
Abstract
Background:
To date, it remains unclear how amyloid plaques and neurofibrillary tangles are related to neural activation and, consequently, cognition in Alzheimer’s disease (AD). Recent findings indicate that tau accumulation may drive hippocampal hyperactivity in cognitively normal aging, but it remains to be elucidated how tau accumulation is related to neural activation in AD.
Objective:
To determine whether the association between tau accumulation and hippocampal hyperactivation persists in mild cognitive impairment (MCI) and mild dementia or if the two measures dissociate with disease progression, we investigated the relationship between local tau deposits and memory-related neural activation in MCI and mild dementia due to AD.
Methods:
Fifteen patients with MCI or mild dementia due to AD underwent a neuropsychological assessment and performed an item memory task during functional magnetic resonance imaging. Cerebral tau accumulation was assessed using positron emission tomography and [18F]-AV-1451.
Results:
Entorhinal, but not global tau accumulation, was highly correlated with hippocampal activation due to visual item memory encoding and predicted memory loss over time. Neural activation in the posterior cingulate cortex and the fusiform gyrus was not significantly correlated with tau accumulation.
Conclusion:
These findings extend previous observations in cognitively normal aging, demonstrating that entorhinal tau continues to be closely associated with hippocampal hyperactivity and memory performance in MCI and mild dementia due to AD. Furthermore, data suggest that this association is strongest in medial temporal lobe structures. In summary, our data provide novel insights into the relationship of tau accumulation to neural activation and memory in AD.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) typically presents with progressive loss of episodic memory and is histopa-thologically characterized by amyloid plaques and neurofibrillary tangles. Amyloid deposition is ass-umed to precede the accumulation of pathological tau protein, followed by neuronal injury, and eventually, the mnestic and cognitive decline [1, 2]. In this context, the interaction between neuronal activation, pathological protein aggregates, and mnestic and cognitive functions remains elusive.
Individuals at risk of developing AD, because of genetic mutations associated with familial AD [3, 4] or apolipoprotein E4 positivity [5, 6], have consistently been demonstrated to exhibit increased neural activation in the hippocampus compared to non-car-riers. While this observation suggests that amyloid-β (Aβ) accumulation may play a role in hippocampal hyperactivation in cognitively healthy individuals, findings regarding the association between Aβ and hippocampal functional magnetic resonance imaging (fMRI) activation in cognitively healthy older adults have been ambiguous [7–11]. Recent multimodal imaging studies measuring amyloid and tau accumulation, as well as hippocampal activation, in cognitively healthy seniors [10, 13] provide a possible explanation: hippocampal hyperactivation may be more closely related to the tau accumulation that follows amyloid build-up, than to the amyloid deposition [12].
At the stage of mild cognitive impairment (MCI), findings regarding changes in the hippocampus’s neural activation are less consistent than in cognitively normal individuals. Using fMRI, several studies re-ported increased hippocampal activation during me-mory tasks compared to cognitively healthy controls [14–18], while others did not [19–21]. This discrepancy has been attributed to different disease stages [22] and the amyloid status [23].
Furthermore, it remains to be investigated which role tau accumulation plays concerning memory relevant neuronal activation in AD as cognitive deficits become apparent. Changes in neural activation could reflect compensation [9, 15] or disease propagation [24, 25]. Given conflicting findings regarding mem-ory-related neural activation in MCI, it is conceivable that tau accumulation continues to be positively correlated with neuronal activation in the presence of cognitive deficits. However, it is also possible that this relationship disappears as an increasing number of neurons is lost.
Using fMRI of a visual item memory task and non-invasive in vivo imaging of tau accumulation with [18F]-AV1451-PET, we, therefore, investigated in a group of AD patients, whether the positive correlation between tau accumulation and hippocampal hyperactivation previously described in cognitively healthy older adults persists in the presence of mnestic and cognitive deficits. Generally, this visual memory task detects the subsequent memory effect (SME) on neural activation, i.e., greater activation for subsequently remembered stimuli than for forgotten stimuli in the hippocampus and the fusiform gyrus and deactivation of posterior midline structures [26]. Using this task, we have previously compared patients with MCI and cerebrospinal fluid (CSF)-biomarkers indicative of AD to age-matched controls: MCI was associated with a smaller SME on activation in the hippocampus and fusiform gyrus and less task-related deactivation in the posterior cingulate cortex (PCC) [27]. However, to date, it is unknown how changes in task-related neural activation are related to local or global tau accumulation.
We hypothesized that entorhinal tau accumulation in AD patients is correlated with episodic memory function and that this tau accumulation is correlated with visual item memory task-induced neural activation. To determine whether the association between tau accumulation and neuronal activation is specific to the medial temporal lobe structures, we also investigated this relationship in the fusiform gyrus, which typically activates during encoding of visual content, and the posterior cingulate cortex, which typically deactivates during encoding of visual content [26–29].
METHODS
Participants
Twenty patients recently diagnosed with MCI or mild dementia due to AD (13 male, 7 female, 55–82 years old), who had undergone an AV-1451-PET during clinical work-up, were recruited. Five patients had to be excluded from further analyses because they were unable to perform the functional task (two) or due to excessive head motion during fMRI (three). Thus, the data of 15 patients entered further analyses (Table 1).
