
Abstract
Across the fields of virology and neuroscience, the role of neurotropic viruses in Alzheimer’s disease (AD) has received renewed enthusiasm, with a particular focus on human herpesviruses (HHVs). Recent genomic analyses of brain tissue collections and investigations of the antimicrobial responses of amyloid-β do not exclude a role of HHVs in contributing to or accelerating AD pathogenesis. Due to continued expansion in our aging cohort and the lack of effective treatments for AD, this composition examines a potential neuroviral theory of AD in light of these recent data. Consideration reveals a possible viral “Hit-and-Run” scenario of AD, as well as neurobiological mechanisms (i.e., neuroinflammation, protein quality control, oxidative stress) that may increase risk for AD following neurotropic infection. Although limitations exist, this theoretical framework reveals several novel therapeutic targets that may prove efficacious in AD.
INTRODUCTION
The consequences of viral infection remain a global priority, as indicated by recent public health challenges [1–3]. Neurotropic viruses pose a particular risk given their capacity to penetrate the blood-brain barrier (BBB), infect cells of the central nervous system (CNS) (e.g., neurons, glia), and persist at low levels as latent infections [4, 5]. Although each maintains unique genetic materials (e.g., single stranded, double stranded; RNA, DNA) encapsulated by one or more distinctive capsid proteins, the persistence of neurotropic viruses can be attributed in part to the integration of viral genomes with the host or their maintenance as extrachromosomal DNA [6]. By utilizing gene regulatory mechanisms to then reproduce infectious particles, these microbes can disrupt various cellular functions and potentially contribute to pathological mechanisms in neurodegenerative diseases [6, 7]. While some neurotropic viruses (i.e., rabies, polio) no longer pose substantial dangers to public health, other neurotropic viruses are emerging as potential modulators of age-related cognitive decline and Alzheimer’s disease (AD), including human herpesviruses (HHVs) [8–11].
The HHV family of viruses are nearly ubiquitous amongst human populations (>90%) and can be distinguished by their unique genetic material, morphology as well as replication mechanisms [7, 12–14]. These include the herpes simplex virus type 1 (HSV-1), herpes simplex virus type 2 (HSV-2), varicella-zoster virus (VZV), Epstein-Barr virus (EBV), cytomegalovirus (CMV), human herpesvirus type 6A (HHV6-A), human herpesvirus type 6B (HHV6-B), human herpesvirus type 7 (HHV-7), and human herpesvirus type 8 (HHV-8), also known as Kaposi sarcoma-associated herpesvirus (KSHV). Neuroinfection of these double stranded-DNA viruses typically occurs during childhood via the infiltration of HHV infected leukocytes across the BBB or by retrograde axonal transport along cervical, motor, and olfactory nerves [15]. While acute infection rarely elicits therapeutic intervention, HHVs instead establish life-long latency accompanied by recurrent reactivation and asymptomatic, persistent viral shedding [7, 15]. Establishment of such reservoirs may be due to HHV integration with host genomes or persistence as extrachromosomal circular DNA [16]. Latent reactivation of HHVs can then be triggered across an individual’s lifespan by a variety of factors, including environmental toxins, psychological stressors, and co-infection with other pathogens [17]. Accumulating evidence also indicates HHVs can modulate neuropathological mechanisms in AD, and are more abundant in individuals displaying age-related cognitive decline and AD, particularly those with AD risk factors (e.g., APOE4) [11, 18–20]. In turn, some investigators have proposed these subliminal yet long-term HHV infections in the CNS may contribute to progressive impairments in neuronal functioning over time, including in AD [21, 22].
Distinguished by its Aβ plaques and tau neurofibrillary tangles, AD is a growing concern across economic, societal, and health indices [23]. In the US, the roughly 5 million AD diagnoses are expected to surpass 13 million by midcentury [24]. Despite its increased prevalence, no available treatments halt or reverse AD progression [25]. Meanwhile, the drug development process is largely unsuccessful, with the vast majority of preclinical results failing to translate into clinical benefits [26, 27]. Although progress in monoclonal immunotherapies provide optimism (e.g., aducanumab), it remains unclear if this pharmacological class will prove effective in large scale, AD cohorts [28]. Therefore, it is increasingly relevant to assess AD risk factors and potential neurobiological mechanisms in AD.
