
Abstract
Given the highly multifactorial origin of Alzheimer’s disease (AD) neuropathology, disentangling and orderly knowing mechanisms involved in sporadic onset are arduous. Nevertheless, when the elements involved are dissected into smaller pieces, the task becomes more accessible. This review aimed to describe the link between c-Jun N-terminal Kinases (JNKs), master regulators of many cellular functions, and the early alterations of AD: synaptic loss and dysregulation of neuronal transport. Both processes have a role in the posterior cognitive decline observed in AD. The manuscript focuses on the molecular mechanisms of glutamatergic, GABA, and cholinergic synapses altered by the presence of amyloid-β aggregates and hyperphosphorylated tau, as well as on several consequences of the disruption of cellular processes linked to neuronal transport that is controlled by the JNK-JIP (c-jun NH2-terminal kinase (JNK)–interacting proteins (JIPs) complex, including the transport of AβPP or autophagosomes.
INTRODUCTION
The study of neurodegenerative diseases has evolved significantly in the last few decades and over time, increasing amounts of evidence are being gathered to understand the mechanisms involved in their appearance. Nevertheless, for now, the enigma of the onset of many of them and which effective therapies may stop their progress, remains unanswered.
One of the most prominent neurodegenerative pathologies is Alzheimer’s disease (AD) [1,2, 1,2]. Initial descriptions of AD established that it was characterized by cognitive affectations caused by neuronal loss [3]. It was also linked to the presence of amyloid-β (Aβ) depositions and hyperphosphorylated tau protein, which resulted in senile plaques and neurofibrillary tangles [3]. Nowadays, although it is still believed that they have a significant relevant function in the pathology, these classical hallmarks are not considered the cause of AD but rather one of its many features [4]. Other alterations include the appearance of metabolic imbalances [5–7] and inflammatory stages’ development, which favor neurodegeneration [8–10]. Several additional metabolic alterations have also been identified in AD pathology, including the appearance of oxidative stress [11, 12], dysregulation of autophagic activity [13, 14], and insulin resistance [15–17], among others. Importantly, all these clinical alterations appear late in the time course of the disease, with the initial onset up to 20 years before the first clinical symptoms [18]. Consequently, new studies need to be run to analyze different potential elements that may be part of the pathology’s early stages. These would presumably identify additional targets involved in the development of effective treatments or preventive strategies.
Considering all these clinical manifestations, it is believed that AD could be a consequence of small changes in cellular functions that build up over time and eventually lead to the loss of synaptic contacts and the disruption of regular axonal transport [19–22], which would lead to the clinical cognitive deficit that is observed in diagnosed AD patients. Considering the wide arrange of elements that seem to take part in the pathology, some research has focused on identifying factors that may behave as cellular master modulators.
The c-Jun N-terminal Kinases (JNK) are a subfamily of the mitogen-activated protein kinases responsible for several intra- and extracellular stimuli [23]. They are subdivided into three different isoforms (JNK1, JNK2, and JNK3), which are expressed from three different genes (Mapk8, Mapk9, and Mapk10) [24–26]. Through their activity, they control a wide arrange of cellular mechanisms and, consequently, they are linked to both physiological and pathological situations [23]. Importantly, JNK regulation of many functions is precisely regulated, both in isoform and tissue manner [23]. Furthermore, gathered data has demonstrated that small and short-lasting changes in JNK activity are enough to regulate cellular responses and the maintenance of homeostatic patterns. Also, it has been described that an increase of JNK activity, either sustained or as acute bursts, leads to the activation of apoptotic mechanisms that occur in pathological stages [27, 28].
Given the significant involvement of the JNKs in many pathological stages, their role in the development of AD has also been studied in the past [28]. It has been described that there is increased activation of the JNKs in AD patient samples both from nervous tissue and cerebrospinal fluid [29]. JNK favors the amyloidogenic pathway of the amyloid-β protein precursor (AβPP) [30–32]. Moreover, JNK directly phosphorylates of tau, a relatively early marker of AD [33]. Moreover, JNK activation has also been linked to the appearance of insulin resistance [34–36], increasing sensibility of neurons to the activation of apoptotic pathways [37, 38]. Furthermore, the evidence shows a correlation between the JNK activation rate and cognitive affectation [29]. In addition, the JNK pathway also regulates microtubule and actin dynamics through the control of cytoskeletal proteins, a crucial process for correct axonal transport and the structure of presynaptic and postsynaptic domains [39, 40] (Fig. 1).
