
Abstract
Background:
Berberine (BBR) plays a neuroprotective role in the pathogenesis of Alzheimer’s disease (AD), inhibiting amyloid-β (Aβ) production and promoting Aβ clearance. Advanced glycation end products (AGEs) promote Aβ aggregation and tau hyperphosphorylation. The activation of mTOR signaling occurring at the early stage of AD has a prominent impact on the Aβ production. This work focused on whether BBR regulates the production and clearance of ribosylation-induced Aβ pathology via inhibiting mTOR signaling.
Objective:
To explore whether BBR ameliorates ribosylation-induced Aβ pathology in APP/PS1 mice.
Methods:
Western blot and immunofluorescence staining were used to detect the related proteins of the mammalian target of Rapamycin (mTOR) signaling pathway and autophagy, as well as the related kinases of Aβ generation and clearance. Tissue sections and Immunofluorescence staining were used to observe Aβ42 in APP/PS1 mice hippocampal. Morris water maze test was used to measure the spatial learning and memory of APP/PS1 mice.
Results:
BBR improves spatial learning and memory of APP/PS1 mice. BBR limits the activation of mTOR/p70S6K signaling pathway and enhances autophagy process. BBR reduces the activity of BACE1 and γ-secretase induced by D-ribose, and enhances Aβ-degrading enzymes and Neprilysin, and inhibits the expression of Aβ in APP/PS1 mice.
Conclusion:
BBR ameliorates ribosylation-induced Aβ pathology via inhibiting mTOR/p70S6K signaling and improves spatial learning and memory of the APP/PS1 mice.
INTRODUCTION
Alzheimer’s disease (AD) is an age-related and insidious neurodegenerative disease that is clinically characterized by progressive cognitive decline accompanied by language, behavioral disorders, social dysfunction, and eventually death [1]. Depending on the age of onset, AD is divided into presenile dementia that before 65 years old and senile dementia that after 65 years old [2]. The main pathological features of AD include the senile plaques formed by the aggregation and deposition of Aβ peptides which is cleaved from amyloid-β protein precursor (AβPP), and neurofibrillary tangles (NFTs) formed by hyperphosphorylated tau protein, as well as loss of synapses in neocortex and hippocampus, neuroinflammation, oxidative stress, and nerve cell death [3, 4]. For many years, AD has become a research hotspot in the neuroscience scientific community. Evidence from clinical and basic studies on Aβ has pointed out that it plays a key role in the onset and development of AD pathogenesis. However, there have been no mature schemes or effective drugs targeted at Aβ for the treatment of AD, suggesting the complexity and challenge of the Aβ hypothesis [5].
In recent years, the role of non-enzyme catalyzed AGEs in the pathogenesis of AD has been drawn more and more attention. AGEs can bind to advanced glycation end products receptor (RAGE) on the surface of nerve cells, and then AGEs and RAGE interact with glial cells to promote Aβ aggregation and tau hyperphosphorylation [6]. Studies have found that D-ribose-induced ribosylation and excessive AGEs production play an important role in tau hyperphosphorylation and the NFT formation via activation of CaMKII [7]. Still and all, the underlying mechanism of abnormal glycosylation in the pathogenesis of AD remains unclear.
Autophagy is an important mechanism of intracellular recycling that delivers cellular metabolize material to lysosomes for degradation, in response to various forms of stress such as nutrient defici-ency, organelle damage, and protein misfolding [8]. According to the process mechanism, autophagy has been identified in three types: macroautoph-agy, microautophagy, and chaperone-mediated auto-phagy. As the most common form, macroautophagy is characterized by the formation of autophagosome after the double membrane structure wraps the substrate protein, and autophagosome fuses with lyso-some to form autolysosome, which releases the substrate protein into lysosome and eventually degrades it under the action of hydrolase, so as to achieve cell homeostasis and organelle renewal [9, 10].
Increasing studies have shown that autophagy is closely related to the occurrence and development of neurodegenerative diseases such as AD, Parkinson’s disease, and Huntington’s disease [11]. Emerging evidence suggests a complex interaction between autophagy and Aβ, which may contribute to the progression of AD. Nilsson and Saido reported that autophagy plays a key role in the Aβ plaque formation, while autophagy deficiency results in intracellular Aβ accumulation and memory impairment of AD mice [12, 13]. The defective autophagy–lysosome pathway not only leads to Aβ accumulation, but also hinders its degradation. A study found that estrogen receptor β (ERβ) interacting with autophagy related protein (Atg) 7 enhanced autophagy activity, promotes Aβ clearance in vitro [14]. A small-molecule enhancer of autophagy, small-molecule enhancers of rapamycin (SMER) 28 decreases the level of Aβ and APP-CTF via Atg5 dependent autophagy, while Beclin1 and Ulk1 are also involved in this process [15].
Berberine (BBR), a natural alkaloid that can be extracted from the roots, rhizomes, stems, and bark of some medicinal plants such as Huanglian (Coptis chinensis), Berberis aristata, Hydrastis canadensis, Tinospora cordifolia, Phellodendron amurense, and so on. As a traditional Chinese medicine with a long history of medicinal use, BBR is widely used for treating diarrhea, gastroenteritis, and type II diabetes [16, 17]. Furthermore, BBR possesses the properties of anti-cancer [18, 19], anti-gynecopathy [20], anti-cardiovascular disease [21], and anti-neurodegenerative disease [22]. A series of studies have focused on BBR and the molecular pathogenesis of AD. BBR could reduce the accumulation of Aβ by activating AMP-activated protein kinase in N2a/APP695sw cells [23], alleviate Aβ induced mitochondrial dysfunction and synaptic loss in primary cultured hippocampal neurons [24]. In addition, BBR promoted autophagic clearance of Aβ via enhancing autophagy activity through the class III PI3K/beclin-1 pathway and inhibited Aβ production by decreasing BACE1 in primary hippocampal neurons and in APP/Tau/PS1 mouse [25].