While the role of viruses in AD has been discussed for decades, contemporary investigations have renewed this debate (Table 1). Genomic analyses of comprehensive brain tissue collections, as well as preclinical examinations of Aβ’s antimicrobial responses, suggest HHVs in particular may contribute to AD [10, 29–31]. The current composition examines these recent advances and considers the potential neurobiological mechanisms that may increase risk for AD following neurotropic infection.
Recent Advances in a Viral Theory of AD
DR, Clinical Dementia Rating; LC-MS, Liquid Chromatography-Mass Spectroscopy; MAP, Rush University’s Memory and Aging Project; Mayo TCX, Mayo Clinic’s Temporal Cortex; MSBB, Mount Sinai Brain Bank; hNPCs, Human Neural Progenitor Cells; hNSCs, Human Neural Stem Cells; RNA-seq, RNA Sequencing; ROS, Religious Orders Study; WES, Whole Exome Sequencing. *Only available for MSBB cohort. **Only 364 ROS/MAP samples were used in ddPCR.
CLINICAL ADVANCES IN AN HHV THEORY OF AD
The role of pathogens in AD has been debated for some time [32, 33]. While investigators have noted a variety of viral, fungal, and bacterial associations with AD, the most abundant evidence implicates HHVs [34, 35]. For instance, aged individuals previously infected with CMV (i.e., for an average of 5 years) display increased risk for AD and faster rates of decline in global cognition during follow-up assessment [36]. Such retrospective cohort studies also indicate increased likelihoods for developing dementia following VZV or HSV (HSV 1 or 2) infection, while this risk is considerably reduced amongst individuals receiving antiviral therapies [11, 38]. Specifically, these neurotropic viruses are thought to induce subacute neurobiological alterations that interact with environmental and/or genetic factors to progressively compromise functioning over time [39, 40]. Such subliminal interactions and chronic alterations in host cells may reflect an adaptive viral response in the CNS, provided that acute, severe infections in the immune-privileged brain might otherwise trigger significant immune responses (e.g., meningitis, encephalitis) and compromise the viability of host cells necessary for viral reproduction [4, 6]. Regardless of its evolutionary relevance, the implications of neurotropic infection are subject to increasing focus in AD etiology.
Contemporary debate was spurred in 2018 with a multiscale analysis of postmortem tissue by Dudley and colleagues, which reported elevated levels of multiple HHVs in the brains of AD individuals [10]. Samples obtained from the Mount Sinai Brain Bank (MSBB) indicated HHV-6A and HHV-7 were significantly more abundant across several regions in AD brains, while expression of these viruses in the anterior prefrontal-cortex in particular was correlated with both AD histopathological (i.e., Braak Score, plaque density) and neuropsychological (i.e., Clinical Dementia Rating; CDR) criteria. Samples from the Religious Orders Study (ROS), Rush University’s Memory and Aging Project (MAP), as well as the Mayo Clinic’s Temporal Cortex consortium (Mayo TCX) yielded similar results, and also indicated an increased rate of HSV-1 infection in AD diagnoses. Moreover, significant associations between host DNA variation and viral quantifications revealed a herpetic regulation of host gene networks that increase AD risk, such as Aβ processing and neuroimmune functioning. Combining genomic, transcriptomic, proteomic, histopathological, and behavioral data into a single meta-nomic analysis, researchers concluded HHVs (i.e., HHV-6A, HHV-6B, HHV-7) were strongly associated with systems-level variation in AD biology [10]. However, these striking findings and the complex multi-network data analysis prompted active debate among fellow researchers.
In response, computational biologists posed several notable concerns [41, 42]. Primarily, authors argued that the bioinformatic approach in the original analysis was skewed and data were not filtered properly, while reanalysis of the same data using alternative statistical tools yielded contradictory results. Following criticism, Dudley and colleagues responded directly, acknowledging that the calculation of viral abundance in comparative transcriptomics can vary depending on analytical strategy, especially when transcripts have low copy number and when group differences are limited. The authors justified their approach by discussing the statistical procedures that accounted for the selective features of the dataset, and went on to validate the sensitivity of their statistical techniques [43, 44].