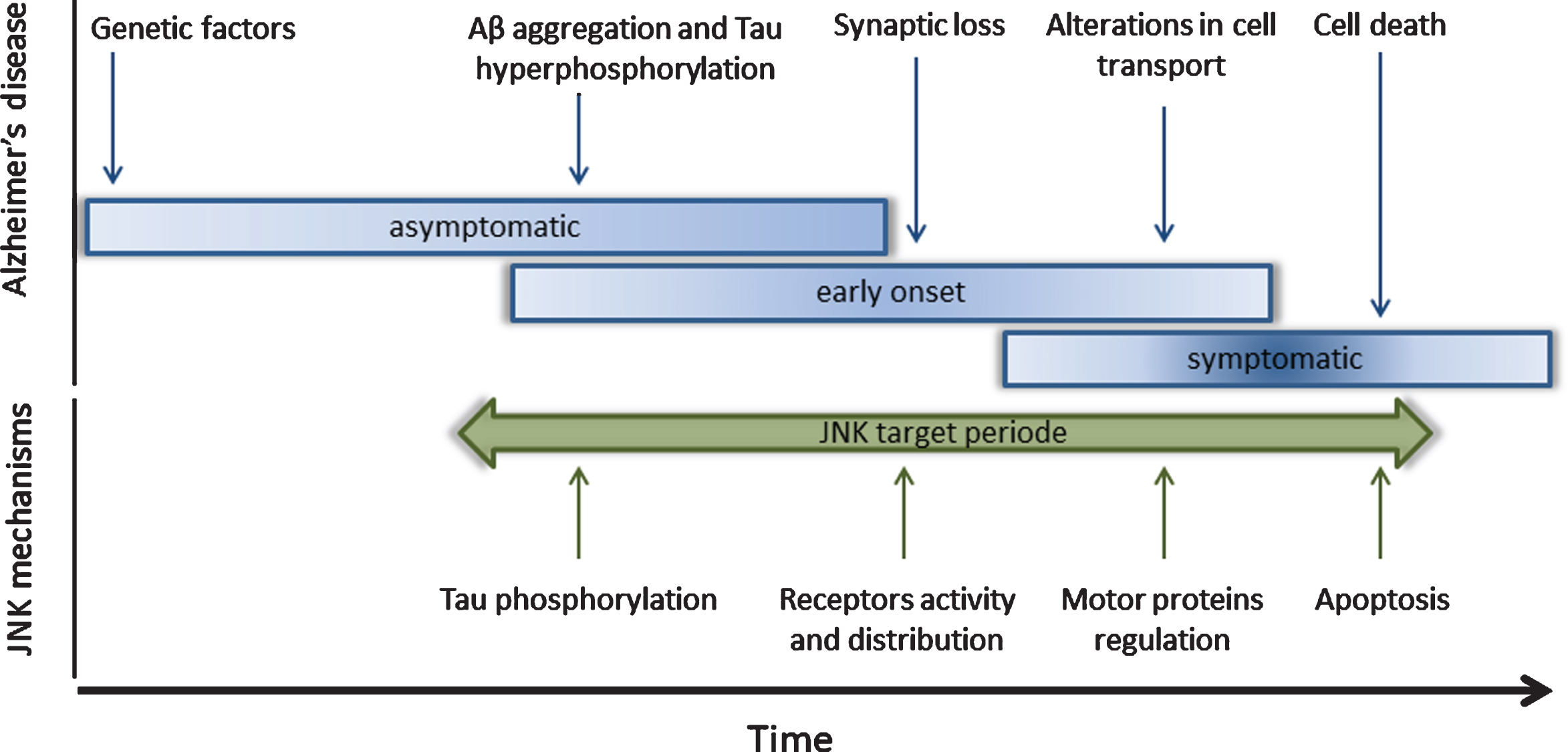
c-Jun N-terminal Kinases (JNK) have been identified as target molecules for Alzheimer’s disease (AD) treatment. In the upper part of the figure, the AD pathology has been subdivided into different molecular stages. At the asymptomatic stage, the genetic factors are important for the beginning and the severity of the disease. During the early onset stage, Aβ aggregates and hyperphosphorylated tau induce synaptic loss and alteration in neuronal transport. These impairments trigger cell death and memory deficits that are observed in the symptomatic stage. In the lower part of the figure is shown the role of c-Jun N-terminal Kinases (JNK) in the different cellular processes that are affected along the AD stages, such as the tau phosphorylation, synaptic maintenance, neuronal transport, and cell viability.
Considering this information, some research gro-ups have already aimed to regulate JNK activity to prevent, revert, or ameliorate some of AD’s pathological features [28, 41]. This review will analyze the link between the JNKs and AD, specifically on the processes of synapse loss and the disruption of neuronal transport (Fig. 1).
