
Abstract
Background:
Epidemiological studies have shown that tooth loss is associated with Alzheimer’s disease (AD) and dementia. However, the molecular and cellular mechanisms by which tooth loss causes AD remain unclear.
Objective:
We investigated the effects of tooth loss on memory impairment and AD pathogenesis in AppNL-G-F mice.
Methods:
Maxillary molar teeth on both sides were extracted from 2-month-old AppNL-G-F mice, and the mice were reared for 2 months. The short- and long-term memory functions were evaluated using a novel object recognition test and a passive avoidance test. Amyloid plaques, amyloid-β (Aβ) levels, glial activity, and neuronal activity were evaluated by immunohistochemistry, Aβ ELISA, immunofluorescence staining, and western blotting. The mRNA expression levels of neuroinflammatory cytokines were determined by qRT-PCR analysis.
Results:
Tooth loss induced memory impairment via an amyloid-cascade-independent pathway, and decreased the neuronal activity, presynaptic and postsynaptic protein levels in both the cortex and hippocampus. Interestingly, we found that tooth loss induced glial activation, which in turn leads to the upregulation of the mRNA expression levels of the neuroinflammation cytokines tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and IL-1β in the hippocampus. We also found that tooth loss activated a stress-activated protein kinase, c-Jun N-terminal kinase (JNK), and increased heat shock protein 90 (HSP90) levels in the hippocampus, which may lead to a glial activation.
Conclusion:
Our findings suggest that taking care of teeth is very important to preserve a healthy oral environment, which may reduce the risk of cognitive dysfunction.
INTRODUCTION
Alzheimer’s disease (AD) is the most common age-associated chronic progressive neurodegenerative disease. AD is pathologically characterized by extracellular deposition of amyloid-β (Aβ) in senile plaques and the intracellular accumulation of the hyperphosphorylated tau protein in neurofibrillary tangles [1]. Additional pathological hallmarks of the disease include inflammatory processes, synaptic and neuronal losses, and cerebral amyloid angiopathy [2]. More than 95%of all AD cases occur in individuals over the age of 60 years and are defined as sporadic AD or late-onset AD. The cause of sporadic AD is likely to be multifactorial, with external or environmental factors interacting with biological or genetic susceptibility to accelerate the manifestation of the disease [3]. The incidence of AD increases markedly with age; however, the cause of sporadic AD remains unresolved and treatment strategies are directed towards its symptoms, and they do not delay the progression of the disease or even prevent its onset [4].
Oral health is essential for maintaining general health and well-being in every stage of life. Tooth loss due to dental caries and periodontitis can trigger or exacerbate diseases such as diabetes, endocarditis, other inflammatory diseases, and AD [5]. Masticatory stimulation has important functions related to physical, mental, and social health [6, 7]. On the basis of findings of animal and human studies, it has been proposed that there is a probable causal connection between masticatory and cognitive functions, which are partly related to the number of functional teeth [8]. In animals, masticatory dysfunction induced by tooth removal inhibits hippocampal neurogenesis and leads to hippocampus-dependent spatial memory and learning deficits [9, 10]. In older men, periodontal disease progression and tooth loss were associated with an increased risk of poor cognitive performance [11]. Taken together, these studies indicated that memory and learning functions are significantly impaired owing to periodontal disease, tooth loss, or feeding on a soft diet which causes masticatory dysfunction. Therefore, mastication appears to play an important role in maintaining hippocampus-dependent cognitive function.
Mastication generates sensory signals, which are transmitted to various regions of the central nervous system (CNS) [12]. According to the enormous body of evidence collected so far, mastication has proved to be effective in transmitting a huge amount of sensory information to the brain and maintaining learning and memory functions of the hippocampus [13]. A reduction of sensory input caused by masticatory dysfunction due to tooth loss decreases the synaptic density in the CNS, which impairs memory function [14, 15]. In addition, masticatory dysfunction acts as a source of chronic stress leading to the increased levels of circulating glucocorticoids, which cause hippocampus-dependent cognitive deficits [16, 17]. These findings suggest that chronic stress induced by decreased mastication due to tooth loss impairs cognitive function.