Summary demographics and neuropsychological performance
MMSE, Mini-Mental-Status Exam; GDS, Geriatric Depression Scale; Verbal memoryDR, delayed recall of the Verbal Learning Memory Test; Verbal memoryLoss, immediate recall –delayed recall of the Verbal Learning Memory Test; ROCFLoss, immediate recall - delayed recall of the Rey Osterrieth Complex Figure Test; ROCFDR, delayed recall of the Rey Osterrieth Complex Figure Test; TMT, Trail Making Test; CSF, cerebrospinal fluid. Either Amyloid-PET or CSF data were available from each participant. Standard deviations are presented in parentheses.
The main inclusion criteria were positive biomarkers indicative of AD pathology and signs of neuronal injury, meeting the criteria for a high likelihood of AD [30, 31]. Evidence of neuronal injury was operationalized as 1) medial temporal atrophy according to the medial temporal atrophy scale [32] on T1-weighted images, 2) temporoparietal and precuneal hypometabolism in fluorodeoxyglucose-PET quantified with 3D-SSP [33], or 3) elevated total tau-protein in CSF (>375 pg/ml). Positive amyloid pathology was defined as CSF Aβ1–42 <550 pg/ml, a tau/Aβ1–42 ratio > 0.52 [34], or a positive amyloid PET during clinical work-up. Importantly, all patients also exhibited strong AV-1451 binding in temporoparietal regions. Exclusion criteria were neurological conditions besides AD, major psychiatric disease, other medical conditions that could affect cognition, inability to give informed consent, MRI exclusion criteria such as claustrophobia, cardiac pacemakers, and other non-MR-compatible implants. Furthermore, patients were excluded if structural lesions were detected on cerebral imaging.
All participants underwent a neurological examination by a neurologist and a comprehensive neuropsychological assessment, as previously described [27]. All patients predominantly had memory impairments, but the majority also had some executive deficits and lesser language impairments.
Independence in activities of daily living was assessed using the history given by caregivers and with the Bayer Activities of Daily Living Scale (cut-off<5). Based on these measures, four patients were demented. The four patients with dementia did not differ significantly from the MCI patients with respect to age (t (13) = 0.112, 95% -CI [–8.714, 9.669]), education (t (13) = 1.122, 95% -CI [–2.123, 6.713]), performance on the MMST (t (13)) = 0.553, 95% -CI [–2.838, 4.793]), or gender (χ2 (1) =0.170, p = 0.680).
Disease severity of the demented patients was defined based on the history given by caregivers and the MMSE [35, 36].
Twelve patients took cholinesterase inhibitors; th-ree patients also took antidepressant medication. All doses of medication had been stable during the weeks before fMRI scanning. The average delay between the Tau-PET and the fMRI was 207.47 days (standard deviation 191.71 days).
The ethics committee of the medical faculty of the University of Cologne approved the study. Written in-formed consent was obtained from all participants before the study following the Declaration of Helsinki.
Procedure
Clinical, neuropsychological, and MRI data were obtained within a month. During fMRI scanning, participants performed a visual item memory task consisting of an encoding (duration: 10 min) and a retrieval phase (duration: 17 min) spaced seven minutes apart (Fig. 1). During encoding, a fixation cross was presented at the center of the screen, where 80 photographs of natural or artificial items were presented. Items were shown for 3.5 s each, with an inter-trial-interval of 1s, in random order. Forty null-events, showing only the fixation cross, were randomly in-terspersed, resulting in variable stimulus-onset asynchronies. Participants were asked to memorize each object and indicate via button press with the index or middle finger of the right hand, whether the object was “natural” such as an animal, fruit, or vegetable, or “artificial”, i.e., human-made or modified. During the retrieval period, a fixation cross was again present at the center of the screen. Eighty stimuli were presented during encoding, and 40 new objects were shown in random order for 3.5 s each, with an inter-trial-interval of 2 s. Stimuli were displayed using the software Presentation (Neurobehavioural systems, Albany, CA, USA) via a screen situated behind the scanner. Participants viewed the screen via a mirror mounted on the head coil. Participants were in-structed to indicate via button press with the middle or index finger of their right hand, whether the object was “old”, i.e., had been presented during encoding, or “new”, i.e., had not been presented previously. During the retrieval period, 60 null-events were interspersed randomly. The task was rehearsed outside and inside the MR scanner before scanning. For this rehearsal, a shortened version of the paradigm and a separate set of stimuli were used.