Most recently, another group of researchers led by Jacobson and colleagues reanalyzed the same tissue samples originally used by Dudley and colleagues, but utilized PathSeq, a bioinformatic algorithm optimized for the detection of microbial RNA. In addition, the authors used digital-droplet-PCR (ddPCR) to assess HHV-6 integrated DNA in samples from the MAP as well the Johns Hopkins Brian Resource Center (JHBRC). Reanalysis of transcriptomic data indicated HHV6 in 4/301 MSBB samples and 2/605 MAP samples, while there was no significant association between PathSeq values and AD diagnoses or criteria (i.e., plaque density, CDR). Similarly, ddPCR indicated HHV-6 was not significantly associated with AD and viral load was low when it was detected [29]. Although these findings did not support a prominent role of HHVs in AD, they did not exclude possible viral contributions to accelerating AD pathogenesis.
PRECLINICAL ADVANCES IN AN HHV THEORY OF AD
Along with observations in clinical cohorts, experiments in cell culture and animal models have renewed the focus on neuroviral contributions to AD. Specifically, investigations have suggested the antimicrobial activities of Aβ triggered by infection may become dysregulated and contribute to neuropathology over time [45, 46]. Particular focus has been placed on HHVs due to a series of seminal studies reporting HSV-1 DNA in amyloid plaques of AD patients [19, 48]. These neuroprotective, innate immune responses have been attributed to Aβ’s inherent antimicrobial properties, including its highly conserved polypeptide sequence and pathogen-relevant structural bioactivity (e.g., oligomerization, fibrillization) [49–51]. In addition, a pair of recent investigations have provided important mechanistic evidence for the potential perturbations in Aβ regulation following HHV infection.
In 2018, researchers led by Moir and colleagues reported the capacity for HHV infection to induce and accelerate Aβ deposition both in vitro and in vivo. Following intracerebral injection of HSV-1, adult 5XFAD mice displayed significantly longer life expectancies compared to wild-type littermates, while adolescent AD mice subjected to infection displayed rapid, premature seeding of Aβ (<48 h) that colocalized with HSV-1 and matured into plaques that morphologically resembled clinical AD samples. Further experiments employing neural cell lines determined Aβ’s antiherpetic capacitates for HSV-1, HHV-6A, and HHV-6B were facilitated by its glycoprotein binding to the viral surface, while subsequent fibrilization inhibited HHV infectivity. Interestingly, researchers expanded their findings to an NPC-generated, 3D neural cell culture system that is genetically engineered to replicate AD plaques and tangles [52]. Upon infection, significantly increased rates of Aβ deposition were observed in comparison to sterile environments, and co-localization with viral particles supported previous findings of viral entrapment by Aβ fibrilization [30]. However, it was unclear if the high titer of virus primarily used in these experiments was indicative of the low grade HHV infection observed in the AD brain.
A separate group of researchers recently provided additional evidence that low titers of HSV-1 can replicate AD-like pathology in vitro and potentially contribute to AD [31]. Employing NSC-generated cultures, Kaplan and colleagues illustrated the capacity for low levels of HSV-1 to trigger the formation of dense plaques positive for Aβ and paired helical filaments positive for pTau. In addition to significantly altered expression in Aβ processing (e.g., APP, BACE1, PSEN1, PSEN2) and pro-inflammatory cytokines (e.g., TNF-α, IL1β, IL-6, IFN-γ), researchers identified reactive gliosis reminiscent of postmortem AD tissue. Notably, such HSV-1 induced alterations were attenuated by treatment with an antiviral medication (i.e., Valacyclovir) that has previously been associated with decreased risk of dementia and is currently being assessed in multiple AD clinical trials [11, 53]. Subsequent analyses utilizing 3D tissue models indicated similar neuronal variations in response to herpetic infection, along with reduced electrophysiological firing rates [31]. Importantly, these results were obtained under physiologically normal conditions (i.e., absence of genetic manipulations), while the minimal virus titers were thought to emulate those viral loads found in AD patients.
In addition to a renewed enthusiasm for a neuroviral theory of AD, contemporary studies highlight important considerations in assessing AD etiology. For example, clinical evaluations measured viral loads primarily in cortical samples, so it is possible AD diagnoses may be tightly correlated with infection prevalence, but in different subcortical structures or other areas of the CNS (e.g., cervical nerve termini). Such sampling errors are particularly relevant given that AD can exhibit heterogeneous neuropathology and HHV distribution in the brain can be focal rather than diffuse [54–57]. These studies also underscore the value of advanced analytic techniques in neurovirology, as well as the growing necessity for translational, interdisciplinary approaches in determining the underlying mechanisms of AD [58, 59]. In addition, the cross-sectional and factorial experimental designs utilized in recent studies encourage future examination of time-dependent variation in vector-host interactions that may overlap with physiological aging processes in the AD brain [60, 61].