Our previous studies have shown that BBR lowers the Aβ level by inhibiting the activity of β/γ-secretases and enhancing α-secretases expression in the hippocampus of AD mice, and improves Alzheimer’s-like cognitive impairment, but whether this effect is through the autophagy pathway has not been studied [26]. Therefore, we propose the hypothesis that BBR improves ribosylation-induced Aβ pathology of AD by regulating the autophagy pathway. To test this hypothesis, in vitro and in vivo experiments were used to study the regulation of BBR on autophagy and Aβ aggregation and whether BBR could regulate the production and clearance of ribosylation-induced Aβ pathology via regulating mTOR signaling and autophagy pathway was explored using N2a cells and transgenic APP/PS1 mice.
MATERIALS AND METHODS
Drugs and animals
BBR (100 mg/tablet) was purchased from Sanofi Minsheng Health Pharmaceutical Co. Ltd (Hangzhou, China). D-ribose was purchased from Sigma-Aldrich (St. Louis, MO, USA) (V900389). Aminoguanidine hydrochloride was purchased from Sigma-Aldrich (St. Louis, MO, USA) (396494). Rapamycin was purchased from Cell Signaling Technology (CST) (MA, Boston, USA) (9904). APP/PS1 transgenic mice were purchased from the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences & Peking Union Medical College. Animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and followed the ethical standards approved by the Research Ethics Committee of Chongqing General Hospital.
Morris water maze test
An escape platform (diameter 8 cm) was placed 1 cm below the surface in the northeast quadrant of a circular tank (diameter 1.2 m and depth 0.4 m, 25±1°C). Mice adapted to the experimental environment were trained 4 times per day for 6 consecutive days as acquisition trials. Each trial began with putting a mouse into the different quadrant and letting it keep swimming freely for 60 s. Each mouse climbed onto the platform and staid for 5 s (or failed to locate the platform within 60 s was given extra 30 s to be guided to the platform), they were put back in their cages and wiped dry. The time taken for each mouse to reach the platform is considered the escape latency, which together with swimming trajectory were recorded by the ANY-maze video tracking software (Stoelting Co., USA). After 1 day’s rest, the mice were performed with probe test without the platform for 60 s. The mice were placed into the quadrant opposite to the original platform quadrant. The time, distance in the original platform quadrant and the number of crossings that occurred in the probe were observed and recorded.
Cell culture and treatments
Mouse neuroblastoma cells, Neuro-2a (N2a) were stored by our laboratory. N2a cells were cultured in Dulbecco’s Modified Eagle Medium Nutrient Mixture F-12 (DMEM/F12, Gibco, Carlsbad, CA, USA) supplemented with 10%fetal bovine serum (FBS, Invigentech, Irvine, CA, USA) and antibiotics (100 U/ml penicillin and 100μg/ml streptomycin), incubated in humidified atmosphere with 5%CO2 at 37°C. N2a cells were treated with 10 mM D-ribose for 24 h, used for constructing hyperribosylation model. For evaluate the effect of BBR on autophagy activity, N2a cells were precoated with 1μM BBR for 12 h, while the control group was treated with 1.5 mM aminoguanidine hydrochloride (AGEs inhibitor) for 12 h after D-ribose tresatment, or 100 nM Rapagymin for 12 h.
Western blot analysis
Cell lysates and the brain tissue of APP/PS1 mice were lysed in RIPA Lysis Buffer containing protease inhibitor PMSF. Extracted total protein was determined by BCA Protein Assay Kit (Beyotime Biotechnolgy, Shanghai, China), and equivalent protein were separated on the corresponding concentration of gels and then transferred to PVDF membranes (Bio-Rad, Hercules, CA, USA). Membranes were blocked and incubated primary antibodies against Phospho-mTOR (Ser2448) (CST, 5536), mTOR (CST, 2983), Phospho-p70 S6 Kinase (Thr389) (CST, 9234), p70 S6 Kinase (CST, 9202), SQSTM1/p62 (CST, 39749), LC3A/B, Beclin-1, Atg3 (Autophagy Antibody Sampler Kit, CST, 4445), LC3B (CST, 3868), BACE (CST, 5606), AGE (abcam, 23722), Presenilin 1 (abcam, 76083), Presenilin 2 (abcam, 51249), and Neprilysin (proteintech, 18008) overnight at 4°C. Then membranes were incubated with secondary antibody for 1 h at room temperature. Protein bands were visualized using Immobilon Western Chemiluminescent HRP Substrate (Millipore Corporation, Billerica, MA, USA) and Tanon-5200 multi chemiluminescence analysis system (Tanon, Shanghai, China).
Paraffin section
The whole brains of APP/PS1 mice were removed and fixed with 4%paraformaldehyde for 48 h. The specimen was cut into 2μm paraffin sections using a Rotary Microtome (HM 340E, Thermo Scientific) along the long axis in sagittal plane. After deparaffinization, antigen retrieval was performed in citric acid solution through microwave oven. Then paraffin sections were used for immunostaining analysis.