SYNAPTIC LOSS AND JNKs
One of the earliest alterations of AD is the loss of synapses in most brain areas, an event that can be linked to the toxicity of Aβ, the hyperphosphorylation of tau, or other molecular dysregulations [42–44]. The characterization of cerebrospinal fluid in cohort studies in patients of AD demonstrated an increase in the levels of circulating synaptic proteins, suggesting the disassembling of synaptic structures in the early non-symptomatic stages [45]. Animals models have corroborated these observations by reporting how synthesis of synaptic proteins is dysregulated [46].
The chronology of synaptic dysfunction in AD is not fully described, but some general hypotheses have been formulated. It is known that early events in AD and other dementias are triggered initially by the accumulation of Aβ oligomers in synaptic terminals, followed by axonal alterations, aberrant sprouting, and deficit of trophic factors [47]. Tau pathology is believed to appear secondarily or as a consequence of damage by oligomeric species of Aβ; late changes involve the loss of synaptic contacts, oxidative stress, inflammation, and neuronal loss [47]. Different studies have associated these molecular alterations with the activity of the JNKs [48, 28].
Glutamatergic synapses
Glutamatergic transmission is especially vulnerable to Aβ toxicity, producing a disruption of excitatory synaptic transmission and neuronal plasticity. Overstimulation of postsynaptic N-methyl-D-aspartate receptors (NMDAR) produces an increase in the levels of Aβ in the extrasynaptic space and hyperexcitability of neurons [49]. This neuronal hyperexcitability has been linked to the epileptic seizures observed in AD patients [50] (Fig. 2A). Moreover, it induces NMDAR internalization and posterior downregulation of extracellular signal-regulated kinase 1/2 (ERK1/2) that alters the synaptic plasticity [51–53].
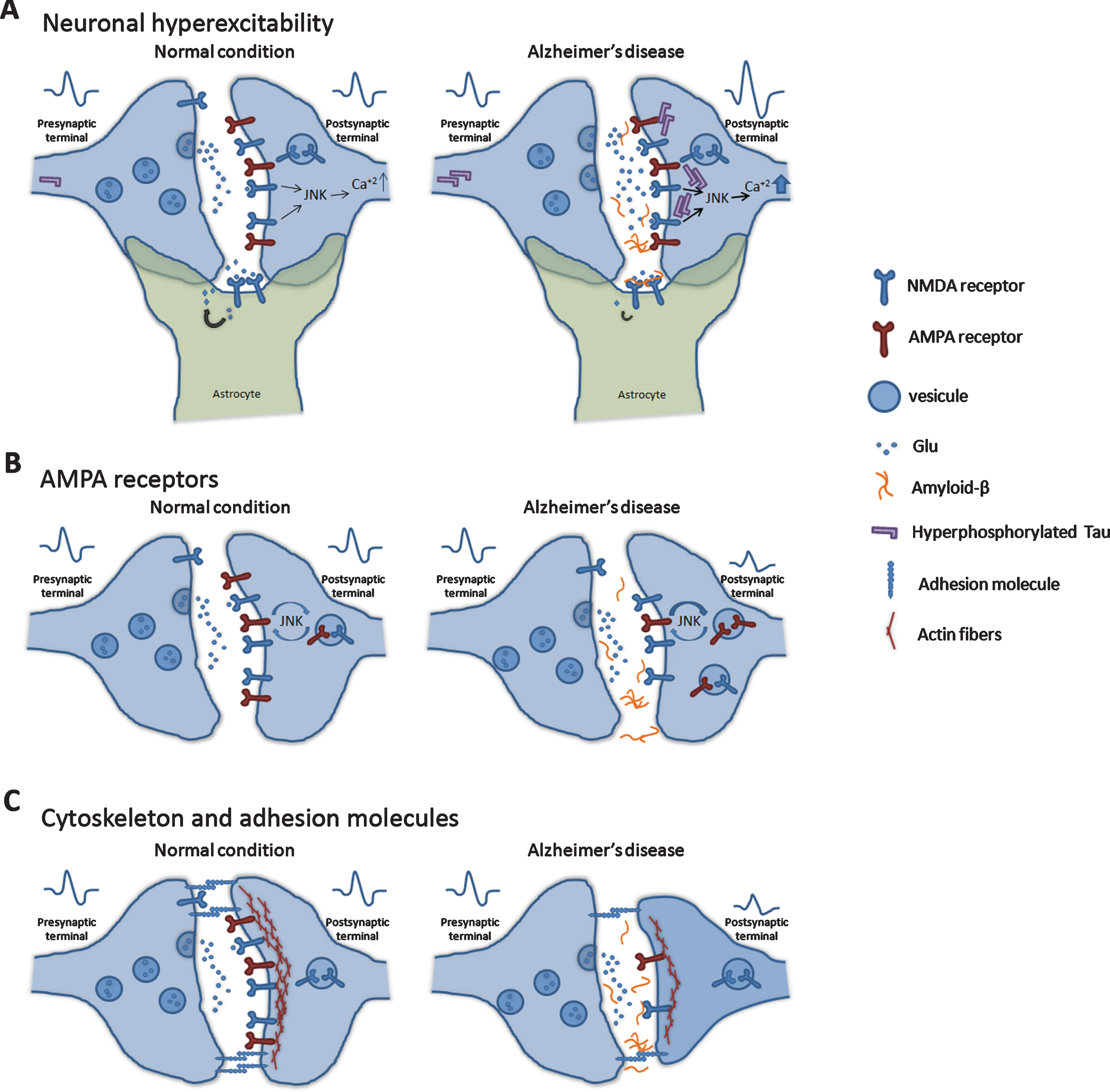
Molecular mechanisms in glutamatergic synapses. One of the earliest alterations of AD is the loss of synapses linked to Aβ and hyperphosphorylated tau’s toxicity. Neuronal hyperexcitability. A) Accumulation of Aβ produces an increase in glutamate levels in the extrasynaptic space, which leads to an overstimulation of postsynaptic NMDAR and the hyperexcitability of neurons. JNKs can regulate calcium influx and synaptic plasticity. Moreover, the presence of hyperphosphorylated tau in synaptic areas alters their structure and neuronal transmission. Also, in astrocytes, Aβ interacts with glutamate transporters and inhibits glutamate clearance from the extrasynaptic space. B) In AD context, AMPAR in the postsynaptic regions has been observed to decrease their density, which induces synaptic depression and reduces synaptic plasticity. The AMPAR density is controlled by JNK signaling. Synaptic structure. D) Aβ species change the localization and expression of synaptic adhesion molecules and alter the actin cytoskeleton, producing a disruption of the cell structure and neuronal network impairments.
