
Abstract
Background:
Alzheimer’s disease (AD) is characterized by amyloid-β (Aβ) deposition. The metabolism of Aβ is critically affected by autophagy. Although rifampicin is known to mediate neuroinflammation, the underlying mechanism by which rifampicin regulates the cognitive sequelae remains unknown.
Objective:
Based on our previous findings that rifampicin possesses neuroprotective effects on improving cognitive function after neuroinflammation, we aimed to examine in this study whether rifampicin can inhibit Aβ accumulation by enhancing autophagy in a mouse model of lipopolysaccharide (LPS)-induced cognitive impairment.
Methods:
Adult C57BL/6 mice were intraperitoneally injected with rifampicin, chloroquine, and/or LPS every day for 7 days. Pathological and biochemical assays and behavioral tests were performed to determine the therapeutic effect and mechanism of rifampicin on the hippocampus of LPS-induced mice.
Results:
We found that rifampicin ameliorated cognitive impairments in the LPS-induced mice. In addition, rifampicin attenuated the inhibition of autophagosome formation, suppressed the accumulation of Aβ1–42, and protected the hippocampal neurons against LPS-induced damage. Our results further demonstrated that rifampicin improved the neurological function by promoting autophagy through the inhibition of Akt/mTOR/p70S6K signaling pathway in the hippocampus of LPS-induced mice.
Conclusion:
Rifampicin ameliorates cognitive impairment by suppression of Aβ1–42 accumulation through inhibition of Akt/mTOR/p70S6K signaling and enhancement of autophagy in the hippocampus of LPS-induced mice.
INTRODUCTION
It is well recognized that accumulation of misfolded proteins and aberrant level of inflammatory mediators in the brain are closely associated with the pathogenesis of Alzheimer’s disease (AD). Substantial evidence has indicated that manipulation of inflammatory responses and autophagic flux could improve and control AD progression [1]. It has been shown that the accumulation of proinflammatory cytokines together with the dysfunctional autophagic pathway causes damage to hippocampal neurons, leading to impairment in learning and memory in a manganese-induced AD model [2].
Macroautophagy/autophagy is a lysosome-depen-dent catabolic process for the turnover of proteins and organelles in eukaryotes. Autophagy plays an important role in immunity and inflammation, as well as metabolism and cell survival [3]. Although originally classified as a type of programmed cell death, autophagy is more widely viewed as a basic cell survival mechanism to combat environmental stressors [4]. Defects in the autophagy machinery or inhibition of autophagy have been reported to be associated with neurodegenerative diseases [5]. For instance, it has been reported that chloroquine (CQ), which inhibits the fusion of lysosome with autophagosome for inhibition of autophagy [6], affects the disease outcome in animal models of AD [7]. Substantial evidence has further shown that enhancing autophagy could promote the clearance of aggregated toxic proteins which may have promising potential for the treatment of AD and other neurodegenerative diseases [8].
Lipopolysaccharide (LPS) is a cell-wall immunostimulatory component of gram-negative bacteria which binds to specific receptors to induce the release of free radicals and various immunoinflammatory cytokines such as TNF-α and IL-1β [9]. Upon exposure to LPS, microglia will be activated and produce various proinflammatory mediators which are potentially neurotoxic [10, 11]. Systemic injection of LPS has been shown to be capable of inducing cognitive impairments [12, 13] which was resemble to the development and progression of AD. In addition, the anti-inflammatory drugs were reported to protect against LPS-induced memory impairment through inhibition of both amyloidogenesis and neuroinflammation [14]. Therefore, systemic LPS-induction is regarded as one of the useful animal models for studying AD.
Microglia is the major immune cells of the CNS that express TLR4, while LPS is a major bacterial TLR4 ligand that activates the innate immune response to infections [15]. In response to LPS, mic-roglia activate the autophagy pathway and suppress the expression of iNOS and IL-6, thereby alleviating cell death [16]. A recent study revealed that the anti-inflammatory phenotype of microglia in TLR4-deficient mice is largely abolished by the activation of autophagy, suggesting that autophagy plays a key role in TLR4-mediated neuroinflammation [17]. These studies demonstrate that TLR4 signaling pathway may inhibit autophagy which is required for optimization of neuroinflammatory responses [18].
Rifampicin is an antibacterial agent that is widely used in tuberculosis and leprosy therapy [19]. Previous studies have demonstrated that rifampicin ex-erted immunomodulatory effects [20]. Our recent finding further indicated that rifampicin inhibited microglial inflammation through inhibition of the TLR4 pathway [21]. When neurons were co-cultured with LPS-induced BV2 microglia, pre-treatment with rifampicin increased neuronal viability and reduced the number of apoptotic cells [22]. Interestingly, recent study has also indicated that rifampicin inhibited rotenone-induced microglia-mediated inflammation via enhancement of autophagy [23]. Although several in vitro studies have shown the neuroprotective effects of rifampicin, the evidence of rifampicin on in vivo animal studies is still insufficient. Therefore, in this study we examined the in vivo effects of rifampicin in LPS-induced mice. We showed that rifampicin can, at least in part, improve memory and cognitive function in LPS-induced mice by enhancing autophagy.