POSSIBLE “HIT-AND-RUN” HHV THEORY OF AD?
Most recent investigations neither prove nor disprove the causal role of HHVs in AD, but suggest long-term implications of viral infection in the CNS. In particular, results do not rule out a possible “hit-and-run” viral theory of AD, whereby transient HHV infection(s) and/or reactivation(s) (i.e., “hit”) trigger(s) chronic changes in cellular functioning that contribute to disease progression, despite a low viral titer observed during latency (i.e., “run”). Here, neuropathological development would be inherently dependent on an individual’s predispositions (e.g., age of initial infection, frequency of reactivation, genetic variation, etc.). Moreover, such pathological contributions may reflect accumulated variation in cellular functioning that results from acute infection, reactivation, latency and/or cyclical alterations in these states of viremia over time. This “hit-and-run” phenomenon can be represented in the context of adenoviral oncogenesis, where a transformed cellular phenotype in response to Ad5 infection can be maintained despite the subsequent absence of viral molecules [62, 63]. Given its implications, this model remains a primary postulation in the etiology of many neurodegenerative diseases (e.g., multiple sclerosis, Parkinson’s disease) and cancers [64–66].
Consideration of bioinformatic sensitivities and the neurotropic characteristics of HHVs reveal that recent clinical results do not exclude a potential “hit-and-run” theory in AD. Specifically, the PathSeq analysis utilized by Jacobson and colleagues was previously applied to postmortem samples from dengue or JCV, microbial infections characterized by severe virion production and PathSeq scores of 5,000 or greater [67, 68]. Conversely, scores of HHV positive samples in their most recent study failed to exceed 40, indicating PathSeq may not be reliable in the context of neurotropic viruses that infect far less numbers of cells in the CNS and establish long-term, latent infections with lower viral loads, such as HHVs [69]. Such sensitivity is reflected in PathSeq’s detection of viruses that have greater neurotropic tendencies, such as CMV and JCV, but its inability to detect HHVs that were otherwise reported by Dudley and colleagues, such as HSV1, HSV2, and HHV7 [10, 29]. Therefore, it remains possible that subliminal CNS infections, such as HHVs, induce long-term alterations in neural functioning that contribute to AD pathogenesis, despite the lack of sensitivity to detect such mild, latent infections during analysis.
Along with clinical findings, assessment of preclinical results in the context of known proteopathic mechanisms indicates possible long-term implications of HHV infection in the CNS. In particular, Moir and colleagues illustrated HSV-1 infection induces distinct conformations in Aβ that are antiherpetic but historically associated with AD pathogenesis, suggesting that the endogenous Aβ plaques found by Kaplan and colleagues under physiologically normal conditions may be reflective of a seeding process induced by initial viral infection [30, 31]. Here, seeding refers to acute, anomalous interactions between specific protein species that can further instigate the assembly and spread of aberrant proteinaceous bodies over time [70]. This postulation is justified given that HHVs, similar to Aβ and pTau, can exhibit distinct interneuronal spreading patterns in areas of the brain where AD neuropathology is commonly observed [71]. Specifically, while Moir and colleagues illustrated Aβ’s unique oligomerization facilitates its antiviral capacities (i.e., binding to viral glycoproteins necessary for host-cell interactions, fibrillization that inhibits viral particles), it also leads to the formation of in vivo plaques reminiscent of the AD brain. Interestingly, Kaplan and colleagues noted a similar plaque morphology in their naïve model system, suggesting low titers of HHVs may trigger innate antiviral conformations of Aβ that lead to time-dependent dysregulation and/or propagation of cytotoxic Aβ in AD.