In astrocytes, Aβ interacts with glutamate transporters and inhibits glutamate clearance from the extrasynaptic space [54, 55]. Over time, this situation causes a drop in the expression of GluN2A and GluN2B subunits of NMDAR and GluA1 and GluA2 subunits of the α-Amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor (AMPAR). Moreover, the density of AMPAR decreases in postsynaptic regions due to the effects of Aβ [56–58]. This reduction induces synaptic depression and downregulates synaptic plasticity [59–61]. Some studies report that the JNKs directly regulate the trafficking of AMPAR, controlling neuronal activity and plasticity [62, 63] (Fig. 2B).
The molecular mechanics that downgrade synaptic function and spines due to JNK activity are still under study but, several targets of JNK are cytosolic substrates that regulate cytoskeletal maintenance in dendrite and axon. In this line, the JNKs, specifically isoforms JNK2 and JNK3 are localized in presynaptic sites and interact with Syntaxin-1, Syntaxin-2, and Synaptosomal-Associated Protein, 25kDa (Snap25), suggesting that JNK activation in presynaptic regions might be related to glutamate neurotransmitter release in the synaptic space [64, 65]. JNK is activated in the postsynaptic space by a wide range of stimuli, including the excitotoxicity produced by NMDA receptors’ overactivation. In AD, JNK inhibition prevents neuronal death in cortical and hippocampal neurons, preventing caspase-3 activation [66] and the loss of glutamatergic synapses [67]. In this line, several studies have evidenced the neuroprotective effect of JNK inhibition in models of epilepsy [51–53] (Fig. 2A).
Finally, Coffey and colleagues described in a recent publication that NMDA-mediated or corticosterone-induced cellular stress in hippocampal neurons in rats exerts rapid reorganization of actin dendritic spine structures through the activity of the JNKs. Furthermore, they supported these results with optogenetic control of the activity of the JNKs, which prevented the alteration of dendritic structures [68].
Cholinergic synapses
Loss of synapsis in basal forebrain cholinergic neurons (BFCNs) is linked to a deficit in stimulation and is involved in cognitive decline and memory loss in AD. In physiological conditions, BFCNs are continuedly maintained by neurotrophic signaling mediators like the brain-derived neurotrophic factor (BDNF) and the nerve growth factor (NGF), promoting cell survival, axon and dendrite maintenance. In AD patients, there is a deficit of these neurotrophic signals causing axonal pruning and degeneration, together with the activation of apoptotic pathways, mediated by the JNKs [69–72] (Fig. 3).