MATERIALS AND METHODS
Animals and experiment procedure
Specific pathogen-free male adult (10–12-week-old, 22–30 g) C57BL/6J mice were purchased from Guangdong Experimental Animal Center. Permit numbers: SYXK (YUE) 2012–0117. The animals were kept under a 12 h light/dark cycle with 45–65% environment humidity and the temperature of 22±2°C. Animals had free access to food and water and were allowed to adapt to the laboratory environment for 10 days before behavioral training. The experiments were conducted under the authorization and supervision of the Ethics Committee of the Experimental Animal Care and Use Committee at Jinan University. Mice were randomized into 1) intraperitoneal injection (i.p.) with saline group (Control), 2) i.p. LPS group, 3) i.p. rifampicin (Rif)+ LPS group, 4) i.p. Rif group, 5) i.p. Chloroquine (CQ) group, 6) i.p. CQ + LPS group, 7) i.p. CQ + Rif + LPS group, and 8) i.p. CQ+Rif group. Each group consisted of fifteen male mice and received 7 consecutive days of injections after training. CQ (35 mg/kg) was injected intraperitoneally 30 min before Rif (30 mg/kg), and Rif was administered intraperitoneally 30 min before LPS (750μg/kg). After training, testing was performed everyday (day 0 to day 7). The experimental flowchart is presented in Fig. 1A.
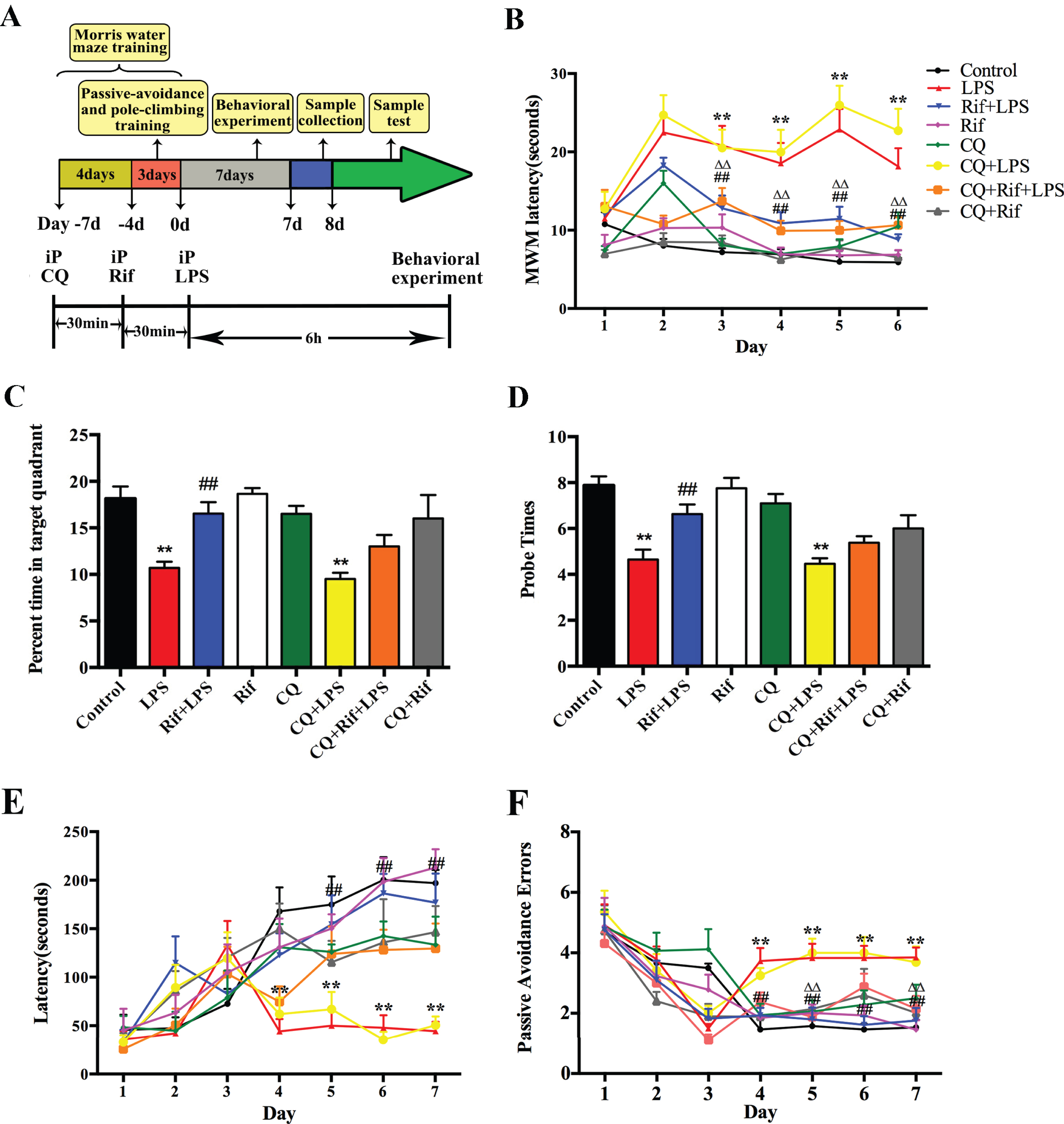
The effect of rifampicin on behavior in LPS-induced mice in the MWM test and PAT test. A) The experimental flowchart. B) Rifampicin can improve LPS-induced memory impairment in mice in the place-navigation test. C, D) Rifampicin can improve the memory function of LPS-induced mice in spatial probe test. E) Rifampicin can improve the step-through latency in Passive Avoidance Test. F) Rifampicin can reduce the error number in Passive Avoidance Test. Each group received 7 consecutive days of injections after training, and all animals were killed and useful tissues were collected on the 8th day of treatments. In the separate experiments, CQ (35 mg/kg) was injected intraperitoneally 30 min before Rif (30 mg/kg), and Rif was administered intraperitoneally 30 min before LPS (750μg/kg). The results are reported as the mean±SEM. * p < 0.05 and ** p < 0.01 versus the control group, # p < 0.05 and # # p < 0.01 versus the LPS group, Δ p < 0.05 and ΔΔ p < 0.01 versus the CQ+LPS group, n = 15.
