the main effector of RAS) in Tg2576 mice and activate ACE2/Ang (1–7). Besides, the ACE2/Ang (1–7)/Mas axis reduces hippocampal Aβ and improves cognition [158]. Further research found that the protective role of DIZE is mediated directly by ACE2 and is related to low soluble Aβ42 and IL1-β levels in the hippocampus. Notably, DIZE restored MasR levels in the hippocampal synaptosomes of Tg2576 mice, which is related to high expression of NMDA NR2B and downstream ERK signaling. Through a meta-analysis, results showed that RAS-targeted antihypertensive drugs achieved better results than other hypertension drugs in reducing the risk of dementia. However, current opinions on ACEIs are different. On the one hand, some researchers find ACEIs not suitable for AD treatment because the clearance process of Aβ may be interrupted by ACE, which is contrary to Aβ pathology [159]. Elsewhere, a study evaluating the effect of antihypertensive therapy on AD pathology and RAS complex found that the level of Aβ in the brain and the level of ACE in hypertensive AD patients were elevated [29]. Many in vitro studies have shown ACE degrades Aβ and converts toxic peptide Aβ42 into less toxic Aβ40. ACEIs may prevent these effects, in vivo, leading to the accumulation of brain Aβ peptides. However, this effect is not consistent, as it is dependent on the treatment time and age of transgenic mice [160]. Subsequent studies in mice have shown that captopril treatment is associated with increased Aβ. of note, the administration of captopril to AD transgenic mice (AβPP) from 28 days to 3 months old neither elevates the levels of Aβ in the brain nor increase plaque deposition. On the contrary, the Captopril administration of Tg2576, 6-month-old AD transgenic mice, significantly increased Aβ42 for 7–11 months, but Aβ40 levels in the cerebral cortex did not increase after 7 months. However, 11 months later, the quantity of amyloid plaques in the cerebral cortex and hippocampus and the number of Aβ42 immuno-positive areas added up by 2–2.5 times, whereas the degree of deposition was negatively correlated with ACE activity [104]. NEP (or neprilysin) have been proved to stimulate catabolism of Aβ40 and Aβ42 peptides, while subcortical endothelin converting enzyme inhibitors (previously developed for enhancing the antihypertensive effect of ACEIs) and neutral endopeptidase inhibitors can prevent the catabolism, leading to an overload of brain Aβ peptide [161]. Compared with ACEIs, AT1RBs have been found to ramp up the expression of insulin-degrading enzymes, which are known to mount up the catabolism of Aβ peptides. As a result, administering valsartan to AD transgenic mice (Tg2576) at 5–11 months old could prevent Aβ peptide overload in the brain and plasma, improve AD spatial cognitive impairment and was related pathology [162].
It has long been suggested that among the elderly at risk of cognitive decline, ATEI should be replaced with AT1RB. Of note, ARBs may provide more advantages than ACEIs in the treatment of AD as follows: First, ARBs can increase the difference effect of ACE2/Ang (1–7)/Mas axis, but ACEIs does not show such a large difference effect [163]. Second, passing the BBB is a challenge in the development of new AD drugs. For instance, the currently widely used ACEI, enalapril, cannot pass through the BBB, and almost all ARBs have acceptable CNS penetration [28]. Notably, a few observational studies and pilot trials have shown that centrally active ACE positively impact on the incidence of AD, regardless of its antihypertensive effect. Other studies have also focused on ARBs and RAS targeted drugs in AD prevention. However, the role of cerebral ACE in cognitive decline and prevention of dementia is unclear, probably because cerebral ACE is not specific for angiotensin-I. Cerebral ACE can also decompose brain peptides and amyloid peptides that enhance cognitive function and convert toxic Aβ42 into less toxic Aβ40 [89, 99]. Therefore, ACEIs may have cognitive enhancement function in the short-term, whereas they may increase Aβ42 brain load and cognitive decline in the long term. The clinical relevance cannot be ruled out in humans, as the ACE gene has been proved to be associated with AD through human genome-wide analysis. To validate the harmful effects of ACEIs on AD in humans, it is necessary first to verify that the DD-ACE gene polymorphism is related to AD protective effects. A preliminary in vitro experiment proved the potential protective effect of polymorphism of the DD-ACE gene on AD. These have been demonstrated in HEK cells expressing familial AD-Swedish mutations of human APP and human ACE. Angiotensin AT1 receptor blockers prevent the amyloid-βosis neuropathic process by overexpressing the amyloid-degrading insulin-degrading enzyme, which may exert a greater cognitive protective effect than ACEIs. This is because they share a cognitive enhancement effect with ACEIs, which is directly related to a common AT1 passivation effect [152]. In vivo, ACEIs cannot be confirmed to increase the Aβ peptide levels in the brain. In addition, ACEIs enhance the formation of angiotensin II and IV, thereby stimulating non-antagonism AT2 and AT4 receptors, notably, the activation of these receptors in the cognitive process has been fully confirmed [165]. Current randomized, placebo-controlled phase II clinical trials in patients with mild to moderate AD are aiming to evaluate the efficacy of ARB losartan in the treatment of AD. At a dose that does not lower blood pressure, losartan reduces amyloid plaque and inflammation in AβPP /S1 transgenic mice. Of note, Ang II can accelerate the deposition of Aβ and cause neuroinflammation, vasoconstriction, and mitochondrial dysfunction, reduction in G protein signaling, and increase in brain endothelial dysfunction [166]. It has been found that the ACEI ramipril reduces the activity of central ACE, and Ang II activity is reduced [167]. At this time, an effective vasoconstrictor may improve cerebral blood flow in the brain, just like in peripheral blood vessels. By increasing the release of acetylcholine, thermophilic cells may produce beneficial AD-related effects [168]. Besides, Ang II participates in the inhibition of the release of acetylcholine, which is a widespread neurotransmitter [168]. Therefore, ACEIs may be vital in AD by reducing Ang II-mediated increase in acetylcholine release [169]. However, all antihypertensive drugs are classified into one category, yet these studies do not provide any potential mechanism in cognitive therapy. Some antihypertensive drug categories may play a blood pressure-independent role. Among the types of antihypertensive drugs, the relationship between ARBs and cognitive ability is the most consistent. Many animal experiments have revealed the role of ARBs in the content of cerebral amyloid [170]. However, the role of ARBs on Aβ has not fully been elucidated. Through in vitro studies, candesartan was shown to lower the production of Aβ, whereas valsartan prevented the oligomerization of Aβ and enhanced the activity of IDE-mediated proteolytic enzymes. Animal studies using a rodent AD model further confirmed the potential therapeutic effect of ARBs [171]. When telmisartan was used in an AD transgenic mouse model, or intracerebroventricular injection of starch-like protein and induced ischemia, its cognitive effect was improved. Telmisartan further reduced ICV streptozotocin-induced experimental dementia in mice [172]. Within the research process, it was found that these treatments were associated with inflammation. It is related to the alleviation of AD. A previous study also found that olmesartan pretreatment could improve cognition and prevent vascular disorders caused by Aβ before the ICV injection of Aβ. This study also showed that ARBs may have additional effects on Aβ oligomerization and hippocampal synaptic plasticity. Notably, these are direct effects, not antihypertensive effects, which further confirm amyloid clearance and metabolism [173]. In a nutshell, these preclinical studies using AD animal models have shown that ARBs have protective effects on cognition, which is related to their anti-Aβ (reducing production and oligomerization, increasing Aβ degradation) and vascular protective properties (improving BBB, restoring endothelial function, abating inflammation and adding up cerebral blood flow) [174]. Moreover, in an ongoing study of the neuropathological analysis of 890 elderly with hypertension, subjects with ARBs showed fewer markers of amyloid deposits when compared with subjects treated with other antihypertensive drugs [175].
CONCLUSION AND PERSPECTIVE
The new coronavirus pneumonia (COVID-2019), caused by SARS-Cov-2, broke out in China at the end of 2019 and spread worldwide later on. The pathway of SARS-Cov-2 invading human body is similar to the severe acute respiratory syndrome coronavirus (SARS-COV), through the spike protein (Spike protein, S protein) on the surface of the virus binds to the receptor ACE2 on the surface of respiratory epithelial cells, and the viral envelope protein is fused with the cell membrane, and the virus releases the genetic material into the cell, which then doubles in replication, causing a series of pathological changes. However, ACE2 is a key component in RAS. Studies have shown that RAS participates in the multi-level response of AD, and ACE2 has a key effect. The highest mortality rate of COVID-19 is the elderly and individuals with weak immune systems, which may also be caused by uneven distribution of medical resources. This is especially true for those affected by neurodegenerative diseases and most of their caregivers.
AD is a common progressive neurodegenerative disease, with abnormal cognition and behavior. It is the most common pathogenesis of dementia. Although there has been some progress in the research on the pathogenesis and pathophysiology of AD, the research on the mechanism of AD is still in the hypothesis stage. At present, the treatment options for AD are limited, and many methods have not been proven effectively [170]. Exploring the link between RAS and AD may be a potential area for multi-target therapy. RAS participates in AD multi-level events such as oxidative stress, inflammation, cerebral blood flow and BBB function, Aβ cascade, mitochondrial dysfunction, neuronal plasticity and survival, synaptic function, and neurotransmission. Currently, the epidemiological evidence of the role of RAS drugs in AD therapy is still insufficient. This review introduces the relationship between the three classic hypothetical mechanisms of AD (neuritis, oxidative stress, Aβ cascade) and RAS contact, how the ACE/Ang II/AT1 axis and ACE2/Ang (1–7)/MasR axis of RAS improve AD and some related drugs in vivo, in vitro, and in clinical trials. RAS targeted antihypertensive drugs have greater advantages than other antihypertensive medicine in reducing AD risk. Excessive activation of ACE/Ang II/AT1 axis can cause neuroinflammation, oxidative stress, and Aβ cascade reaction, which induced apoptosis and neurodegeneration and further development into AD. The activation of the ACE2/Ang (1–7)/MasR axis regulatory pathway directly promotes memory and learning while downregulating the ACE/Ang II/AT1 axis, providing an alternative, exciting and novel approach to AD therapy. We hope that this review will help deepen the understanding of the relationship between AD and RAS, thereby improving the quality of future epidemiological and clinical studies. Inclusion criteria should be defined clearly in future research, solve brain penetration problem, and define the cumulative effects and cognitive outcomes of drugs. It will be particularly valuable to examine the effects of genetic polymorphisms, long-term follow-up and middle-aged interventions.
Footnotes
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (No.81574040, 81873351), Clinical basic research project of State Administration of traditional Chinese Medicine (No.JDZX2015001), the Key Project Foundation of Support Program for the Excellent Young Faculties in Universities of Anhui Province in China (No.gxyq ZD2018053), and the Project of High-Level Talents in AHUTCM (2019RCZD001). Distinguished Young Scholars Project of Natural Science Foundation of Anhui Province in China(grant No.1908085J27).