Immunofluorescence staining
N2a cells cultured on coverslips were fixed with 4%paraformaldehyde, permeabilized with 0.1%Triton X-100 in PBS. N2a cells or 2μm paraffin sections were blocked with 5%goat serum albumin and incubating with primary antibodies Phospho-mTOR (Ser2448), LC3B, BACE1, Presenilin 2, or amyloid-β 1–42 (abcam, 10148) at 4°C overnight. After incubation with secondary antibodies, Alexa Fluor® 488-conjugated antibodies (ZSGB-BIO, Beijing, China) for 1 h in the dark, the nucleuses were stained using DAPI (Beyotime). The coverslips were installed onto glass slides using Antifade Mounting Medium (Beyotime), and a laser scanning confocal microscope (Leica TCS-SP2, Germany) or NEXCOPE microscope (NE900, USA) was used for detection.
Statistical analyses
All the experimental data were analyzed using GraphPad Software. ANOVA and Independent-sample t test were applied to compare the differences of measurement data between each group. The data were represented as means±standard error of mean (SEM) and p < 0.05 was considered statistically significant.
RESULTS
BBR promotes autophagy by inhibiting the activation of mTOR signaling pathway in N2a cells
To examine the effect of BBR on mTOR and autophagy signaling, Rapamycin was used as a positive control, which stimulates autophagy by inhibiting mTOR. N2a cells were treated with 1μM BBR or 100 nM Rapamycin for 24 h. After exposure to BBR, the level of p-mTOR and p-p70S6K decreased compared to the control group, which was the same effect as the treatment with Rapamycin (Fig. 1A, B). Similarly, after BBR treatment, the expression of autophagy-related proteins LC3B, Atg3, and Beclin1 significantly increased, synchronously, the elective autophagy receptor SQSTM1/p62 was degraded (Fig. 1C, D). Then we verified these results using immunofluorescence. As illustrated in Fig. 1E, compared with control group, BBR treatment group showed the enhanced fluorescence intensity of both p-mTOR and LC3B proteins, which was consistent with the effect of Rapamycin.
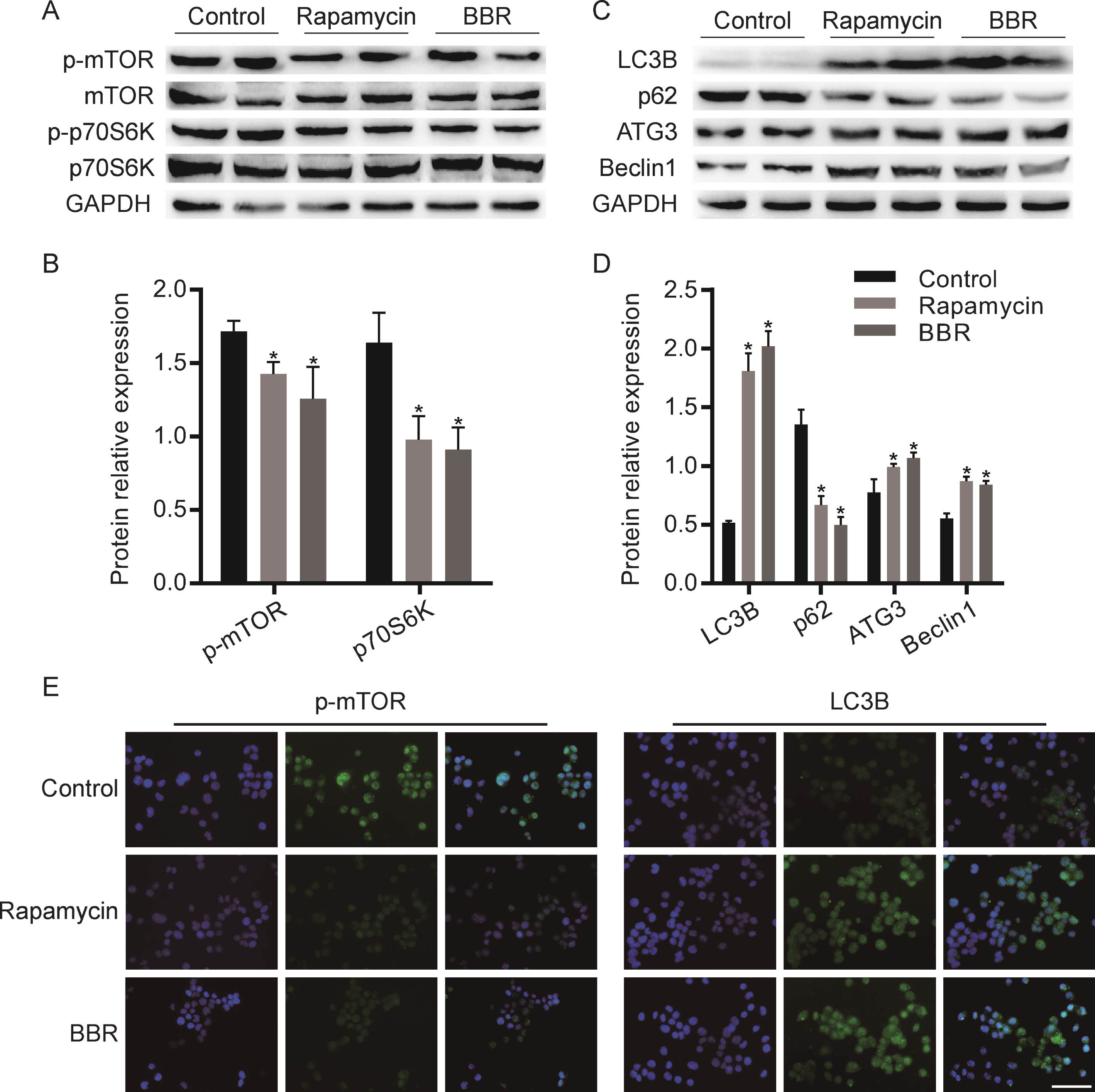
BBR promotes autophagy by regulating mTOR signaling pathway in N2a cells. A) Western blot analysis the expression of p-mTOR and p-p70S6K in Rapamycin or BBR treated cells. Quantification is presented in B. C) Western blot analysis the expression of autophagy related proteins in Rapamycin or BBR treated cells. Quantification is presented in D. E) Immunofluorescence detected the expression of p-mTOR and LC3B. The data are shown as mean±SEM. *p < 0.05. Scale bars = 100μm.