Morris water maze test
The Morris water maze (MWM) is a classic device for detecting mouse hippocampal-dependent learning, including acquisition of spatial memory and long-term spatial memory [24]. All mice were tested in the MWM program and equipment (purchased from Chengdu Technology & Market Co, LTD). A pool with a 1.2-m diameter was filled with water made opaque with whole milk at 23±2°C. A hidden platform 5 cm in diameter was placed 1.5 cm under the water in a specific position; the location of this platform remains constant from trial to trial. On training trials, the mice were placed in the pool randomly facing the pool wall in the northeast, southeast, southwest, or northwest direction. Each mouse performed 3 trials per day for 7 consecutive days and each trial lasted for 60 s. After training, we administered LPS, Rif, or CQ for 6 h prior to the beginning of the test. On the last day of the test, the mice received a 60-s trial without the platform to assess target quadrant localization.
Passive avoidance performance test
The Passive Avoidance Test (PAT) is a fear-motivated test for the study of long- and short-term memories in an associative manner [25]. It takes advantage of the natural dark preferences of mice. The test was using a “step-through” apparatus (Che-ngdu Technology & Market Co, LTD.) composed of two compartments isolated with a retractable door. One compartment lit with a bright cold house light as the safe compartment while the other made from dark opaque walls and roof as unsafe side. The floor in unsafe side is connected to the 39V electric shock. The mice were placed in the safe but bright side for the training trials during the first three days. All of the mice were placed in the bright side after 6 h of the daily injection and got shocked when they moved completely into the dark side. The latency to enter the dark unsafe side for the first time and total entry error times was observed and automatically recorded. In this test, a 300 s cut-off time was established if the mice did not cross the border.
Immunofluorescence staining
Brains were immersion-fixed in fresh 4% par-aformaldehyde (PFA) in 0.1 M sodium phosphate buffer (PBS) for 24 h, and chilled sequentially in 10% (wt/vol), 20%, and 30% sucrose in 0.1 M PBS at 4°C. After embedding in optimal cutting temperature compound (OCT; SAKURA, Japan), brains were sectioned vertically at 10-μm thickness on a freezing microtome (Leica CM 1850; Leica Microsystems, Seoul, Korea) and incubated at 37°C overnight. Brain sections were blocked in 1% normal bovine serum albumin (BSA) solution for 1 h and incubated overnight at 4°C respective with 1:100 dilution of an antibody against microtubule-associated protein 2 (MAP-2, Millipore Corp, Billerica, MA, USA) and 1:100 dilution of an antibody against amyloid beta 1–42 (Abcam, Cambridge, UK). On the following day, the sections were incubated in a 1:200 dilution of a TRITC donkey anti-rabbit secondary antibody and FITC donkey anti-mouse secondary antibody for 1 h at room temperature. Then the sections were stained with DAPI staining solution for 10 min. Finally, fluorescence images were obtained by using fluorescence microscopy (Leica Microsystems, Wetzlar, Germany). The numbers of MAP 2 and Aβ1–42 positive cells were manually counted and assessed in thirty adjacent sections of the hippocampus by an observer who was strictly blinded to treatment status.
Observation of autophagosome by transmission electron microscopy
To confirm the occurrence of autophagy in the mouse brains, transmission electron microscopy was used to image the ultrastructure of hippocampus. Brain tissues were extracted from cold 4% paraf-ormaldehyde and 2.5% glutaraldehyde solution, followed by rinsing with 0.1 M phosphoric acid rinse solution, and then fixed with 1% osmium acid fixative for 2-3 h. After fixation, brain tissues were dehydrated with ethanol and acetone. Following dehydration and embedding, brain tissues were cut by LKB-1 ultrathin slicing machine. Finally, the samples were imaged under the transmission electron microscope. Morphological criteria used for identification of autophagosome/autophagic vesicles were based on their ultrastructural characteristic as described [26]. They included circular vesicles with 300–900 nm in diameter, a contrast with structures that were white or lighter than the cytoplasm, a complete cytoplasmic content and a double limiting membrane, closed single membrane-bound vacuoles containing partially disintegrated contents.
Western blot analysis
Mice were sacrificed and the brains were taken out on the ice plate quickly. The brain tissues were homogenized in RIPA lysis buffer (Bioteke Co, Beijing, China) containing 1 mM phenylmethylsulfony fluoride. After centrifugation for 15 min at 12,000×g at 4°C, the protein content of the supernatant was determined with a BCA Kit (Bioteke Co, Beijing, China). Aliquots of protein were denatured by incubation in Laemmli buffer (Sigma) for 10 min at 100°C and subjected to SDS-polyacrylamide gel electrophoresis (PAGE) using a 6–15% gradient gel (BioRad, Cressier, Switzerland). Proteins were transferred onto a 0.22μm polyvinylidene fluoride (PVDF) membrane (Biorad) and blocked in 5% non-fat dried milk for 1 h. The blots were incubated overnight at 4°C with specific antibodies against LC3, Bcl-2, Beclin1, P70S6K, p-P70S6K, Akt, p-Akt, mTOR, p-mTOR (1:1000 dilution, Cell signaling Technology Inc, MA, USA), then the membranes were treated by 1:5000 dilution of horseradish perox-idase-conjugated secondary antibody (Cell signaling Technology Inc, MA, USA) for 1 h at room temperature. Finally, signals were measured with an enhanced chemiluminescence kit (ECL, Millipore, USA) on a gel imaging system (Millipore, Billerica, MA, USA), and the results were visualized using Quantity One software.