The development of an in vivo model of recurrent HSV-1 infection and its correlation with AD-phenotype further supports potential “hit-and-run” neuroviral contributions to AD. Following extensive validation of their techniques, De Chiara and colleagues recently established a mouse model of recurrent HSV-1 infection through the repeated application of thermal stressors, thus emulating the herpetic reactivation observed in the clinical population [72]. Compared to uninfected controls, as well as infected yet unstressed counterparts, animals subjected to multiple reactivations displayed progressive accumulation of AD hallmarks across multiple brain regions of interest, including Aβ deposition, pTau, amyloid aggregates, and indicators of neuroinflammation (i.e., astrogliosis, IL-1β, IL-6). Remarkably, such neuropathogenic measures were consistently correlated with impairments in memory capacities and proportional to the frequency of viral reactivation (i.e., frequency of thermal stress applications) (Table 1). Consistent with a potential “hit-and-run” scenario, these results suggest viral reactivation over time, rather than primary infection and/or latency, may induce long-term changes in cellular functioning that contribute to increased risk for AD pathogenesis.
POTENTIAL NEUROVIRAL MECHANISMS IN AD
Given the potential increased risk for AD due to HHV infection, the neurobiological mechanisms that may facilitate their interaction have been increasingly scrutinized [73, 74]. Considering a mild state of chronic inflammation is a pervasive feature of the aging brain, viral infection may interact with or exacerbate neuroinflammatory processes and contribute to AD [75, 76]. Indeed, such aberrant neuroinflammation in aging results in significantly greater risk for neurodegenerative conditions in the elderly, most commonly AD [77–79]. For example, exaggerated production of pro-inflammatory cytokines in aging can exert detrimental effects on neurons and correlate with behavioral deficits across multiple cognitive domains (e.g., working memory, sustained attention) [80–82]. As AD pathogenesis is associated with altered neuroinflammatory profiles, and neurotropic viruses can alter host cell immunity, latent or recurrent HHV infection may contribute to or accelerate AD neuropathology.
Several immune mechanisms may facilitate HHV contributions to AD, including cell surface expression of immune receptors, cytokine signaling, and the regulation of peripheral immunity. As one of the most abundant and versatile classes of immune receptors, disruptions in MHC/HLA signaling is associated with AD susceptibility and other viral infections characterized by neurocognitive deficits, including HIV-Associated Neurological Disorders (HANDs) [83–85]. In an attempt to evade host defense mechanisms and persist as latent reservoirs, infection by HSV-1, HSV-2, HHV-6A, HHV-6B, and HHV-7 similarly results in the modulation of MHC/HLA receptors [86–89]. In addition, elevations in several pro-inflammatory cytokines, particularly TNF and IFN, accompany the neuronal and cognitive dysfunction observed in AD as well as other neurotropic viruses (e.g., HIV) [90–93]. Interestingly, evidence indicates HSV-1, HSV-2, HHV-6, and HHV-7 significantly perturb host cell regulation of TNF [94–97]. Furthermore, such HHVs can impair IFN signaling as well [96, 98–100]. Along with variation in immune receptors and cytokine signaling, elevated inflammatory processes originating in the periphery (e.g., pro-inflammatory chemokines and cytokines, infiltrating T lymphocytes and monocytes) are thought to exacerbate underlying inflammation within the CNS, thus furthering neuropathology in AD as well as HANDs [101–104]. Given their trophic capacities for immune cells outside the CNS, HHVs may also augment systemic inflammation that subsequently contributes to infiltrating immune processes in AD [105].
In addition to inflammation, HHV infections may advance AD by perturbing subcellular protein quality control (PQC) [106]. The failure to maintain intracellular protein homeostasis is a molecular hallmark of aging and AD, particularly as it relates to the degradation of malformed or aggregated proteins via the autophagic-lysosomal pathway [107, 108]. Here, double membrane structures called autophagosomes sequester otherwise unwanted proteins and subsequently fuse with lysosomes to ensure catabolism of their substrates [109]. Given the dependence on a well-regulated proteome in post-mitotic cells (i.e., neurons), PQC impairments in the AD brain result in a net loss of function across a multitude of neural processes and homeostatic mechanisms [110, 111]. Similarly, viral infection and virion production are associated with impaired proteostasis in host cells, including neurons [112–114]. Thus, neuroviral infection may contribute to impaired PQC in AD pathogenesis, particularly by disrupting autophagy.