BBR decreases BACE1 and γ-secretase and enhances Aβ-degrading enzymes Neprilysin in N2a cells
We further studied the effect of BBR on Aβ associated kinase in N2a cells. As shown in Fig. 2A, B, compared with the control group, BBR treatment inhibited the expression of β-secretase, BACE1 and γ-secretase, PS1 and PS2, and enhanced the expression of Aβ decomposition enzyme, Neprilysin. This effect was similar to Rapamycin. Next, immunofluorescence staining was used to detect the effect of BBR on β-secretase, BACE1 and γ-secretase, PS2. As shown in Fig. 2C, BBR treatment decreased the fluorescence intensity of BACE1 and PS2 proteins, which was consistent with the effect of Rapamycin.
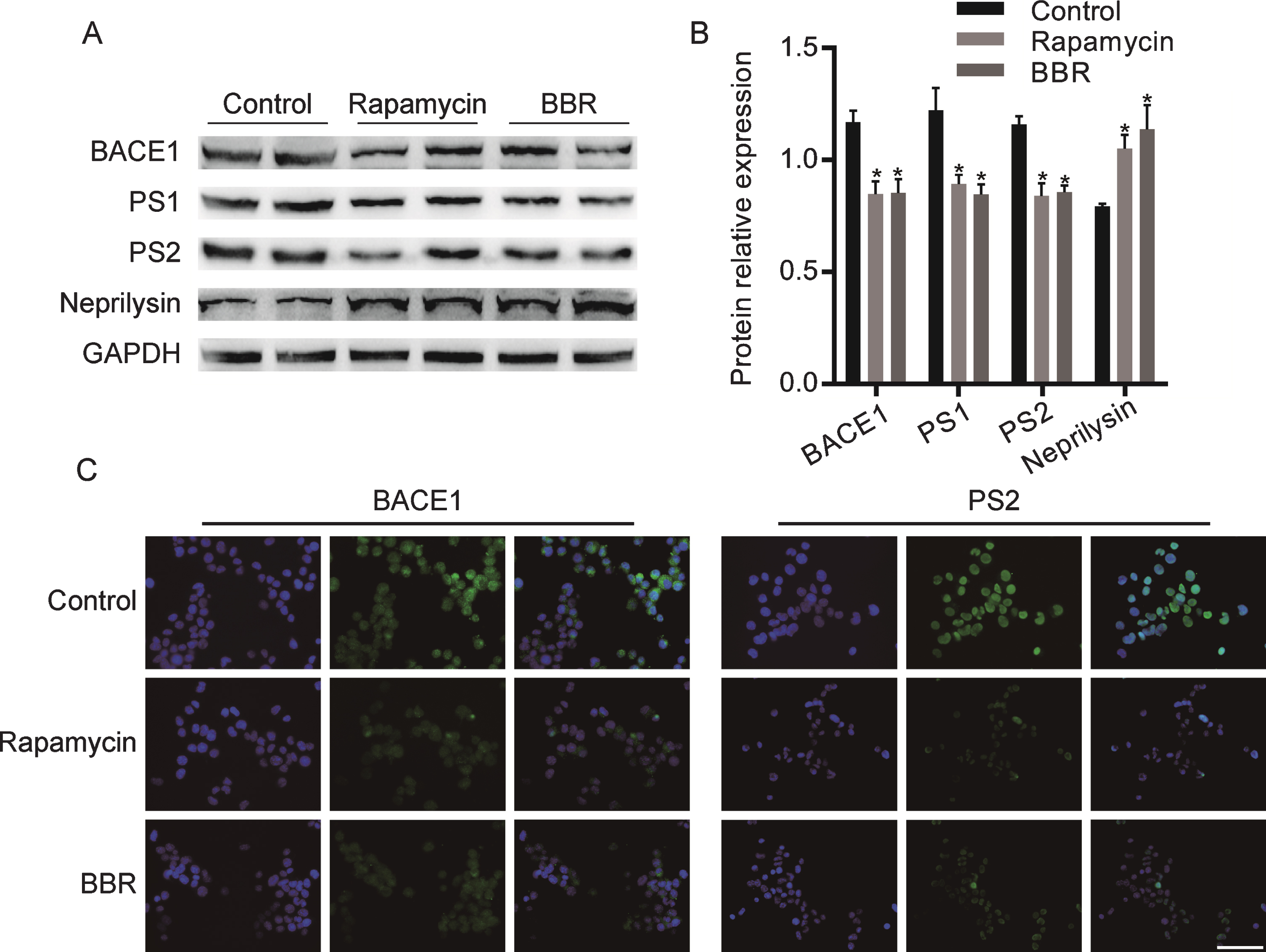
BBR regulated the expression of Aβ associated kinase in N2a cells. A) Western blot analysis the expression of BACE1, PS1, PS2, and Neprilysin in Rapamycin or BBR treated cells. Quantification is presented in B. C) Immunofluorescence detected the expression of BACE1 and PS2. The data are shown as mean±SEM. *p < 0.05. Scale bars = 100μm.