Statistical analysis
Data analysis was performed using SPSS 19 statistical software. Data are presented as the mean±SEM and were analyzed using one-way ANOVA followed by Tukey’s honest significant difference test. Statistical significance was accepted at p < 0.05.
RESULTS
Rifampicin ameliorates cognitive impairments in LPS-induced mice
To validate our LPS-induced cognitive impairm-ent in mouse model, we conducted the MWM test to evaluate the learning and memory functions of the animals (Fig. 1A). As shown in Fig. 1B, escape latency in LPS-induced group was significantly longer than that in control group since day 3 of the acquisition phase (p < 0.01), suggesting the success of the model. Emerging evidence has revealed that autophagy plays a critical role in neuroinflammation and AD [2]. To verify the role of autophagy in the observed LPS-induced cognitive functional deficit, we treated the animal with CQ, a known inhibitor of autophagy [6], before LPS injection as described in the Materials and Methods. The result of MWM test showed that the performance of CQ+LPS group was worser than LPS group, suggesting that autophagy played a positive role in protection against LPS-induced cognitive deficit. To examine the possible ef-fect of rifampicin in this model, we pre-treated the LPS-induced mice with rifampicin. Remarkably, the escape latency was significantly reduced (p < 0.01), indicating that rifampicin could improve memory impairment. To determine whether autophagy mediated the observed effect of rifampicin (Rif), we pre-treated mice with CQ and found that the escape latency of CQ+Rif+LPS group was longer than that of Rif+LPS group. These results suggest that autophagy is required for the rifampicin-mediated protective function against LPS-induced cognitive deficit. In the testing trial on day 7, the mice of LPS group and CQ+LPS group spent significantly less time in the quadrant of the platform than those of the control group (Fig. 1C, D). In the Rif+LPS group, the number of platform crossings and time spent in the target quadrant increased significantly compared with the LPS-treated groups in the probe trial. In addition, treatment of CQ reduced the memory recovery function conferred by rifampicin treatment (Fig. 1C, D). These results suggest that rifampicin improve memory function in an autophagy-dependent manner.
We further confirmed the beneficial effect of rifampicin on memory improvement by the Passive Avoidance Test. As expected, the LPS-induced mice showed a significantly reduced step-through latency compared with those in control group (p < 0.01), and the passive avoidance error number was significantly increased from day 3 onward (Fig. 1E, F). We found that treatment of mice with rifampicin significantly improved the step-through latency in the Passive Avoidance Test and CQ abolished the observed effect of rifampicin (Fig. 1E, F). These results suggest that rifampicin significantly improve memory function in the LPS-induced mice, and that the protective effect of rifampicin is dependent on autophagy.
Rifampicin attenuates the neuronal damage and accumulation of Aβ1–42 in the hippocampus of LPS-induced mice
The hippocampus plays a key role in memory and cognitive function [28]. It is reported that cholinergic neurons in the hippocampus are vulnerable to LPS treatment [27]. To explore the effect of rifampicin on LPS-induced neuronal impairment in the hippocampus, we examined whether LPS induced neuronal cell loss by MAP2 immunofluorescence staining. As shown in Fig. 2, LPS induced a dramatic reduction of neuron number in the hippocampus. However, the neuronal loss induced by LPS could be significantly reduced by rifampicin pre-treatment (Fig. 2). This result indicates that rifampicin is neuroprotective in the LPS-induced mice.

Rifampicin effects on LPS-induced hippocampus neuron impairment in the hippocampus. Immunostaining of MAP2 (red) protein in the hippocampus with primary antibody, quantified images of n = 30 per group. Statistical analysis of the percent of MAP2 positive cells in the hippocampus. * p < 0.05 and ** p < 0.01 versus the control group, # p < 0.05 and # # p < 0.01 versus the LPS group.
We have previously found that LPS induces Aβ1–42 accumulates in the hippocampus [29]. Therefore, we asked whether Aβ1–42 was involved in the neuroprotective effect of rifampicin. We first con-firmed that LPS prominently increased Aβ1–42 accumulation in the hippocampus (Fig. 3). Interestingly, rifampicin treatment reduced Aβ1–42 to a level similar to the basal level (Fig. 3). This result demonstrates that rifampicin prevents the toxic Aβ1–42 accumulation in the hippocampus induced by LPS.