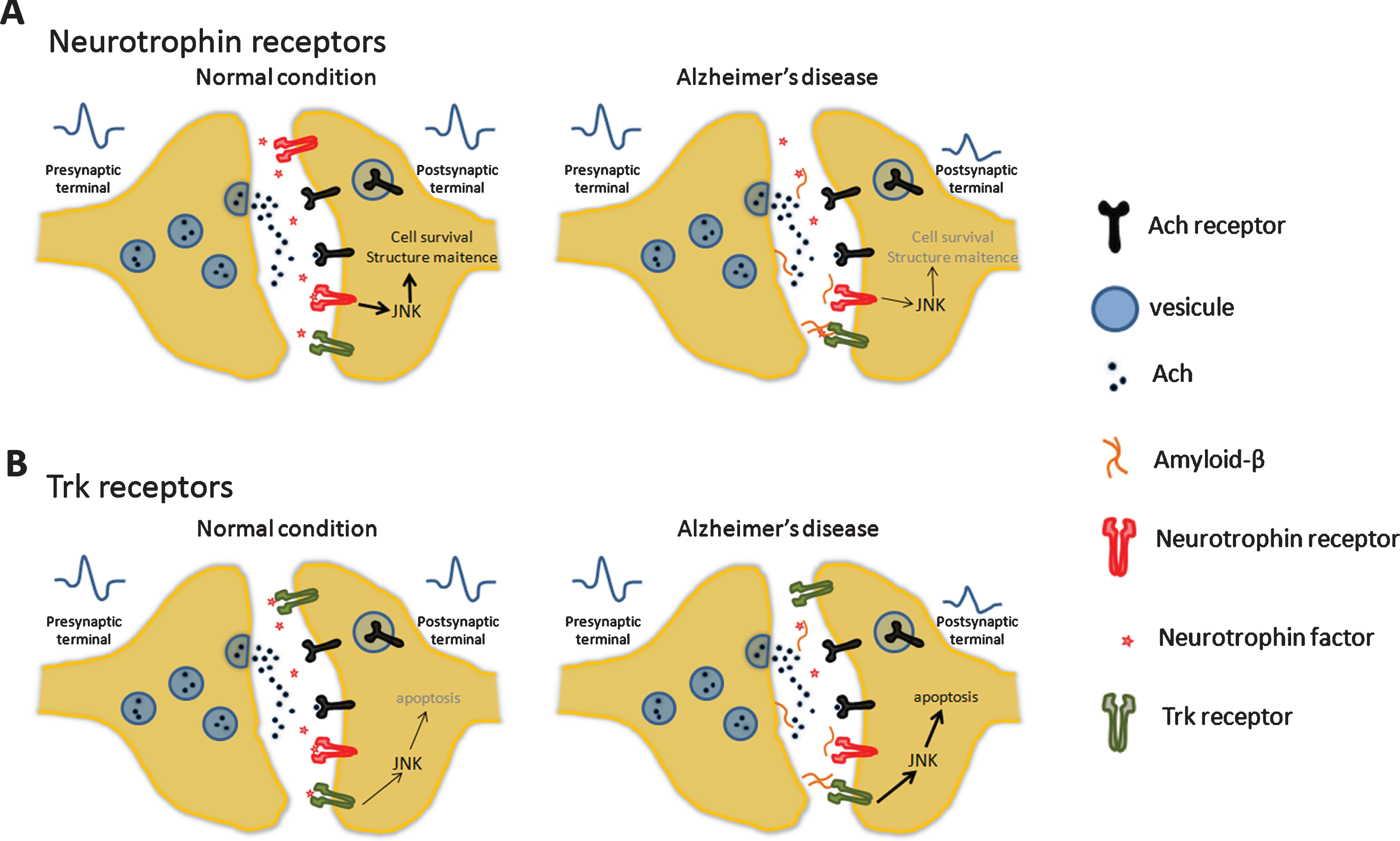
Cholinergic synapses. A) In basal forebrain cholinergic neurons, brain-derived neurotrophic factor (BDNF) and the nerve growth factor (NGF) promote cell survival and axon and dendrite maintenance. B) In AD, these processes have a dysregulation and occur an axonal pruning and degeneration. Change in neurotrophic levels reduced the Trk activation that enhances the apoptosis via JNK pathway.
Furthermore, increased expression of p75 in AD brains also favors neurotrophic signaling dysfunction [73, 74]. This mechanism has also been linked with an increase in the amyloidogenic pathway activity since p75 can interact with the beta-site amyloid precursor protein cleaving enzyme 1 (BACE1), an essential enzyme in the production of Aβ. This step in the formation of Aβ from AβPP has also been linked to the activity of the JNKs [75].