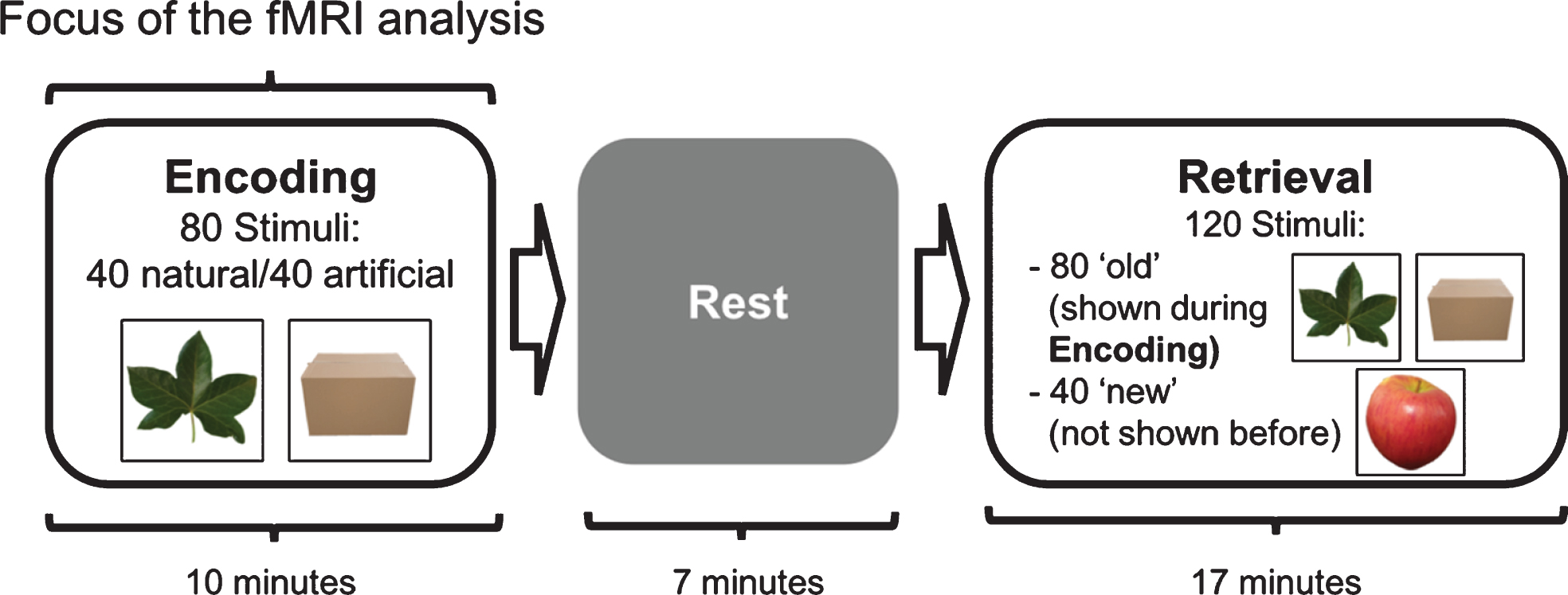
Schematic representation of the visual item memory task illustrating the order of task phases. The encoding and retrieval phases were separated by a 7-minute rest period. Also depicted are example visual stimuli. During encoding, participants were instructed to memorize stimuli and indicate whether the stimuli are natural or artificial via a button press. During retrieval, participants indicated via a button press if the stimuli were ‘old’ (presented during encoding) or ‘new’ (not previously presented).
Behavioral data analysis
Trials with reaction times less than 400 ms or more than two standard deviations greater than the mean were considered outliers and classified as invalid trials together with missed trials. The analysis focused on items that were correctly classified during the retrieval session. The performance was assessed using the sensitivity index d’, the difference between hit rate (H; previously seen items classified as “old” divided by all previously seen items) and false-alarm rate (F; new items classified as “old” divided by all new items) in standard deviation units [37]: d’=Φ–1(H’)–Φ–1(F’). Since the performance in this task is not solely dependent on hippocampal memory formation but also stimulus processing [26], we additionally derived two modality-independent measures of memory performance from the neuropsychological assessment: 1) Performance on the delayed recall of memory tests as a parameter most sensitive to deficits in episodic memory [38] and, 2) the number of items forgotten during the delay period (information initially reproduced minus information reproduced in the delayed recall) as an estimate for memory consolidation and resilience to interference [38–40]. Composite delayed recall and memory loss scores were computed from visual and verbal memory scores to generate measures independent of the type of content. Specifically, raw test scores from the VLMT (the German version of the Rey Auditory Learning Test [38]), and the Rey-Osterrieth Complex Figure Test [41] were z-transformed and averaged.
Neuropsychological, demographic, and ROI measures were normally distributed as assessed with Shapiro-Wilks tests. The delay between PET and MRI measurements, as well as medication, were not normally distributed. All statistical analyses not performed at the voxel level were conducted with SPSS (Version 24.0, IBM Corp., Armonk, NY). In the present sample, none of the neuropsychological measures or values from ROIs that entered statistical analyses, except for the cortical thickness of the entorhinal cortex, were correlated with age. To account for the large number of correlations that were computed, correction for multiple comparisons was performed by controlling the false discovery rate (FDR) [42]. Correlations significant after FDR-correction (p < 0.0068) for multiple comparisons are highlighted in the Results section.
Significant correlations of tau accumulation with fMRI or cognitive measures were also computed as partial correlations, correcting for the delay between MRI and PET measurements and psychoactive medication. Likewise, significant correlations between cognitive and fMRI measures were computed as partial correlations, corrected for psychoactive med-ication. Statistical values for correlations that were not significant can be found in Supplementary Table 1.
All statistical tests were performed using robust methods with bootstrapping to account for the small sample size and non-normal distribution of a subset of variables. Results are, therefore, presented with 95% confidence intervals (CI).
MR and PET acquisition
The PET scans were collected using a PET-CT Siemens Biograph mCT Flow 128 Edge (Siemens). A low-dose transmission scan was performed for attenuation correction before PET scanning. PET scans were acquired in list mode over 15 min, 90 min after intravenous injection of a mean dose of 230 MBq of 18F-AV-1451. The scans were iteratively reconstructed using a 3D-OSEM algorithm of four iterations and 12 subsets. Finally, they were smoothed with a Gaussian filter of 5 mm full-width at half-maximum on a 128×128 matrix.