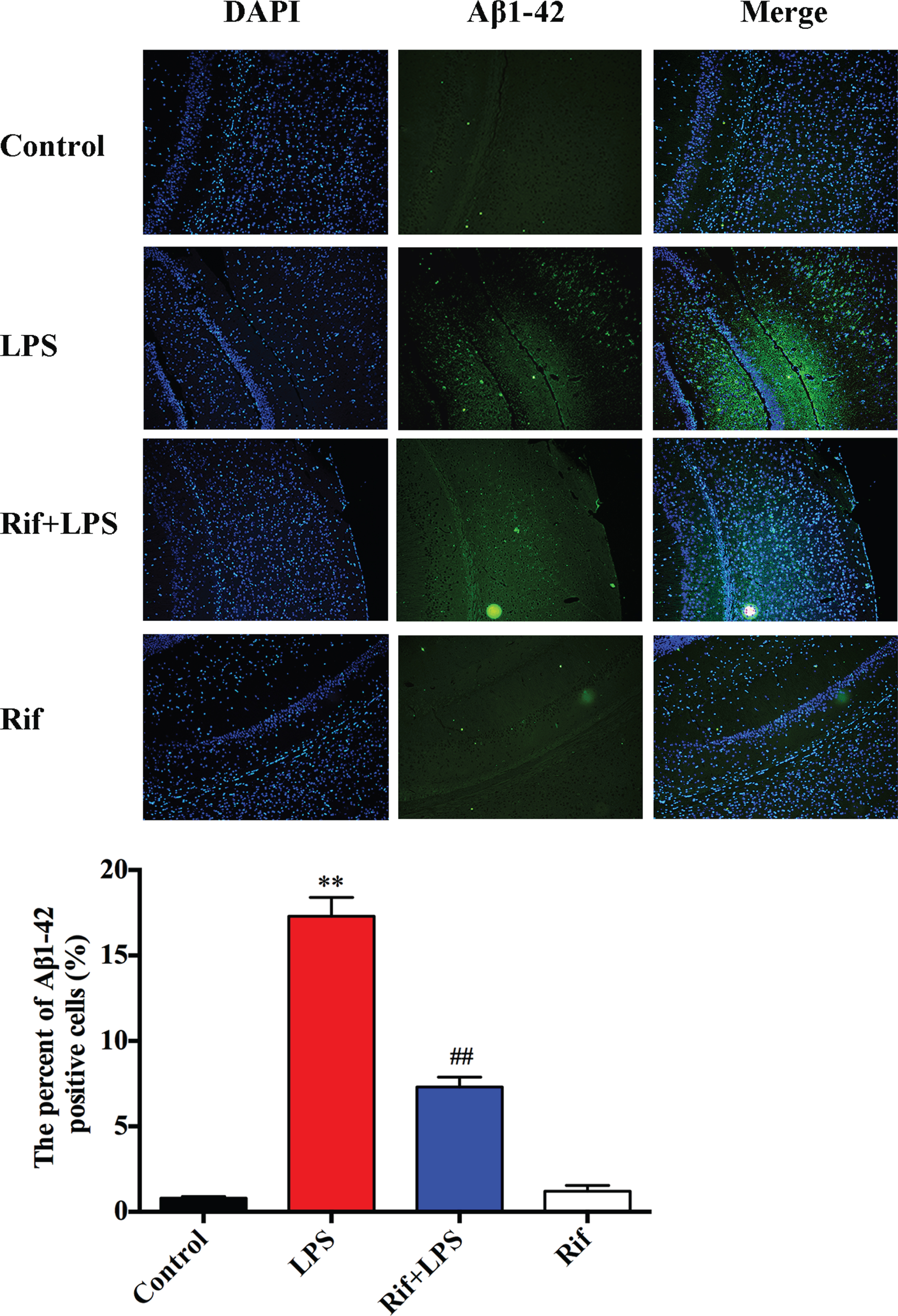
Rifampicin effects on LPS-induced Aβ1–42 accumulation in the hippocampus. Immunostaining of Aβ1–42 (green) protein in the hippocampus with primary antibody, quantified images of n = 30 per group. Statistical analysis of the percent of Aβ1–42 positive cells in the hippocampus. * p < 0.05 and ** p < 0.01 versus the control group, # p < 0.05 and # # p < 0.01 versus the LPS group.
Rifampicin restores the autophagosome formation in the hippocampus of LPS-induced mice
A large body of evidence has shown that activation of autophagy is crucial for removing neurotoxic substances including Aβ1–42 [8]. To determine whether autophagy played a role in the effect of rifampicin, we used electron microscopy to examine autophagosome formation in hippocampal neurons. Under electron microscopy, autophagic vacuoles/autophagosomes were defined according to the criteria including circular vesicles with 300–900 nm in diameter, a contrast with structures that were white or lighter than the cytoplasm, intact cytoplasmic content and a double limiting membrane, closed single membrane-bound vacuoles containing partially disintegrated contents [26]. As shown in Fig. 4, we observed the autophagosomes in hippocampal neurons in control mice. However, we hardly found any autophagosome in the LPS-treated mice. Interestingly, mice treated with rifampicin promoted the formation of autophagosomes in hippocampal neurons (black solid arrow). Based on the morphology, it seemed that rifampicin particularly promoted macroautophagy. These observations suggest that LPS blocks normal autophagic process while rifampicin ameliorates the inhibitory effect of LPS on autophagy.

Rifampicin enhances LPS-induced mice autophagic flux in hippocampus. Transmission electron microscopy was employed to detect the ultrastructure of cells in hippocampus. The black dotted arrow points to the structure of autophagosomes, quantified images of n = 5 per group.
Rifampicin restores autophagy inhibited by LPS in the hippocampus
The enhancement of neuronal survival, reduction of Aβ1–42 accumulation and restoration of auto-phagosome formation by rifampicin in the hippocampus of mice suggested that its neuroprotective effect may be mediated by the activation of autophagy. To test this hypothesis, we determined the autophagic flux in hippocampus by examining the expression of LC3 and p62 after different treatments. Autophagic flux can be determined by the upregulation of LC3-II expression and downregulation of P62 expression [30]. Compared with control, the expression of LC3-II in LPS group was significantly decreased while the level of p62 was increased, suggesting a reduction of autophagic flux (Fig. 5A, B). Strikingly, treatment of rifampicin dramatically enhanced LC3-II level and reversed P62 expression level to basal level (Fig. 5A, B). This result suggested that rifampicin restored the basal autophagic flux in the LPS-induced mice. We further found that this effect of rifampicin was abolished by CQ (compared CQ-Rif-LPS with Rif-LPS groups) (Fig. 5B). Therefore, these results are consistent with our electron microscopic observations, and further support the notion that rifampicin abolishes the autophagy-inhibitory effect by LPS, and that rifampicin reverses the LPS-induced effect in an autophagy-dependent manner.