GABAergic synapses
Recent studies in AD patients and mouse models detected decrease expression on gamma-Amino-butyric acid (GABA) subunits receptor, indicating deficiencies in the inhibitory synaptic activity and neuronal transmission in the brain [76–79]. Changes in the inhibitory transmission enhance neuronal hy-perexcitability, producing the epileptic seizures observed in AD [80]. Moreover, GABAergic impairment has been associated with depressive behavior in AD patients. Different studies in AD mouse models have indicated that abnormal levels of Aβ oligomers, an increase in tau hyperphosphorylation and aggregation interfere with GABA inhibitory activity (Fig. 4). This aberrant brain activity in the hippocampal and septohippocampal circuits has been correlated with cognitive impairment detected in AD mice models [81–84].
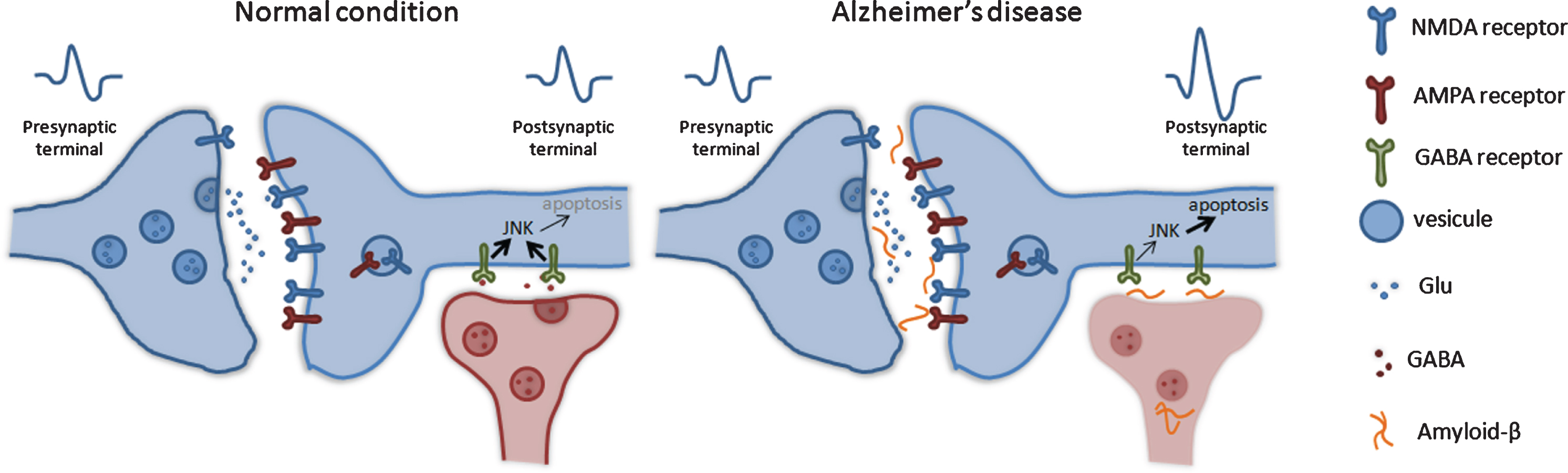
GABAergic transmission. GABA is the main inhibitory neurotransmission in the brain and crucial for learning tasks. In AD patients, significantly lower GABA neurotransmitter levels were observed, indicating synaptic function and neuronal transmission deficiency in AD. The changes in the inhibitory transmission enhance the neuronal hyperexcitability, which produces the epileptic seizures and apoptosis. JNK may regulate these processes.
As in the previously described alterations, the JNKs also seem to have a relationship with GABA neurotransmission. In several pathological environments, it has been described how GABA activity inhibits JNKs, providing neuroprotective profiles by reducing the activation of inflammatory and apoptotic responses [85–88].
Tau hyperphosphorylation
The presence of hyperphosphorylated tau in synaptic structures alters their physiological conditions affects the localization of receptors and the distribution of postsynaptic density proteins needed for normal synaptic plasticity. Furthermore, species of hyperphosphorylated tau cause changes in synaptic structure and transmission. It has been observed that tau recruits Fyn to NMDAR/PSD95 complexes and is required to mediate excitotoxicity induced by Aβ and excessive glutamate levels (Fig. 2A) [89, 90]. Also, tau limits the binding of Synaptic Ras GTPase-activating protein 1 (SynGAP1) at postsynaptic regions, promoting local negative regulation of the excitotoxic ERK1/2 pathway [91–93]. Furthermore, several studies have described that tau hyperphosphorylation is linked to JNK activity [33]. In fact, a recent study by He B and colleagues describes how downregulation of the activity of the JNKs reduced tau hyperphosphorylation rates [94].