BBR counteracts the activation of mTOR signaling pathway, inhibition of autophagy, increase of BACE1 and γ-secretase, and the decrease of Neprilysin induced by D-ribose in APP/PS1 mice
Based on in vivo data, we extended the N2a cell studies to an in vivo experimental environment. It has been reported that AGEs can bind with RAGE and then interact with glial cells to promote the aggregation of Aβ and the hyperphosphorylation of tau protein. D-ribose inducing ribosylation and the generation of excessive AGEs are an important cause of tau hyperphosphorylation and the formation of NFTs. To clarify the mechanism by which BBR regulating mTOR signaling pathway to promote autophagy, we first analyzed the effect of BBR and D-ribose on mTOR signaling pathway. D-ribose intraperitoneal injecting increased the expression of AGE, p-mTOR, and p-p70S6K protein in APP/PS1 mice brain. When mice injected with D-ribose were given BBR intraperitoneally, the increase of AGE, p-mTOR and p-p70S6K protein expression was inhibited, which was similar to the effect of AGE inhibitors (Fig. 3A, B). Then we examined the effect of BBR and D-ribose on autophagy-related proteins. D-ribose intraperitoneal injecting decreased the expression of Beclin1, Atg3, and LC3B, and increased p62 protein in APP/PS1 mice brain. After additional intraperitoneal administration of BBR, the decrease of Beclin1, Atg3, and LC3B, and the increase of p62 protein expression was inhibited, this was consistent with the effect of AGE inhibitors (Fig. 3C, D). Then we investigated the effect of BBR on Aβ associated kinase in APP/PS1 mice. As shown in Fig. 3E, F, compared with the control group, D-ribose treatment increased the expression of β-secretase, BACE1 and γ-secretase, PS1 and PS2, and decreased the expression of Aβ decomposition enzyme, Neprilysin. When the D-ribose treated mice were given BBR intraperitoneally, the expression of BACE1, PS1 and PS2 reduced significantly, concurrently, the expression of Neprilysin increased dramatically.
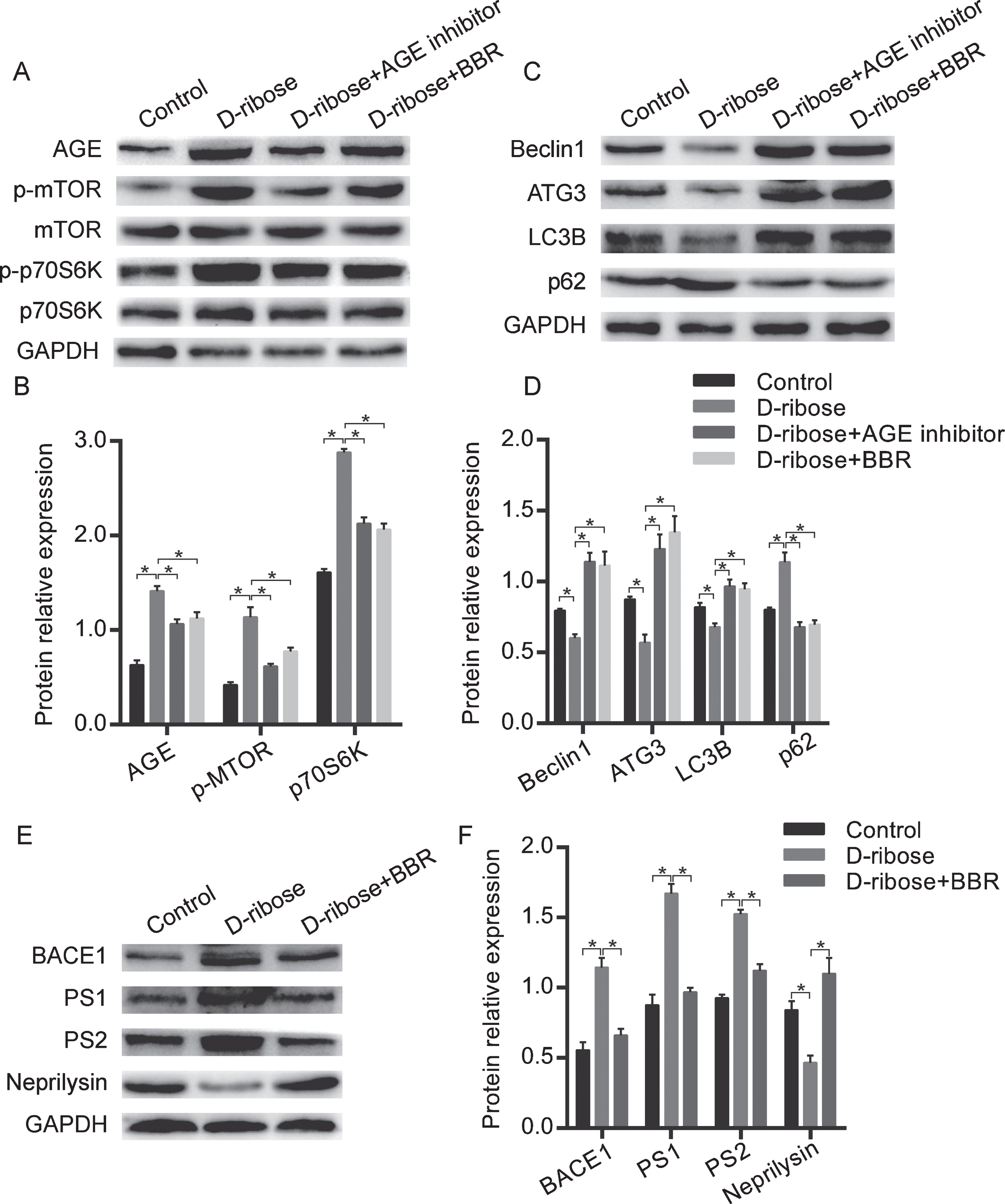
BBR blocked the mTOR signaling pathway, regulated autophagy and Aβ associated kinase in APP/PS1 mice. A) Western blot analysis the expression of AGE, p-mTOR, and p-p70S6K in D-ribose group, D-ribose supplementary AGE inhibitors group, and D-ribose supplementary BBR group. Quantification is presented in B. C) Western blot analysis the expression of autophagy related proteins in D-ribose group, D-ribose supplementary AGE inhibitors group and D-ribose supplementary BBR group. Quantification is presented in D. E) Western blot analysis the expression of BACE1, PS1, PS2, and Neprilysin in D-ribose group and D-ribose supplementary BBR group. Quantification is presented in F. The data are shown as mean±SEM. n = 10. *p < 0.05.