MRI scanning was performed using a 3T MAGNETOM Trio with a custom-built BrainPET insert in the bore of the magnet (Siemens, Erlangen, Germany). Vacuum cushions were used to reduce head motion.
T1-weighted images (MPRAGE) were acquired with the following parameters: repetition time (TR) 2250 ms; echo time (TE) 3.03 ms; flip angle 9°; 176 sagittal slices; resolution 1.0×1.0×1.0 mm3). FMRI scans were acquired using an EPI sequence with the following parameters: TR = 2400 ms, TE =30 ms, field of view = 210 mm, 36 slices, matrix size = 64×64, in-plane resolution = 3.3×3.3 mm2, slice thickness 3 mm, distance factor = 10%, flip angle = 90°.
During the encoding session, 250 images were acquired. The field of view was angulated parallel to the medial cerebellar tentorium, to reduce susceptibility artifacts in the medial temporal lobe [43, 44].
Processing of MRI data
Each participant’s T1-image was segmented and parcellated using FreeSurfer (Version 6, https://surfer.nmr.mgh.harvard.edu) [45–47]. Following this process, cortical thickness, hippocampal volume, and total intracranial volume were derived. Hippocampal volume was divided by the total intracranial volume to account for differences in head size in subsequent analyses. The first eleven images of the encoding fMRI time series, during which instructions were presented to the participants, were discarded from analysis to ensure that the MR signal had reached a steady state.
Consequently, 239 images entered the analysis. fMRI data analysis was performed with FSL (FMRIB’s Software Library, Version 5.0, http://www.fmrib.ox.ac.uk/fsl), employing different modules of the FSL-software package, specified as follows: Non-brain tissue was removed using BET [48]. Each time series’ EPI images were spatially realigned with MCFLIRT [49] to correct for head movements between scans. Resulting fMRI time-series were spa-tially smoothed using a Gaussian kernel with FWHM = 8 mm, and a high-pass temporal filter (125 s) was applied. Based on the realignment parameters, participants with excessive head motion (maximum relative displacement of > 3 mm) were excluded from further analysis. In the remaining data sets, FSLMotionOutliers was used to compute DVARS (D stands for the temporal derivative of timecourses, and VARS refers to root mean squared (RMS) variance over voxels; [50]), a measure of head motion based on the rate of change in the BOLD (blood oxygen level-dependent) signal across the whole brain at each timepoint.
Furthermore, FSLMotionOutliers was used to identify timepoints corrupted by head motion and enter them in a confound matrix included in the gen-eral linear model (GLM) to censor these timepoints. In addition to this confound matrix, global signal, white matter signal, the signal of the large arteries at the base of the brain, CSF signal, and the six motion parameters were included as covariates of no interest. GLM time-series statistical analysis of individual data sets was carried out using FILM (FMRIB’s Improved Linear Model) with local autocorrelation correction [51].
Trials during the encoding phase can be divided into three types of events: trials where items presented during encoding were later correctly identified as “old” (OC), trials where items presented during encoding were later falsely identified as “new” (OF), and invalid trials (INV; i.e., misses and outliers). We primarily investigated neural activation during successful encoding, i.e., the OC trials. Additionally, neural activation underlying encoding can also be operationalized as the fMRI signal that is greater for stimuli that were later remembered than that for those later forgotten: the subsequent memory effect (SME). OC trials were contrasted against OF trials to analyze the SME.
A constant epoch regressor consisting of a boxcar function beginning at stimulus onset and lasting 3.5 s (stimulus duration) was defined for each event type. Additionally, for each event type, a variable epoch regressor with the identical timecourse as the respective constant epoch regressor, but variable boxcar width defined by the respective trial’s response time was constructed. The variable epoch regressor was orthogonalized to the constant epoch regressor to account for the effects of response time variability, significantly influencing the ampli-tude of the hemodynamic response [52, 53]. The re-sulting six regressors were convolved with a double gamma hemodynamic response function and ente-red into a first-level GLM analysis, together with the covariates listed above. Resulting statistical maps were transformed into the space of the T1- image. For voxel-wise analyses, the statistical maps were normalized to standard space using non-linear deformations obtained using the CAT12 tool-box (Computational AnatomyToolbox 12, http://www.neuro.uni-jena.de/cat/) implemented in SPM12 (https://www.fil.ion.ucl.ac.uk/spm/software/spm12). Second-level analyses of the fMRI data were perfo-rmed using non-parametric testing as implemented in the FSL module randomize. The non-par-ametric approach was chosen because of its sta-tistical robustness in small sample sizes. Mean framewise displacement was included as a covariate of no interest. Framewise displacement was not correlated with any of the activation values used in the region of interest (ROI) analyses. Voxel-wise second-level analyses were constrained to gray matter. Threshold-free cluster enhancement [56] and family-wise error (FWE)-correction were employed to correct for multiple testing. Voxels with p < 0.05 are reported as significant.
For correlation analyses, parameter estimates (betas) for the constant epoch regressors of OC trials (remembered items) and OF trials (forgotten items) were extracted from the ROI described below.