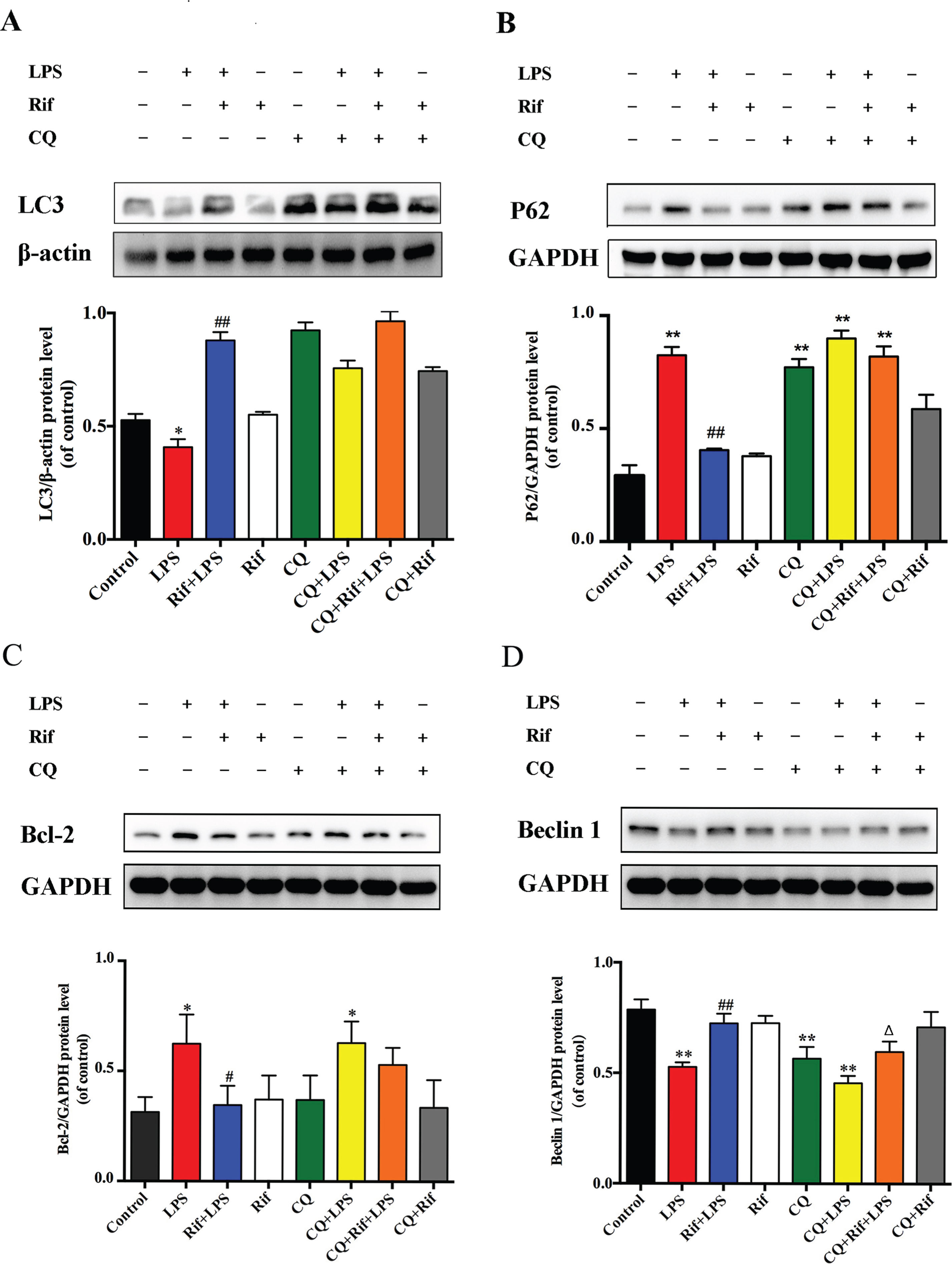
Rifampicin effects on LC3, P62, Bcl-2, and Beclin 1 proteins expression in the brain of LPS-induced mice. Representative western blot bands of LC3, P62, Bcl-2, and Beclin 1 protein expression in the brain. The LC3 protein has two bands: the upper band is LC3-I and the lower band is LC3-II. Quantitation of LC3, P62, Bcl-2, and Beclin 1 protein expression in the brain of mice in each group. The vertical axis represents the ratio of protein to β-actin or GAPDH gray levels. Data are presented as the mean±SEM. * p < 0.05 and ** p < 0.01 versus the control group, # p < 0.05 and # # p < 0.01 versus the LPS group, Δ p < 0.05 and ΔΔ p < 0.01 versus the CQ + LPS group, n = 6.
Beclin 1 and Bcl-2 are involved in rifampicin-regulated autophagy
Beclin 1, a yeast homologue Atg6, is a multifunctional protein that is required for the autophagic process [31]. It has been reported that the antiapoptotic protein Bcl-2 inhibits autophagy through acting on Beclin 1 protein [32]. To examine whether Beclin 1 and Bcl-2 were involved in LPS-regulated autophagy, we analyzed their protein expression levels in the hippocampal tissue. As shown in Fig. 5C and D, the LPS group indeed showed significantly higher level of Bcl-2 and lower level of Beclin 1 expression than the controls. Importantly, rifampicin pretreatment restored Bcl-2 and Beclin 1 levels in the hippocampal tissue of mice induced by LPS, and these changes were abolished by CQ treatment. Collectively, these results suggest that Beclin 1 and Bcl-2 are involved in the regulation of rifampicin-mediated autophagy restoration in LPS-induced mice.