HHVs influence several rate limiting steps in autophagy, potentially reflecting vector-host interactions that are necessary for latent neurotropic viruses to evade immune detection [115–117]. For example, HSV-1, HSV-2, HHV-6, and HHV-7 infections are each associated with blunted levels of the anti-autophagic Bcl-2, suggesting the activation of autophagy in host cells is necessary for viral integration and subsequent viral proliferation [118–121]. Interestingly, Bcl-2 remains a genetic risk factor in AD and is downregulated by Aβ, while similar mitigation of its expression is observed in animal models of HAND [122–125]. Although the targeting of lysosomal degradation pathways by HSV-1, HSV-2, HHV-6, and HHV-7 may reflect an initial flux in autophagy following infection, it is unclear how its chronic modulation could precipitate lysosomal deficiencies [126–129]. However, it is notable that lysosomal storage dysfunction is characteristic of many neurodegenerative conditions, including AD and Parkinson’s disease, and is thought to facilitate HANDs [130–133]. HHVs may also disrupt PQC and contribute to AD by inhibiting autophagosome formation (i.e., Beclin-1 antagonism) or inhibiting subsequent fusion with lysosomal membranes (i.e., accumulation of LC3 I/II) [129, 134–136]. Interestingly, such impairments in autophagosome dynamics and functionality are reminiscent of observations made in AD as well as in vitro investigations of viral proteins implicated in HAND [130, 137–139].
Along with inflammation and PQC, HHV infection may facilitate AD by contributing to increased levels of oxidative stress in the aged CNS. Elevations in reactive oxygen species and reactive nitrogen species, as well as the adverse consequences of such elevations, are characteristic of the aged brain and contribute to pathogenic processes in AD [140–143]. Although these imbalances in reactive oxygen species and reactive nitrogen species are correlated with age across tissue types, the properties of the CNS render it especially vulnerable to the accumulation of these species. In addition to the post-mitotic origin of neurons, the CNS retains decreased antioxidant defenses (i.e., compared to other organs), while its increased energetic demand and decreased energetic reserve is compensated by a high degree of vascular turnover, which itself can generate oxidative stress due to increased heavy metal exposure [144]. Moreover, such accumulation can be augmented due to a number of age-dependent factors, including tonic microglial activation, decreased mitochondrial efficiencies, increased BBB permeability, and further mitigated antioxidant mechanisms [142, 146].
Similar to other neurotropic viruses (e.g., HIV), accumulating data indicate HHV infection results in increased oxidative stress, while such increases can potentially contribute to disease progression in the CNS [147]. Indeed, elevations in markers of oxidative stress following herpetic infection have been illustrated across a variety of animal and in vitro models, including neural cell systems [134, 148–152]. Utilizing the recently developed in vivo model of recurrent HSV-1 infection, viral reactivations in the CNS and subsequent oxidative stress resulted in functional impairment of key a protein (i.e., GRP78) previously linked to AD pathogenesis [153]. However, while markers of reactive oxygen species/reactive nitrogen species dysregulation are associated with most HHVs, further investigation of HHV-7 in particular appears warranted [154–159].
In addition to their antimicrobial activities, HHVs may facilitate AD neuropathogenesis by inducing the deposition and accumulation of pathologically relevant proteins. Across primary rodent neuronal cultures, NSC models, and an in vivo model of recurrent infection, HSV-1 induces the phosphorylation and cleavage of AβPP, intracellular accumulation of Aβ, as well as subsequent impairments in neuronal functioning (e.g., synaptic transmission, viability, differentiation) [72, 160–163]. In addition to the deposition of toxic Aβ, HSV infection can also induce the phosphorylation and accumulation of pTau [164, 165]. Such induction of Aβ and pTau was similarly reported in PBM-derived microglia infected with HHV6A, suggesting the aberrant regulation of pathologically relevant proteins in AD may be maintained amongst HHVs [166].
CONCLUSION, FUTURE DIRECTIONS, AND IMPLICATIONS FOR TREATMENT
As exemplified by contemporary public health challenges, the burdens posed by viral infection as well as AD remain prominent. Despite continued expansion in our aging cohort, the lack of effective treatments for AD warrants further assessment of its etiology. Genomic analyses of comprehensive brain tissue collections, as well as preclinical examinations of Aβ’s antimicrobial responses, have renewed focus on the potential role of latent neurotropic viruses in AD, particularly HHVs [10, 29–31]. Consideration of these advances does not conclude HHVs cause AD, nor do they exclude a role of neurotropic viruses in contributing to or accelerating AD pathogenesis [22]. However, current data reveal a possible “Hit-and-Run” viral theory of AD, as well as potential neurobiological mechanisms that may increase risk for AD following HHV infection (Fig. 1).