Aβ accumulation
Besides modulating synaptic NMDA and AMPA receptors’ activity, Aβ species change the localization and expression of synaptic adhesion molecules, producing a disruption of the neuronal network (Fig. 2B). These proteins, such as Neural cell adhesion molecule 2 (NCAM2) and N-cadherin, are crucial for forming and maintaining synapses. Significantly, N-cadherin disruption by Aβ induces JNK activation in mice neuronal culture [95–100].
Aβ species also disrupt the actin cytoskeleton, producing impairments in the structure of dendritic spines and altering neuronal transmission [101–103] (Fig. 2B). Moreover, another study shows that Aβ activates the Rho-associated protein kinase (ROCK) pathway, which results in phosphorylation of cofilin-1, which changes actin dynamics [104].
Other cellular mechanisms controlled by JNKs, such as energetic imbalances, mitochondrial and en-doplasmic reticulum stress, or inflammatory res-ponses are potentially involved in synaptic loss [16, 105–109].
ALTERATIONS IN NEURONAL TRANSPORT AND JNKS
JNK-interacting proteins (JIPs) control axonal transport
JIPs are scaffold proteins that recruit JNKs family members through JNK-binding domain (JBD) [110] and MAPK phosphatases through another subdomain [111]. These scaffolding proteins, depend on the stimuli, are specifically modulated and differentially located in the cell [112, 113]. JIPs are subdivided under four different members: JIP1/Islet-Brain 1 (IB), JIP2/Islet-Brain 2 (IB2), JIP3/stress-activated protein kinase-associated protein 1(JSAP) and JIP4/JNK-Associated Leucine-Zipper Protein (JLP). JIP1 and JIP3 are adaptors of molecular protein motors and regulators of synaptic vesicle movement and axonal growth [114, 115]. JIP3 regulates the transportation of phosphorylated AβPP in Threonine 668 by JNK in neurites. The deficit of JIP3 causes accumulation of phospho-AβPP in the soma that leads to neurotoxic effects [116, 117].
JIP and JNK control the autophagosomes movement
In neurons, autophagosome biogenesis may happen in distal axons and exhibit bidirectional movement (Fig. 5A) [118]. Sustained retrograde movement is needed to transit from the distal axon to the proximal axon or soma to fuse with lysosomes. Thus, the bidirectional movement of autophagic vacuole occurs and is regulated by a putative JNK activity that may impede the early exit of the autophagosomes. Phosphorylation of S421 of JIP1 by JNK may switch the direction of autophagosomes [119, 120]. Misfunction of the autophagy pathway has been related to AD since failures in this cellular process may contribute to a reduction in the clearance of Aβ aggregation (Fig. 5B) [118]. Therefore, JNK could be a target in the control of the autophagosomes movement.

Neuronal transport alterations. A) In physiological conditions, a: APP is transported in synaptic vesicles that contain TrkA, GABAB receptors, BACE1 and PSEN1 (SV-APP vesicles). Vesicles are transported by kinesin-1 and scaffold protein JIP1/3, through microtubule roads in anterograde directions. b: The autophagic process degrades AβPP as it occurs with aged-organelles, misfolded protein and other intracellular material. Endosomes and autophagosomes are transported retrogradely by the cytoplasmic dynein motor protein. B) In pathological conditions, c: JIP1/3 phosphorylation by JNK is necessary to control the anterograde direction; dysfunction in the axonal transport leads to disassembling synaptic vesicle prompts the amyloidogenic pathway. d: Dysfunction of retrograde transport of lysosomes and autophagosome leads to axonal swelling and sustained activation of JNK3. This sustained activity might induce the amyloidogenic pathway and favor tau pathology.
Edwards and colleagues described a relevant role for JIP3 and JNK in the control of axonal transport. They found that in motor neurons of C. Elegans, UNC-16 (ortholog of JIP3) regulates the organelle entrance at the axon initial segment, controlling axon organelle composition. Thus, unc-16 null mutants accumulate high levels of organelles in their axons. Moreover, these mutants exhibit alterations in organelles’ bidirectional transport, mainly in lysosomes located in synaptic regions. The jnk1 C. Elegans null mutants also accumulate lysosomes in axons in lesser extension, supporting that UNC-16 and JNK-1 act as a part of the same process and regulate lysosomal transport [121].