PET processing
The PET images were processed as follows: 1) coregistration to the corresponding T1-image, 2) normalization to the lower cerebellar gray matter, 3) identification of unspecific binding sites, and 4) partial volume correction using the Rousset approach. This resulted in partial volume corrected SUVR values for each cortical and subcortical ROI that were entered in the subsequent statistical analyses. The processing pipeline is described in detail in Baker et al., 2017 [57]
ROI analyses
The FreeSurfer ROI used in the partial volume correction of the AV1451 data served as the basis for the ROI analyses. Based on previous functional imaging studies of memory-related neural activation in cognitively healthy subjects [10, 58], patients with AD [29, 59], and patients with AD under cholinergic stimulation [27, 28], the hippocampus, fusiform gyrus, and the PCC were selected as regions of interest.
The volume-weighted mean of the partial volume corrected SUVR of the cortical ROI corresponding to Braak stages I through VI [60] was computed as a global measure of cortical tau accumulation.
RESULTS
Demographics and behavioral performance
Demographic and neuropsychological data for the sample that entered the imaging data analyses are presented in Table 1. Performance on the item memory task, quantified as d’, was 1.208±0.713 (minimum = 0.057, maximum = 2.692) and correlated with the delayed recall composite (r (15) =0.636, 95% -CI [0.154, 0.913]), but not the memory loss composite (r (15) =-0. 363, 95% -CI [–0.755, 0.127]).
fMRI: whole-brain activation
Neural activation during encoding of subsequently remembered items was observed in an extended network encompassing the fusiform gyrus, the lateral occipital cortex, temporoparietal areas, and primary sensory and motor cortices (Fig. 2, Table 2). During trials with subsequently remembered items, deactivation was observed in a pattern overlapping with the so-called “default mode network”, including posterior midline structures, the anterior cingulate, lateral occipital cortices, as well as lateral temporal areas (Fig. 2, Table 3). An SME was not observed at the whole brain or ROI level.
Local maxima for the group average of activations associated with subsequently remembered items
p < 0.05, FWE-corrected with cluster free threshold enhancement. Coordinates are reported in MNI-space.
Local maxima for the group average of deactivations associated with subsequently remembered items
p < 0.05, FWE-corrected with cluster free threshold enhancement. Coordinates are reported in MNI-space.
Whole-brain AV1451-uptake
Tau accumulation, i.e., AV1451 uptake divided by the uptake in cerebellar gray matter, was most pronounced in the lateral temporal and the parietal lobe, with less retention in the frontal lobes (Fig. 3). This pattern is consistent with the histopathological staging, according to Braak and colleagues [61] and has been reported in several PET studies of AD [60, 62–64].
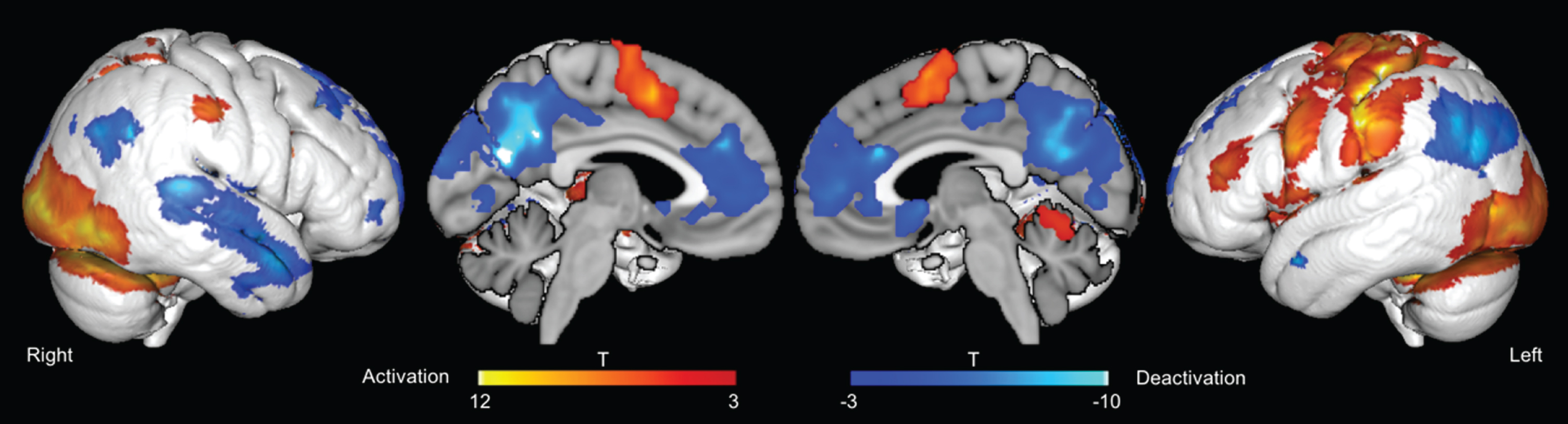
fMRI activation and deactivation for subsequently remembered stimuli. p < 0.05, FWE-corrected with cluster free threshold enhancement.
ROI analyses
Entorhinal tau accumulation was positivel correlated with hippocampal activation for remembered items (r (15 0.670, p = 0.006, 95% -CI [0.429, 0.826]; Fig. 4A). This correlation remained significant when correcting for delay between PET and MRI measurements (r (12) 0.670, 95% -CI [0.264, 0.880]), and medication (r (12) 0.644, 95% -CI [0.246, 0.856]), and after accounting for multiple comparisons. En-torhinal tau accumulation was not correlated with hippocampal activation for subsequently forgotten items (Fig. 4A, B).