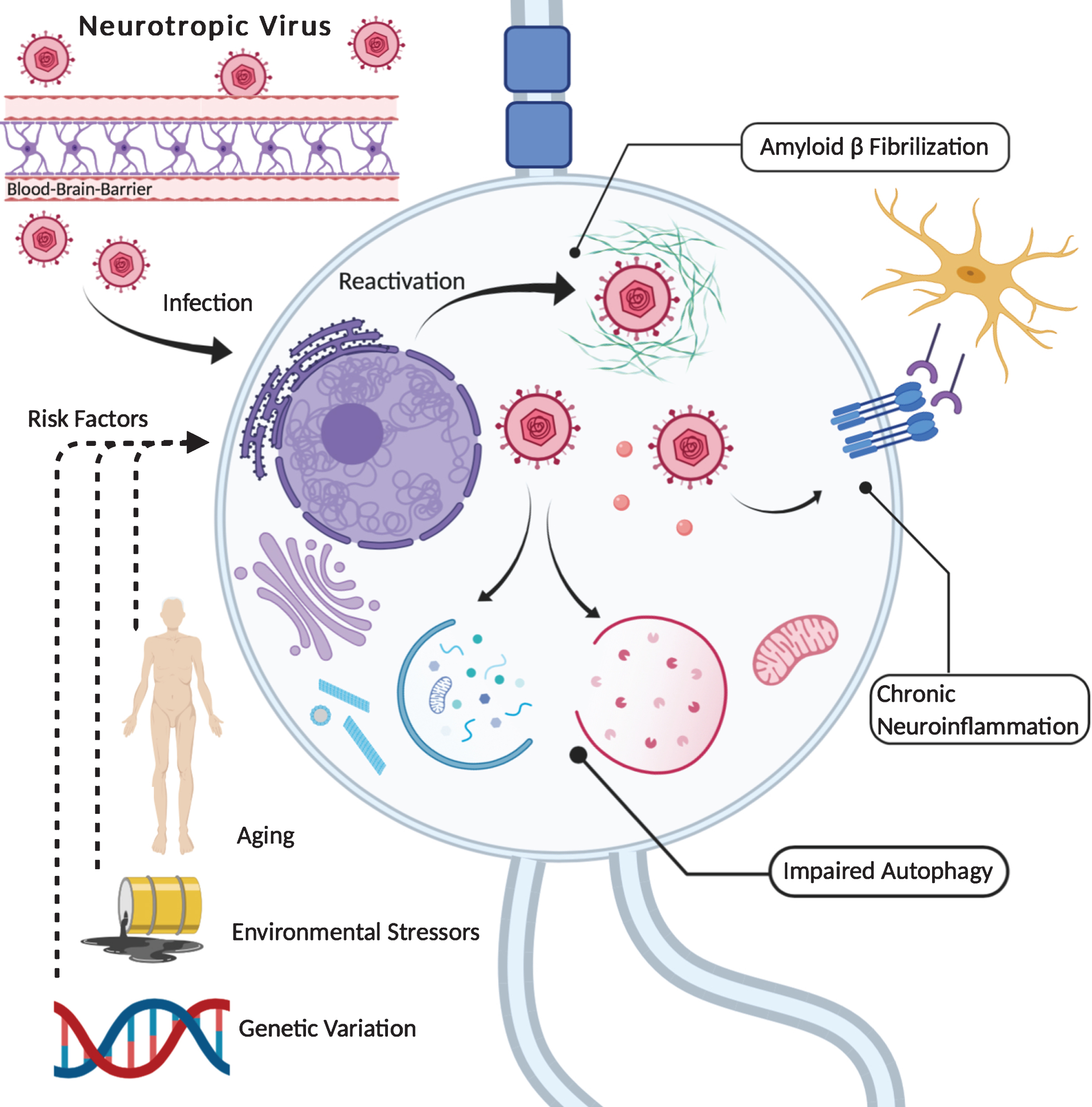
Neurotropic viruses, such as HHVs, penetrate and infect cells within the CNS. Here, they can establish life-long latency accompanied by recurrent reactivation. Along with triggering antimicrobial activities of Aβ (i.e., fibrilization), HHV infection and replication is associated with altered neuroinflammatory profiles (e.g., expression of immune receptors, cytokine signaling) as well as variation in autophagic PQC (e.g., autophagosome dynamics, lysosomal degradation). In addition, infection by herpetic viruses can result in the phosphorylation of pathologically relevant polypeptides (Tau, AβPP), trigger the accumulation of these proteins (pTau, Aβ) and induce elevations in oxidative stress. Interestingly, such effects are similar to observations made in AD, suggesting possible mechanisms by which long-term HHV infection interacts with pathogenic processes in the AD brain. Moreover, reactivation of latent HHVs may be facilitated across an individual’s lifespan by a variety of factors (e.g., environmental toxins, psychological stressors), while genetic predispositions can increase susceptibility to HHV infection as well as AD (e.g., APOE4). Thus, such variables constitute important risk factors in considering a potential HHV theory of AD. Created with Biorender.com. Potential HHV Theory of Alzheimer’s Disease.