BBR improves spatial learning and memory and inhibits the Aβ42 expression of APP/PS1 mice
Above results indicated that BBR may ameliorate ribosylation-induced Aβ pathology via promoting autophagy. Then the Morris water maze test was used to assess whether BBR could affect the spatial learning and memory of APP/PS1 mice followed the schematic diagram of experimental process (Fig. 4A). On the 5 days space navigation training, the four experimental groups all showed a time dependent decrease of escape latency. Compared with the D-ribose supplementary AGE inhibitor group and the D-ribose supplementary BBR group, the D-ribose treatment group had longer escape latency from day 2 to day 5 (p < 0.05) (Fig. 4B). After removing the platform, the time in the platform location, the times across the platform, and the retention time in the target quadrants of D-ribose treatment group had significantly decreased compared with control group, which was abrogated in the D-ribose supplementary AGE inhibitor group and the D-ribose supplementary BBR group (Fig. 4C-E). These differences were visualized in the representative mouse trajectories taken by APP/PS1 mice during the space exploration task (Fig. 4F). Immunofluorescence staining was used to intuitively observe the effect of BBR on Aβ in APP/PS1 brain. Compared with D-ribose group, BBR treated group showed significant decreases in the immunoreactive region of Aβ42 in APP/PS1 mice hippocampal (Fig. 4G). These results indicated that BBR improved spatial learning and memory and promoted the degradation of Aβ42 in the hippocampus of APP/PS1 mice.
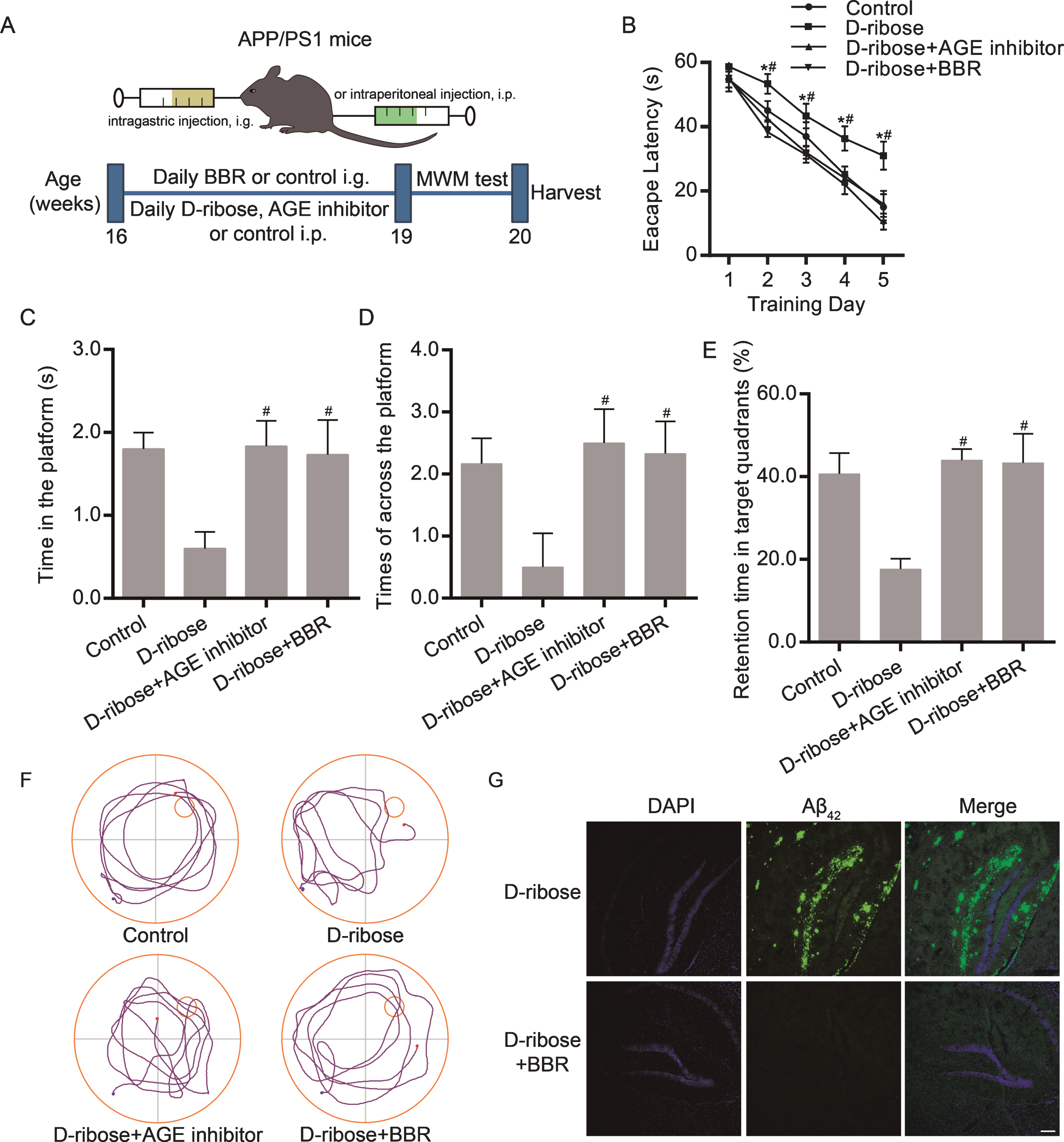
BBR regulated the spatial learning and memory and the Aβ42 expression of APP/PS1 mice. A) Experiment and timeline procedure. 16 weeks APP/PS mice were divided into 4 groups: control group treated with vehicle, D-ribose group, D-ribose supplementary AGE inhibitor group, and D-ribose supplementary BBR group. B) Average escape latency of the 5 days space navigation training. The time in the platform location (C), the times across the platform (D), the retention time in the target quadrants (E), and the representative mouse trajectories (F) were observed during the space exploration task after removing the platform. G) Immunofluorescence detected the expression of Aβ42 in the hippocampus of D-ribose group and D-ribose supplementary BBR group. The data are shown as mean±SEM. n = 10. *p < 0.05 versus the escape latency of day 1. #p < 0.05 versus the D-ribose group. Scale bars = 150μm.
DISCUSSION
BBR has been reported to play a neuroprotective role in the pathogenesis of AD, inhibiting Aβ production and promoting Aβ clearance. This study has demonstrated that BBR improves spatial learning and memory of APP/PS1 mice. BBR promotes autophagy by inhibiting the activation of mTOR/p70S6K signaling pathway, reduces the activity of BACE1 and γ-secretase induced by D-ribose, and enhances Aβ-degrading enzymes, Neprilysin. Thus, BBR regulates autophagy through mTOR signaling pathway, and inhibits ribosylation-induced Aβ production and promotes Aβ clearance (Fig. 5).