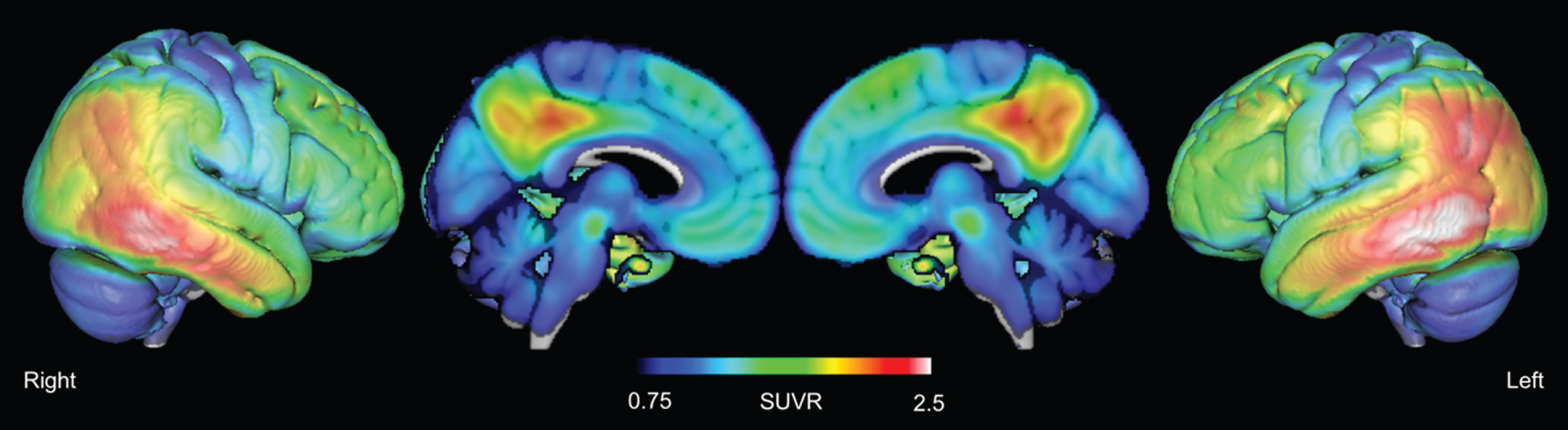
Average AV-1451 retention, normalized to cerebellar grey matter.
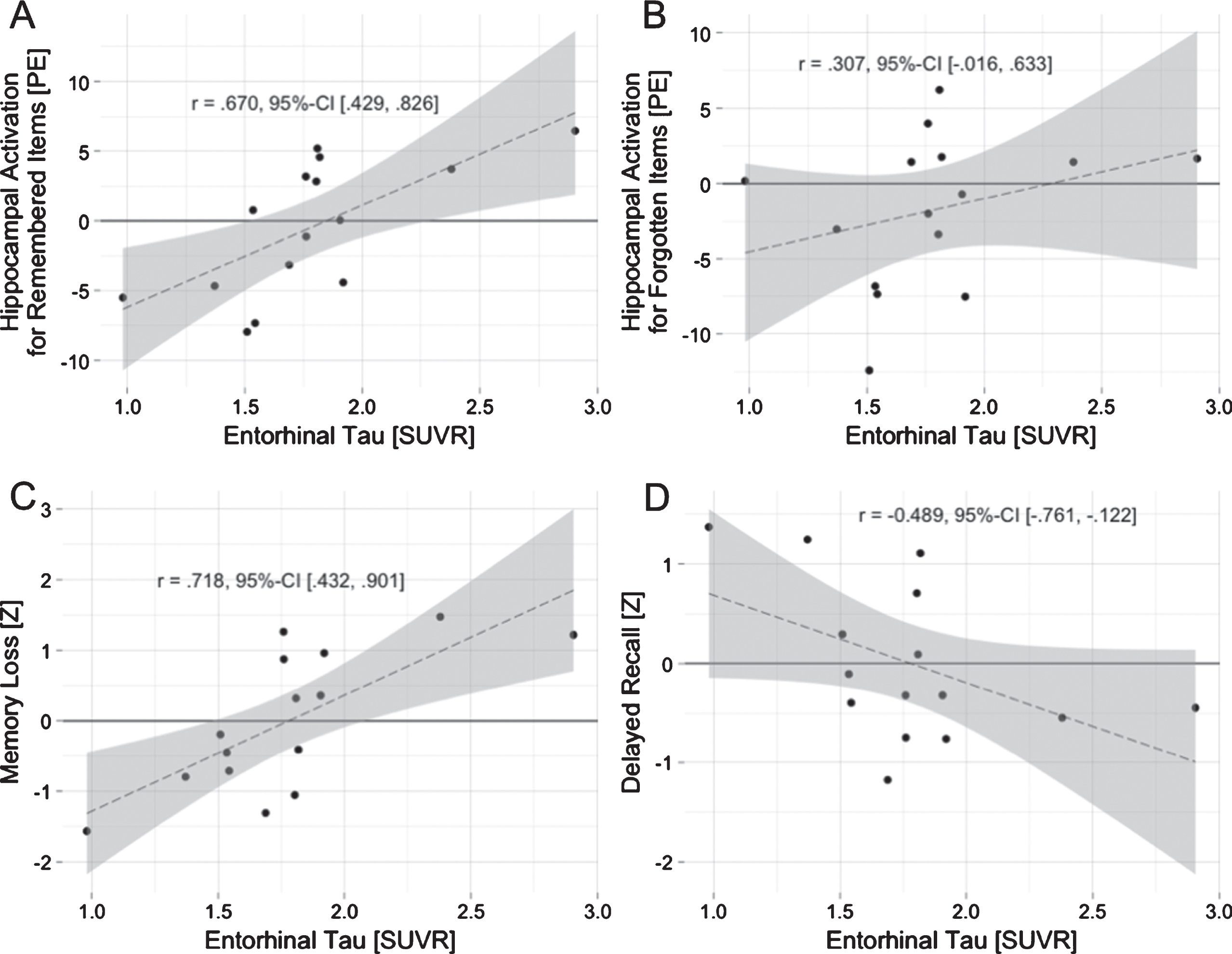
Entorhinal tau accumulation is associated with hippocampal activation and poorer memory performance. Entorhinal tau accumulation correlates with hippocampal activation during encoding of subsequently remembered items (A), but not with activation for subsequently forgotten items (B). Entorhinal tau accumulation also correlates with memory loss after a delay (C) and shows a trend towards a negative correlation with delayed recall performance (D). PE, parameter estimates; SUVR, standardized uptake value ratio.