Reactive gliosis is observed under various conditions, such as infection, ischemia, trauma, and neurodegenerative diseases [18]. Neuroinflammation and neurodegeneration are common features of familial and sporadic AD. In the brains of AD and transgenic mouse models that overexpress mutated human amyloid-β protein precursor (AβPP), reactive microglia and astrocytes are found around Aβ plaques and can play beneficial or harmful roles in disease progression [19, 20]. For example, reactive microglia and astrocytes can contribute to the clearance of Aβ [21, 22]. Conversely, neuroinflammatory cytokines such as tumor necrosis factor (TNF-α), interleukin-6 (IL-6), or IL-1β produced following glial activation are harmful and toxic to neurons [23, 24]. AβPP processing and Aβ accumulation result in the activation of microglia and astrocytes [25]. It has also been reported that inflammation occurs before the accumulation of the Aβ protein and plaque deposition in AβPP transgenic mice [26], suggesting that inflammation is an early response in this AD model. However, the mechanism by which masticatory dysfunction due to tooth loss induces glial activation has not yet been fully elucidated. Although we previously demonstrated that tooth loss in AβPP transgenic mice (Tg2576 mice) showed memory deficits and neuronal cell loss in the hippocampus [10], the pathological factors including neuroinflammation, glial activation, and chronic stress were not evaluated.
In this study, we determined the effects of tooth loss on memory impairment and AD pathogenesis, including its effects on Aβ levels, neuronal activity, synaptic loss, glial activation, and the stress-inducible proteins pJNK and HSP90 by using App-KI mice, AppNL-G-F.
MATERIALS AND METHODS
Experimental animals
The original lines of AppNL-G-F mice with a C57BL/6J genetic background were obtained from RIKEN Center for Brain Science (Wako, Japan). These mice express humanized Aβ with familial APP mutations [Swedish (NL), Beyreuther/Iberian (F), and Arctic (G)] [20]. AppNL-G-F mice develop no obvious deficits in spatial learning and memory up to 6 months of age [27], and Aβ plaque deposition starts at 2 months and saturates at around 7 months [20]. AppNL-G-F mice were divided into the experimental (male = 4 and female = 4) and control (male = 4 and female = 4) groups. In the experimental group, maxillary molar teeth on both sides were extracted from 2-month-old mice under general anesthesia induced with the combination of midazolam (4 mg/kg), butorphanol (5 mg/kg), and medetomidine (0.3 mg/kg) injected intraperitoneally. In the control group, the maxillary molar teeth on both sides remained intact. After tooth extraction, the experimental and control mice were fed with powder diet for 2 months. All mice were housed under a 12 h light/dark cycle and had access to food and water ad libitum. All animal experiments were performed in accordance with the Guidelines for Animal Experiments of the Animal Experimentation Committee of Nagoya City University.
Novel object recognition test
The novel object recognition test was conducted on 4-month-old mice. This test is based on the innate tendency of rodents to explore novel objects over familiar ones and is believed to measure episodic memory. The experimental procedure was as described previously with some modifications [28, 29]. Briefly, the novel object recognition test consisted of three sessions: habituation, training, and retention. Each mouse was individually habituated to a box (40×40 cm2 and 40 cm high) by allowing it to explore the box without objects for 5 min for 3 days (habituation session). Twenty-four hours after the last habituation session, the mouse underwent a 5 min training session of exposure to two identical objects in an open field box. The time spent exploring each object was recorded by a video camera. After the training session, the mouse was returned to its home cage. After an interval of 24 h, the mouse was returned to the same box containing two objects, one identical to the objects in the training session but previously unused and one novel object. The mouse was allowed to explore for 5 min in the retention session, during which the amount of time in exploring each object was recorded. Throughout the experiments, the objects used were matched in terms of their physical complexity and emotional neutrality.
Passive avoidance test
The step-through passive avoidance test was performed as described previously [10, 30]. Briefly, the apparatus for this test consisted of a lighted compartment (90×115 mm2 and 150 mm high) and a dark compartment (140×175 mm2 and 150 mm high) with a steel rod grid floor connected to a shock generator. For the acquisition trial, each mouse was placed in the light compartment and allowed to explore freely. The mouse was allowed to move to the dark compartment. The latency time until the mouse completely entered the dark compartment was measured. The door separating the lighted and dark compartments was closed as soon as the mouse entered the dark compartment. The mouse upon entering the dark compartment completely received a foot shock of 0.1 mA for 3 s through a steel rod grid. After the acquisition trial, the mouse was returned to its home cage. Forty-eight hours later, the mouse was placed again in the apparatus. When the mouse stayed in the lighted compartment, it is considered that it retained the memory or remembered the aversive stimulus. When the mouse entered the dark compartment, it is considered that it had an impaired memory of the aversive stimulus. The maximum cut-off latency time was set at 300 s.