Schematic diagram of the BBR possible mechanisms. BBR reversed the effect of D-ribose by inhibiting the expression of AGEs, which upregulated autophagy by inhibiting the expression of p-mTOR/p-p70S6K and promoting the expression of Beclin1. Meanwhile, the inhibition of BBR on AGEs decreased the expression of β/γ-secretase and increased the protein level of Neprilysin.
Amyloid plaques resulting from the deposition of Aβ peptide are the typical hallmark of AD pathology. In the amyloidogenic pathway, transmembrane protein AβPP is cleaved by α-secretase, β-secretase (BACE1), and γ-secretase which mainly contains four subunits: presenilin (PS1, PS2), nicastrin, APH-1 and PEN-2 [27]. α-Secretase cleaves AβPP producing a soluble α-fragment (sAβPPα) and an 83 residue C-terminal fragment bound to membrane (C83), which will be cleaved by γ-secretase to a peptide fragment called P3 and a AβPP intracellular domain (AICD). This process is called nonamyloidogenic pathway. On the contrary, in the amyloidogenic pathway, BACE1 cleaves AβPP, releasing a soluble β-fragment (sAβPPβ) and a C-terminal fragment (C99), which will be processed by γ-secretase and lead to the formation of Aβ and AICD [28, 29].
BBR is a natural isoquinoline alkaloid isolated from Rhizoma coptidis and other herbs, whose working mechanism still needs to be further explored. Studies have shown that BBR has the therapeutic potential for central nervous system (CNS) diseases, such as AD, cerebral ischemia, mental depression, schizophrenia, and anxiety [30]. A series of in-depth studies have been carried out on animal models of AD or in vitro experiments. In HEK293/tau cells, BBR attenuates calyculin A induced cytotoxicity and tau hyperphosphorylation [31]. Durairajan et al. demonstrated that BBR ameliorates Aβ pathology, gliosis, and cognitive impairment in AD transgenic mouse model [32]. Moreover, BBR against the altered intrinsic properties of the CA1 neurons induced by Aβ neurotoxicity in Aβ treatment rats [33]. Panahi et al. reported that BBR treatment could restore Al maltol-induced behavioral derangements by regulating the physiological abilities, histological changes, and BACE1 activity in the rabbit model of AD [34]. In addition, as an autophagy regulator, BBR might be able to regulate autophagy in response to the pathological mechanisms of various diseases [35]. Our previous research found that BBR lowers the Aβ level by inhibiting the activity of β/γ-secretases and enhancing α-secretases expression in the hippocampus of AD mice and improves cognitive impairment [26]. In this report, we demonstrated that BBR decreases the expression of β-secretase, BACE1 and γ-secretase, PS1 and PS2, enhances Aβ-degrading enzymes Neprilysin in N2a cells and AD mice.