Tau protein accumulation was not significantly associated with local neural activation in the PCC or the fusiform gyrus.
To determine if the association between entorhinal tau depositions and hippocampal neural activation was specific to these tau depositions, we correlated neural activation for subsequently remembered items with tau depositions averaged across the six Braak stages. Hippocampal activation for subsequently remembered items was not correlated with global tau accumulation. Furthermore, hippocampal volume and entorhinal cortex thickness were not significantly associated with hippocampal activation for subsequently remembered items.
Neural activation, tau accumulation, and cognition in the medial temporal lobe
To further elucidate the possible consequences of the association between entorhinal tau accumulation and neural activation, we examined how local activation and tau accumulation were related to memory function measures.
The memory loss composite score was positively correlated with entorhinal tau accumulation (r (15) 0.718, 95% -CI [0.432, 0.901]) (Fig. 4C). This correlation was significant after accounting for the delay between MR and PET measurements, medication, and multiple comparisons. The memory loss composite score also correlated positively with hippocampal activation (r (15) 0.478, 95% -CI [0.079, 0.783]), but missed significance when accounting for medication (r (12) 0.442, 95% -CI [–0.101, 0.788]).
The delayed recall composite correlated negatively with entorhinal tau accumulation (r (15) –0.489, 95% -CI [–0.761, –0.122]) (Fig. 4D). This correlation was not significant when accounting for the delay between PET and MRI measurements (r (12) –0.394, 95% -CI [0.024, –0.736]) or medication (r (12) –0.502, 95% -CI [0.125, –0.821]).
The delayed recall composite was not correlated with hippocampal activation. d’ was not correlated with entorhinal tau accumulation or hippocampal activation.
DISCUSSION
We demonstrate that entorhinal tau deposition is associated with greater hippocampal activation during the encoding of information in AD patients at the stage of MCI and mild dementia. Furthermore, we observed that the amount of entorhinal tau accumulation and, to a lesser degree, hippocampal activation are positively correlated with memory loss over time. These associations were specific to the medial temporal lobe structures; neural activation in the fusiform gyrus and the PCC was not significantly associated with local or global tau accumulation.
Tau accumulation and neural activation
Entorhinal tau accumulation correlated positively with hippocampal activation for subsequently remembered items. While this has not previously been investigated using tau PET and fMRI in a patient sample, similar findings have been reported in normal cognitive aging [10, 13]. Maass et al. observed greater neural activation in the anterior temporal lobe and the hippocampus in tau-positive than tau-negative cognitively healthy participants. Marks and colleagues described a correlation between tau accumulation in Braak I/II areas and hippocampal activation for items subsequently misidentified, while Huijbers et al. observed hippocampal “encoding success activity” to be correlated with inferior temporal tau accumulation. In analogy to Huijbers and colleagues, we found hippocampal activation for subsequently remembered items associated with entorhinal tau accumulation. A possible explanation for tau accumulation association with different activation patterns is that Marks et al. used a pattern separation-completion paradigm.
In contrast, Huijbers and colleagues used a face memory encoding task, more similar to our visual item memory task. The most parsimonious explanation for the discrepancy regarding tau accumulation location associated with hippocampal activation between our study and Huijbers et al. are differences in the disease stage and tau burden, which were substantially greater in our sample. Huijbers and colleagues did not observe an association between tau accumulation in the entorhinal cortex and hippocampal activation and specially selected the inferior temporal cortex ROI as a proxy for the spread of tau accumulation to the neocortex since entorhinal tau accumulation is commonly observed in advanced age [64]. In our sample, however, entorhinal tau accumulation was much higher than in the samples reported by Huijbers and Johnson [12, 64].
In cognitively healthy seniors, hippocampal hyperactivity has been linked to tau accumulation, while amyloid and APOE status appear to play a smaller role [12]. Amyloid status has been discussed to explain the equivocal findings regarding changes in hippocampal activation in MCI [23]. While all patients in our sample were amyloid positive, we cannot directly link amyloid load to hippocampal activation, since we lack a standard quantitative amyloid measure that allows comparisons across all participants. However, based on the cascade of the pathology in AD and its association with neurodegeneration and cognition [2, 65–67], tau accumulation is more likely to drive hippocampal activation at the stage of MCI or mild dementia than amyloid.