Rifampicin inhibits the activation of Akt/mTOR/p70S6K signaling pathway in the brains of LPS-induced mice
The results above suggested that rifampicin improved LPS-induced cognitive impairments by enhancing autophagy. We next attempted to explore a more detailed underlying regulatory mechanism. Since the Akt/mTOR/p70S6 pathway is well-known to inhibit autophagy [33], we examined the effect of rifampicin and LPS on this pathway. As shown in Fig. 6, we found that the level of phosphorylated Akt (p-Akt) in the hippocampal tissue of LPS-induced mice were elevated compared with control. In addition, the expression of p-mTOR and p-p70S6K, the downstream signaling components of Akt pathway, were significantly increased (Fig. 6). However, pretreatment with rifampicin significantly restored the expression of p-Akt, p-mTOR, and p-p70S6K (Rif+LPS group), indicating that rifampicin may act on or upstream of Akt for the inactivation of this pathway. Altogether, these results suggest that rifampicin plays a neuroprotective role by inhibiting the activation of Akt/mTOR/p70S6K signaling pathway to enhance autophagy in LPS-treated mice.
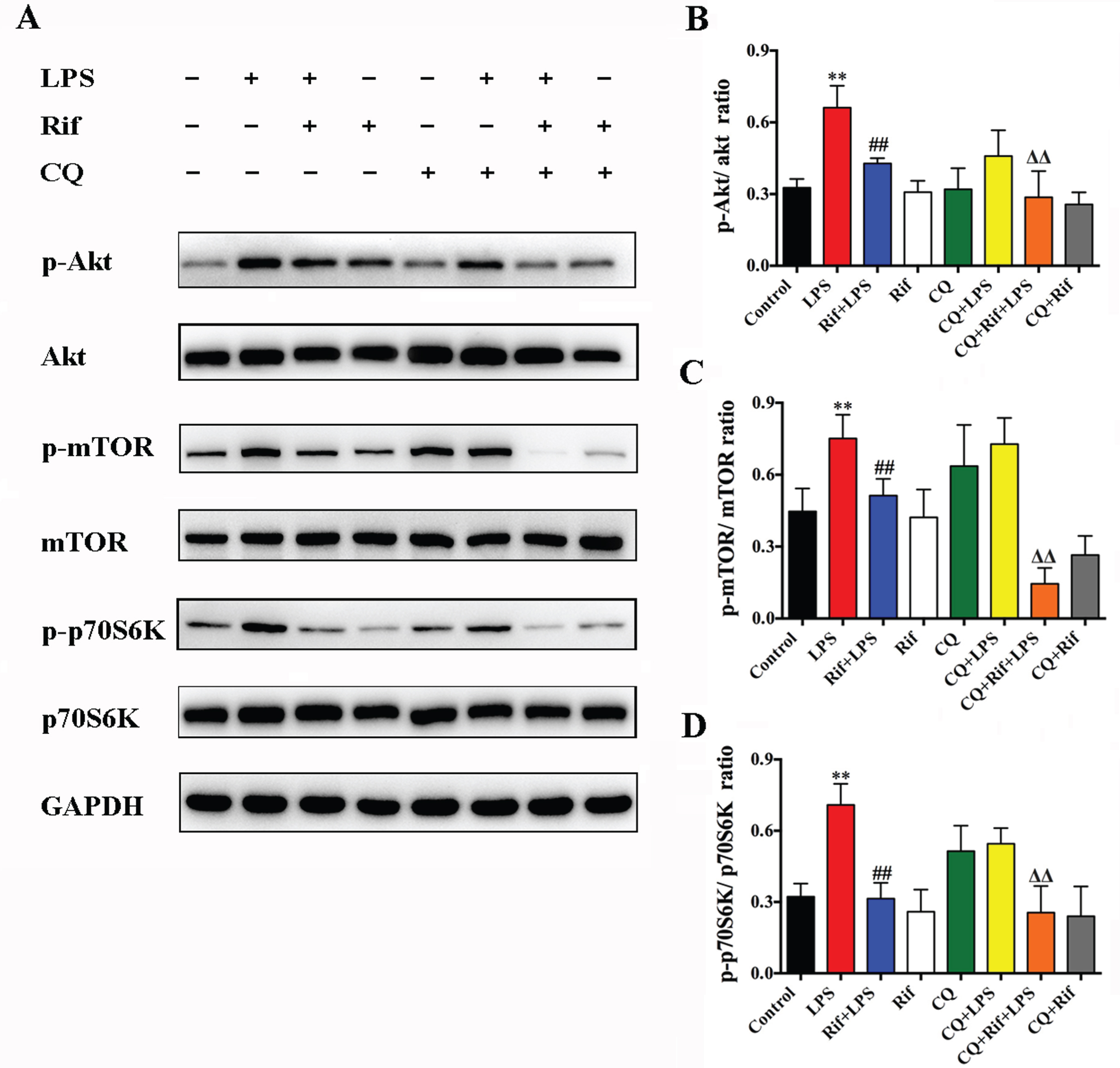
Rifampicin enhanced autophagy was associated with the inhibition of the Akt/mTOR/p70S60k pathway. The levels of the phosphorylated and total Akt, mTOR, and p70S6k proteins were examined. GAPDH was used as a loading control. Data are presented as the mean±SEM. * p < 0.05 and ** p < 0.01 versus the control group, # p < 0.05 and # # p < 0.01 versus the LPS group, Δ p < 0.05 and ΔΔ p < 0.01 versus the CQ+LPS group, n = 6.