The relevance of JIP3 in axonal transport was also reported in sensory axons of null mutant Jip3 zebrafish larvae. In this model, phospho-JNK3 tended to accumulate in axonal swellings with lysosomes. Other retrograde cargoes, such as autophagosomes or late endosomes, were not affected by lacking Jip3 [122]. Moreover, transportation of lysosomes and JNK3 seems to be independent because transfection of partial full Jip3 (lacking JBD) only restored lysosome accumulation but not the swelling and accumulation of phospho-JNK3 [122].
JNK3 induces accumulation of phospho-AβPP products in neuronal soma
AβPP protein is best known as the precursor of Aβ molecules. However, it is an integral membrane protein that can be found in presynaptic and postsynaptic areas where it forms trans-synaptic contacts that, in physiological conditions, contribute to the maintenance and stability of synaptic structures [123]. Proteomic studies of the presynaptic active site revealed that AβPP is an important molecule forming a part of the scaffold machinery that stabilizes the docking and release of synaptic vesicles into the synaptic cleft [124, 125]. Furthermore, AβPP takes a role in trafficking and localization of GABA-B receptors (GBRs) in the presynaptic space [126, 127] (Fig. 5A).
After synthesis, AβPP sorts inside synaptic vesicles (SV-AβPP) to the trans-Golgi network from where they are transported to both dendrites and axons. A deficit in AβPP axonal transport has been suggested to be part of AD pathology’s early event. SV-AβPPs are co-transported jointly with other components such as BACE1, Presenilin-1 (PS1), Tropomyosin kinase receptor A (TrkA, Growth Associated Protein 43 (GAP43), Synapsin I (Fig. 5A) [123, 128]; and evidence points out that missorted or stagnated AβPP vesicles might promote the activation of the amyloidogenic pathway, favoring the production of Aβ aggregates [129].
Yoon et al. clarified the role of JNK3 and the endocytic pathway in the reduction levels of the full-length AβPP on the cell surface [130]. They observed that JNK3 is the responsible isoform of AβPP phosphorylation at the threonine 668 site, favoring the internalization of AβPP in endosomes leading to neurotoxic effects (Fig. 5B) [130]. This study shows that in 5×FAD mice (transgenic Alzheimer’s disease model mice carrying five mutations in APP and PSEN1), phospho-JNK was colocalized with phospho-AβPP (threonine 668 site) in neuritic process hinting that activated JNK3 induces AβPP phosphorylation. In addition, tau hyperphosphorylation was found in the dystrophic axon, suggesting that JNK3 activation is a common event and may one of the first signals in triggering the brain pathology (Fig. 5B).
In this way, the neurotrophic factor NGF impedes the AβPP phosphorylation by the JNKs through the interaction with Isoform C of the SH2-containing sequence adaptor (ShcC). This effect increases the non-amyloidogenic pathway activity and promotes the localization of AβPP in the Golgi apparatus and the membrane cell surface [131]. Above observation has been supported at pharmacological level using peptide (D-JNKI1) that inhibits JNK activity in the brain parenchyma. Thus, the AβPP phosphorylation in Threonine 688 is blocked, reducing the formation of Aβ species, CTFβ fragments and Aβ oligomers in favor of the non-amyloidogenic pathway that prevents the loss of synaptic function in hippocampal neurons [32]. Therefore, the inhibition of JNK3 would be a potential target to block the amyloidogenic pathway’s development.
CONCLUSIONS
After reviewing evidence, it can be concluded that JNKs have an essential role in controlling initial events that lead to AD’s development. Their activation alters synaptic activity and neuronal transportation of AβPP and vesicles from endosomal recycling pathways (lysosomes and autophagosomes). Consequently, JNK dysregulation leads to loss of synaptic contacts, cell death, and amyloidogenic pathway upregulation. The importance of the JNK3 isoform in all these cellular processes has been highlighted.
For all that, the specific inhibition of JNK isoforms in precise neuron subpopulations could be a candidate strategy to restore axonal transport and physiological synaptic activity by preventing the amyloidogenic pathway’s consequences other alterations. Accordingly, to understand the role of JNK isoform in neuronal transport, synapsis formation and maintenance would be a useful tool to design new therapeutic approaches for AD.
Footnotes
ACKNOWLEDGMENTS
The research group is partly supported by funds from the Spanish Ministerio de Economía y Competitividad (SAF2017-84283-R to AC), the Generalitat de Catalunya (2014SGR-525 to AC) (2017 SGR 625 to CA) and CIBERNED (Grant CB06/05/2004 to AC). Fellowship Grodman Academic International Specialization Stays 2018 B (University of Guadalajara, Foundation USA) to RDCT.