Hippocampal activation and cognition: Aberrant hippocampal activation?
Hippocampal activation was not associated with performance on the item memory task, quantified with the measure d’. Huijbers and colleagues also did not find a correlation between d’ and any imaging measure. This lack of correlation may result from the fact that performance in visual item memory and face encoding tasks is not solely dependent on medial temporal function, but also requires visual processing, reflected in the strong activation in higher-order visual areas such as the fusiform gyrus [12, 26]. In late MCI and mild dementia, these areas are often also affected by hypometabolism, tau deposition (cf. Fig. 3), and atrophy, but not as severely and consistently as the medial temporal lobe [63, 69]. To obtain a true measure of memory (dys-) function and account for confounding effects of content type and stimulus processing, we computed a memory loss composite from verbal and visual memory scores.
This memory loss composite correlated highly with entorhinal tau accumulation and weakly with hippocampal activation. In other words, entorhinal tau, and, to a lesser degree, hippocampal activation during encoding, are associated with imperfect memory, but tau accumulation better explains memory deficits than hippocampal activation. Hippocampal activation may still be contributing to more deficient memory formation since it was positively correlated with the amount of information that was forgotten, i.e., worse memory.
Since hippocampal activation associated with encoding success was correlated with entorhinal tau accumulation, this activation could have been interpreted as compensatory. However, this is unlikely as compensatory activation is expected to be related to improved performance [70] and not worse memory, as in our case. Furthermore, evidence from an-imal models, individuals at risk of developing AD, and individuals at preclinical stages indicates a pat-hological role of increased hippocampal activation rather than a compensatory mechanism [71, 72]. Hippocampal hyperactivity may also be the consequence of disinhibition resulting from a disconnection from cortical inputs [73]. Ultimately, an experimental intervention would be required to determine if the causal effects of neuronal activation on cognition are compensatory or detrimental. There are reports that amelioration of task-related hippocampal hyperactivation by low doses of the antiepileptic drug levetiracetam can improve memory performance in MCI [17, 18]. However, patients in those studies were recruited based on clinical criteria. Therefore, it is unknown how the observed hyperactivation was related to AD pathology or if AD pathology was even present in all participants
In summary, while we could demonstrate that entorhinal tau accumulation is positively correlated with hippocampal activation, further studies combining biomarkers of AD pathology and an experimental intervention will be necessary to determine the nature of tau-linked hippocampal activation.
Limitations
The study is limited by its relatively small sample size, a general problem in clinical populations [74, 75]. However, we observed strong associations, arguably because we investigated a patient cohort, where disease burden is expected to be much greater than in cognitively healthy samples [10, 13]. The strength of the observed associations is all the more striking considering the delay between fMRI and PET measurements (207.47 days, standard deviation 191.71 days), which was a little higher, but still in the same order of magnitude as in studies investigating cognitively normal subjects [10, 12]. Nonetheless, our key findings, the positive correlations between entorhinal tau accumulation, hippocampal activation, and memory loss over time, remained significant when accounting for the delay between measurements and psychoactive medication in a subset of patients. However, when interpreting these findings, it has to be taken into account that they may still have influenced the results despite the statistical correction for these factors.
To reduce the complexity of the fMRI task, a two- choice task was used. However, this is also a limitation, because control stimuli with scrambled images would have allowed for differentiation between the visual effect of stimulus presentation and stimulus encoding. This needs to be considered when interpreting the activation pattern, especially for visual areas.
Arguably, this study is also limited by the absence of a control group. However, it was performed this way, because we specifically sought to investigate in patients the association between tau accumulation and fMRI activation, which has consistently been reported in cognitively normal samples. Since the same fMRI task has been used to compare MCI pat-ients to an age-matched control group [27], no additional insights were expected from the inclusion of a control group. In light of this, it was ethically problematic to perform AV1451-PET in a cognitively normal sample.
A further limitation is that a subset of patients (4 out of 15) had a formal dementia diagnosis. Given that early dementia and MCI are adjacent diagnoses along the continuum of AD, and the fact that this sub-set did not differ significantly from the remaining patients in demographics and overall cognition, it is highly unlikely that fundamentally different pathophysiological mechanisms drove the observed effects. Finally, this study is also limited by its cross-sectional design. Any inference regarding causality, therefore, needs to be regarded with caution.
Conclusions
The present in vivo data demonstrate that tau accumulation in the entorhinal cortex is associated with neural activation in the hippocampus. Especially entorhinal tau accumulation is negatively correlated with memory performance in symptomatic AD, extending previous findings in cognitively healthy aging [10, 13]. Our data provide evidence that the positive correlation between hippocampal activation and entorhinal tau accumulation is also present in the early symptomatic stages of AD. However, hippocampal activation measured using fMRI does not appear as the primary determinant of memory function. The association between local tau accumulation and neural activation was specific to the medial temporal lobe, indicating that tau accumulation does not generally increase or attenuate neural activation locally at this disease stage. Thus, the present findings contribute to our pathophysiological understanding of the relationship between tau accumulation, neural activation, and memory in AD at the stage of MCI and mild dementia and may help inform future longitudinal and interventional studies.