DISCUSSION
In this study, we used LPS-induced neuroinflammatory in vivo model to demonstrate for the first time that rifampicin improves memory and cognition functions by enhancing macroautophagy and reducing Aβ accumulation in the hippocampus. In recent years, autophagy has gained considerable attention in neurodegenerative diseases because of its involvement in multiple key biological processes. For example, dysregulation of autophagy has been linked to the accumulation of misfolded proteins that are associated with neuronal death in these disorders [34]. Emerging evidence has further indicated that autophagy and neuroinflammation are closely correlated and there may be a crosstalk between them [35]. With respect to rifampicin, numerous reports have shown that this anti-inflammatory drug exhibits prominent immunosuppressive effects [19–22 , 37]. Recent findings further revealed that rifampicin could inhibit Aβ aggregation and decrease reactive oxygen species and has potential for AD treatment [38]. Tomiyama et al. also reported that rif-ampicin-mediated inhibition of Aβ aggregation and neurotoxicity involves scavenging of free radicals [39]. In addition, upregulation of LRP1 and P-gp at the blood-brain barrier by rifampicin enhances brain Aβ clearance, and this effect could explain, at least in part, the protective effect of rifampicin against AD [40]. In addition to AD, Liang et al. provided evidence that rifampicin exerts neuroprotection against rotenone-induced microglia inflammation, partially through the autophagy pathway, and suggested that modulation of autophagy by rifampicin is a novel therapeutic strategy for Parkinson’s disease [23].
Rifampicin has previously been shown to ameliorate lithium-pilocarpine-induced damage in the hippocampus and memory deficit in rats [37]. Rifampicin-treated AlCl3 rats exhibited significant attenuation in memory deficits [41]. Our previous study has shown that LPS treatment leads to cognitive impairment in mice which is accompanied by microglia activation [29]. Together with our present study, these results indicate that microglia-mediated neuroinflammation and systemic autophagy inhibition could induce cognitive impairment, and rifampicin reduces LPS-induced cognitive impairment by enhancing autophagy in the hippocampus.
Compared with other brain regions, microglia in the hippocampus shows a higher proliferative capacity in response to LPS-induced neuroinflammation [42]. Epidemiological studies have revealed that patients with leprosy treated with rifampicin showed a significant reduction of Aβ deposition and lower incidence of dementia than patients without receiving rifampicin treatment [43]. Here, we showed that rifampicin significantly reduces the Aβ1–42 deposition and neuronal loss in LPS-induced mice. We further demonstrate that autophagy plays a key role in the mechanism of rifampicin-mediated improvement of cognitive disorders. Obviously, the underlying mechanisms by which rifampicin reduces Aβ1–42 deposition is complex and is likely involve multiple signaling pathways for the regulation of cellular processes include but not limit to inflammatory responses and autophagy. Further detailed investigation is required for fully elucidate the mechanisms.
LC3 and its family of proteins play an important role in the extension and closure of the isolation membrane. These proteins bind to the surface of autophagic membranes and thus they are important markers of autophagosomes [44]. They exist in the inner and outer membranes of the autophagosome at various stages. The LC3-II/β-actin ratio can reflect changes in autophagy [45]. On the other hand, P62/SQSTM1 is the selective cargo receptor for autophagy to degenerate misfolded proteins, which is metabolized through autophagic lysosome pathway. Its changes can indicate the activity of autophagic process [46]. By analyzing LC3 and P62 expression, our findings indicated that rifampicin promotes autophagy in the brain of LPS-induced mice (Fig. 5). Consistent with our findings, it has been reported that rifampicin significantly enhances the ratio of LC3-II/LC3-I in BV2 cells [41]. Together with our electron microscopic observation (Fig. 4), rifampicin seems to alleviate macroautophagy inhibition in LPS-induced mice. Mechanistically, we showed that Beclin and Bcl-2 are involved in the observed rifampicin’s action on autophagy regulation. Whether rifampicin directly interacts with these proteins and also mediates other types pf autophagy requires further investigation in the future.
Akt/mTOR/p70S6K signaling pathway is a major negative regulatory pathway of autophagy and is also a key homeostatic regulatory pathway of cell growth, proliferation, and survival [33]. Akt normally exists in the cytoplasm, and its upstream factor PI3K is mainly regulated by growth factors. Akt regulates a wide range of functions, such as autophagy, proliferation, death, and oxidation through its downstream proteins include mTOR, Bad, and Caspase 9. mTOR is a sensor for cell nutrient status, stress, and growth factor that plays an important role in autophagy [47]. When mTOR is activated, it stimulates the activation of downstream p70S6K to form phosphorylated p70S6K, which then promotes protein synthesis, proliferation, and growth, accelerates cell metabolism, and inhibits autophagy [48]. In this study, we found that in the LPS-treated mice, Akt/mTOR/p70S6K signaling activity in the brain tissue is regulated by rifampicin. Currently, the exact molecular targets of rifampicin in different brain cells remained to be elucidated. Understanding the underlying mechanisms in more detail in the future studies would be important for fully exploring the therapeutic potential of rifampicin in diverse neurological disorders.
CONCLUSION
Rifampicin can promote the clearance of Aβ1–42, rescue the neurons, and improve the neurological performance in the LPS-induced mice. The protective mechanism may be related to inhibition of Akt/mTOR/p70S6K signaling pathway and enhancing autophagy. The results obtained in this study may further support the therapeutic potential of rifampicin in the treatment of neuroinflammation and neurodegenerative diseases such as AD.
Footnotes
ACKNOWLEDGMENTS
This study was supported by grants from the Natural Science Foundation of China (Nos.81200930, 82071568, 81974210), the Training program for outstanding young teachers in higher education institutions of Guangdong Province (Nos. YQ2015024), the Guangdong Natural Science Foundation (2019A1515010671) and the Fundamental Research Funds for the Central Universities (Nos. 21617482).