Although there are investigations indicating an association between HHV infection and AD, some studies do not support this finding, while others have documented seropositive individuals who do not develop AD [47, 167–172]. To address this discrepancy, future studies are encouraged to assess the role of specific HHV strains and AD, provided that non-pathogenic viral strains can obscure otherwise reliable associations between infection and disease progression [173, 174]. Furthermore, as assessment of viral levels can vary across studies (i.e., host immunoglobulins, integrated DNA, viral transcripts, etc.), and these inconsistent measurements may fail to precisely distinguish states of viremia between studies (i.e., acute infection, reactivation, latency and/or multiple reactivations), it behooves future investigations to employ parallel techniques across analyses in order to enhance the reliability of conclusions [10, 175]. In addition, the cross-sectional designs employed by some investigations may confound the assessment for AD risk following herpetic infection, provided that HHV reactivation(s) over time may facilitate neuropathological progression to AD [71, 153]. Consistent with this postulation, some data indicate elevated HSV immunoreactivity among MCI patients compared to those diagnosed with AD, while significant associations between viral infection and AD risk may only become apparent after extended durations [35, 176]. Thus, the adoption of longitudinal study designs is encouraged to more accurately capture the heterogeneous trajectories and potentially time-dependent risks of AD due to HHVs.
Additionally, some studies do not report proportional variation in cognitive deficits as a result of HHV infection [175]. Given the vast prevalence of HHVs across populations, as well as the asymptomatic nature of latent neurotropic infections, future investigations should screen for infected yet non-demented individuals who might otherwise be categorized as controls and potentially confound neurocognitive assessments [39]. Similarly, investigators should consider if the diversity and abundance of gastrointestinal microbiota contributes to AD among HHV infected persons who otherwise fail to display behavioral impairments [177]. Furthermore, as the vast majority of mechanistic data is currently derived from cell line in vitro models, the inclusion of neuronal and in vivo models is encouraged in order to increase the external validity of results and enhance drug development in AD [178, 179].
The potential role of neurotropic viruses in AD reveals several novel therapeutic targets that merit consideration, including polymerase inhibitors, transcriptase inhibitors, and transferrin-targeting delivery vehicles. Among HSV-1 and HSV-2 infected individuals, the implementation of a DNA polymerase inhibitor (i.e., valacyclovir) is currently being assessed in a pair of Phase II clinical trials for AD. Distinguished by their country of origin (USA versus Sweden), duration (18 months versus 4 weeks) and dosage (4.0 g/day versus 500 mg/day), these investigations will provide pertinent information for AD treatment, especially as adherence to such therapeutics reportedly reduces the risk of dementia by 10-fold in HSV-1 infected individuals and preserves neuronal viability otherwise compromised following HSV reactivation [11, 180]. Examination of nucleoside reverse transcriptase inhibitors may also prove efficacious, provided that the antiretroviral medication lamivudine can mitigate age-related neuroinflammation that is implicated in AD and HHV infection [181]. Such genomic-regulatory approaches in treatment seem appealing, as the delivery of select transcription factors was recently shown to revert aging and AD-associated mechanisms [182]. Bioengineering advances that enhance CNS drug delivery should also be considered, given that the efficacy of antiretrovirals in the CNS is inherently dependent upon penetration across the BBB [183]. In particular, the development of transferrin-targeting immunoglobulins may serve as a reliable drug delivery vehicle to the brain and enable the amelioration of neuroviral contributions to AD [184, 185].
DISCLOSURE STATEMENT
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-0814r2).