Immunohistochemistry
After the behavioral experiments, the mice were anesthetized and perfused transcardially with cold phosphate-buffered saline (PBS: 137 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4, and 1.5 mM KH2PO4, pH 7.4), and brains were quickly removed. One hemisphere of each brain was fixed in 4%paraformaldehyde in PBS at 4°C overnight, cryoprotected with increasing concentrations of sucrose (20%and 30%) in 0.1 M phosphate buffer (0.038 M NaH2PO4 and 0.162 Na2HPO4, pH 7.4) at 4°C for 2–3 days. Serial sagittal sections were cut on a vibratome (40μm thickness, Leica Microsystems) and stored at –20°C until histological analysis. The cortical and hippocampal tissues of the other hemisphere of each brain were dissected and frozen in liquid nitrogen and stored at –80°C for biochemical analysis. For Aβ immunohistochemical staining, antigen retrieval was performed by heating sections in citrated buffer (pH 6.0, 10 mM trisodium citrate) on slides in a pressure cooker for 5 min, and then the sections were incubated with an anti-Aβ antibody (82E1, IBL) overnight at 4°C to detect both Aβ40 and Aβ42. Immunopositivity signals were visualized using an ABC Elite kit (Vector Laboratories Inc.). Images were obtained using a microscope (Carle Zeiss). Aβ deposits were quantified as the percentage of immunostained area (positive pixels) divided by the examined area (total pixels) using ImageJ software (NIH, Bethesda, MD, USA). For immunofluorescence staining, the sections prepared with a cryostat were washed in PBS and blocked in 5%skim milk in PBS-T (0.25%Triton X in PBS) for 1 h at room temperature (RT) following antigen retrieval. Sections were incubated with appropriate primary antibodies, namely, mouse monoclonal anti-c-Fos (EnCor Biotechnology), rabbit polyclonal anti-Iba1 (Wako), mouse monoclonal anti-GFAP (Sigma), and mouse monoclonal anti-NeuN (Sigma) antibodies, at 4°C overnight. The sections were washed with PBS-T and incubated at RT for 1 h with appropriate secondary antibodies, namely, goat anti-mouse Alexa Fluor 488 or 555 and goat anti-rabbit Alexa Fluor 555 (Thermo Fisher Scientific). Nuclei were stained with DAPI (Vector Laboratories Inc). Images were obtained using an Axio Observer (Carle Zeiss) or a confocal fluorescence microscope SpinSR10 (Olympus). Fluorescence intensity was quantified using ImageJ software. To quantify the Iba1+, GFAP+, and c-Fos+ cells, we randomly selected 4 fields (1×1 mm2 square per field) of the cortex and 2 fields (1×1 mm2 square per field) of the hippocampus, and the mean was used for statistical analysis. Measurements were taken from sections per mice and all immunostaining analysis was performed blinded.
Aβ ELISA
The cortex and hippocampus of the brain were homogenized in 19 volumes of ice-cold Tris-buffered saline (TBS; 10 mM Tris and 150 mM NaCl, pH 7.6) containing a protease inhibitor cocktail (Roche, Mannheim, Germany). The resulting homogenates were centrifuged at 100,000 rpm for 20 min at 4°C. After removing the supernatants, the pellets were washed with ice-cold TBS, and then 10 volumes of 6 M guanidine hydrochloride were added to the pellets. The samples were sonicated and incubated at RT for 1 h. The homogenates were centrifuged at 100,000 rpm for 20 min at 4°C. The resulting supernatants were transferred to a new tube and stored at –80°C until analysis and used for insoluble Aβ determination. The amounts of insoluble Aβ40 and Aβ42 were assayed using ELISA kits (Wako Pure Chemical Industries, Osaka, Japan). Aβ levels were normalized to brain tissue weight.