Autophagy plays an important role in the pathogenesis of AD. A large number of studies have found that the autophagic lysosomal pathway obstacle not only leads to the production of toxic Aβ, but also is an important reason for the failure of the clearance of Aβ, finally leading to the accumulation and deposition of Aβ in the brain [36]. Therefore, autophagy is considered to be another degradation pathway of Aβ, which is similar to the Aβ degradation kinase [37].
Autophagy is controlled by a highly regulated process. mTOR is a highly conserved serine/threonine kinase that controls cell growth and proliferation mainly by regulating the metabolism of amino acids, glucose, nucleotides, fatty acids and lipids, and by suppressing autophagy [38]. mTOR activates its downstream serine/threonine kinase p70 ribosomal S6 kinase (p70S6K) in response to intracellular state change [39]. Studies have found that mTOR pathway is not only involved in regulating autophagy termination and lysosomal recombination in NRK cells [40], but also considered as an essential homeostatic protein for interneuron development in the brain [41]. Beclin1 is another key regulator of autophagy, which regulates the synthesis and maturation of autophagosome [42]. During the formation of autophagosome, microtubule-associated proteins light chain 3A (LC3A) is activated by autophagy related protein 7 (Atg7), transferred to Atg3, then conjugated with phosphatidyl ethanolamine to form LC3B. SQSTM1/p62 is a key factor in the formation of autophagosome and is selectively degraded in autolysosome system [43]. Our recent study has found that autophagy disorder enhanced the production of Aβ by enhancing the activity of the γ-secretase complex. Autophagy inhibitor could significantly activate the expression of PS1, Nicastrin, and pen-2 in the γ-secretase and promote the production of Aβ [44]. In this study, we first examined whether BBR promoted autophagy by regulating the mTOR signaling pathway, thereby reducing the expression of Aβ and its related kinases of APP/PS1 mice and N2a cells. Our results indicate that BBR reduced the activation of mTOR/p70S6K, promoted autophagic lysosomal pathway and followed by reducing Aβ generation in APP/PS1 mice and N2a cells.
AGE has been evidenced as a major source of neurotoxicity in AD via promoting Aβ aggregation and tau hyperphosphorylation. AGEs interacting with RAGE is associated with AD pathology and regulates AβPP processing through increasing the expression of cathepsin B and asparagine endopeptidase, or conjunction with the reactive oxygen species, which increased Aβ formation [45, 46]. In this study, D-ribose caused the overexpression of AGEs, promoted the intensity of mTOR signaling pathway, and inhibited autophagy. Then we detected whether BBR regulates the expression of AGEs and thus influences the mTOR signaling pathway and autophagy. After adding BBR, the express of AGEs and mTOR/p70S6K signaling were inhibited and autophagy was enhanced. In addition, BBR also counteracted the increase of BACE1 and γ-secretase and the decrease of Neprilysin induced by D-ribose. These results reveal that BBR may enhance autophagy by inhibiting the expression of AGEs and the activation of mTOR/p70S6K signaling pathway and reduce the level of Aβ by inhibiting the expression of β/γ-secretase and increasing the protein level of Neprilysin.
Furthermore, we intuitively detected the effect of BBR on expression of Aβ in the hippocampus of D-ribose treated mice. The results showed that BBR significantly inhibited the signal intensity of Aβ. Moreover, Morris water maze experiments showed that BBR improved cognitive impairment of AD mice. It should be noted that there is not definitive link of Aβ pathology with changes in cognitive performance beyond temporal correlation in this report. In addition, the presence of other transcription factors underlying AGEs and autophagy interacting to induce the downregulation of Aβ remains unknown, and the factors through which BBR exerts its effect should be further investigated.
Ribosylation-induced Aβ pathology has been well accepted across all animal species, and it is therefore expected that seeking a selective inhibitor limiting ribosylation-induced Aβ pathology will be an important defense against internal Aβ pathological injury. The discovery provides a powerful pathway to help to meet these needs, which BBR ameliorates ribosylation-induced Aβ pathology via inhibiting mTOR/p70S6K signaling.
In conclusion, we demonstrated that BBR restrains Aβ generation in the APP/PS1 mice and N2a cells. Firstly, BBR promotes autophagic lysosomal pathway by suppressing mTOR/p70S6K signaling and promoting the expression of autophagy-related protein Beclin1, Atg3, and LC3B, and promotes Aβ clearance. Moreover, BBR also inhibits ribosylation-induced Aβ production and promotes Aβ clearance in APP/PS1 mice and N2a cells. Together, these findings reveal ameliorating ribosylation-induced Aβ pathology by BBR via inhibiting mTOR/p70S6K signaling and imply that it may be possible to develop BBR as a new medication of anti-Aβ pathology, by which BBR both inhibits ribosylation-induced Aβ generation and promotes Aβ clearance, and consequently help to moderate the strong drive to relieve Aβ pathology.
Footnotes
ACKNOWLEDGMENTS
This work was supported by the Chongqing Natural Science Foundation (cstc2018jcyjAX0602), the Science and Technology Planning Project of Yuzhong District of Chongqing (20180104), the Medical Science and Technology Innovation Fund project of Chongqing General Hospital (Y2018MSXM03) and the Chongqing Health and Family Planning Scientific Research Project (ZY201702042).