Western blot analysis
The cortex and hippocampus of the brain were homogenized in RIPA buffer [50 mM Tris-HCl (pH 7.6), 150 mM NaCl, 1%Nonidet P-40, 0.5%sod-ium deoxycholate, 0.1%SDS] containing a protease inhibitor cocktail (Roche) and a phosphatase inhibitor cocktail solution 1 (Wako). The resulting homogenates were incubated on ice for 30 min and then centrifuged at 12,000 rpm for 30 min at 4°C. The protein concentrations in the supernatants were determined using a BCA protein assay kit (ThermoFisher Scientific). Equal amounts of protein were separated by SDS polyacrylamide gel electrophoresis (SDS-PAGE), and separated proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Millipore). They were then blocked with 5%skim milk in TBS-T buffer for 1 h at RT. These membranes were then incubated with appropriate primary antibodies, namely, anti-GFAP (1/1000, Sigma), anti-Iba1 (1/1000, Wako), anti-syna-ptophysin (SYP, 1/20,000, Abcam), anti-PSD95 (1/1000, Cell Signaling), anti-phospho (p)-JNK [p-SAPK/JNK (T183/Y185), 1/1,000, Cell Signaling], anti-HSP90 (1/1,000, Cell Signaling), anti-JNK (1/1,000, SAPK/JNK, Cell Signaling), anti-CREB (1/1,000, Cell Signaling), anti-pCREB (1/1,000, Cell Signaling), ERK (1/1,000, Cell Signaling), pERK (1/1,000, Cell Signaling), and anti-actin (Proteintech Group) antibodies at 4°C overnight. The membranes were washed with TBS-T and then incubated with appropriate secondary antibodies conjugated to horseradish peroxidase. Immunoreactive bands were visualized using Immunostar Zeta or Immunostar LD (FUJIFILM Wako Pure Chemical Corporation) and analyzed using Amersham Imager 680 (GE Healthcare Life Science). Signal intensity was quantified using ImageJ software.
Quantitative RT-PCR (qRT-PCR) analysis
Total RNA was isolated from the hippocampus and cortex using Trizol (Invitrogen) following the manufacturer’s instructions. Reverse transcription was performed using a ReverTra Ace qPCR RT kit (TOYOBO, Japan). Quantitative real-time PCR (qPCR) was carried out using the KOD SYBR qPCR Mix (TOYOBO) and 7500 Fast Real-Time PCR System (Applied Biosystems). The expression levels of mRNA were normalized with the corresponding amount of GAPDH mRNA using the comparative threshold cycle method following the manufacturer’s protocols. Amplification was performed using the following primers (sense and antisense): TNF-α (5’-GGGCTTCCAGAACTCCAGG-3’ and 5’-GCTCTCCACTTGGTGGTTT-3’), IL-6 (5’-TGATGGATGCTACCAAACTGGAT-3’ and 5’-CTGTGACTCCAGCTTATCTCTTGGT-3’), IL-1β (5’-GAAGCACCAGCACATTGCTTT-3’ and 5’-GGAGCCTCATGGCCCAATTT-3’), IL-10 (5’-CAGAGAAGCATGGCCCAGAA-3’ and 5’-GCTCCACTGCCTTGCTCTTA-3’), TGFβ (5’-ACTGGAGTTGTACGGCAGTG and 5’-GGGGCTGATCCCGTTGATTT-3’), GAPDH (5’-GCATCTTCTTGTGCAGTGCC-3’ and 5’-GAGAAGGCAGCCCTGGTAAC3’).
Statistical analysis
Statistical analysis was performed using a statistical package, GraphPad prism software (GraphPad Software, San Diego, CA). Data are presented as the mean±SEM from at least three independent experiments. Statistical significance was analyzed by one-way ANOVA or Student’s t-test. Data were considered significant when p < 0.05.
RESULTS
Tooth loss induces memory impairment
AD is commonly associated with memory impairment and tooth loss is known as a risk factor for AD. To determine whether tooth loss could induce memory impairment in AppNL-G-F mice, we extracted the maxillary molar teeth on both sides from 2-month-old mice (experimental group) and then conducted the novel object recognition test to evaluate short-term memory and recognition ability at 4 months of age. AppNL-G-F mice exhibit memory and learning deficits starting at 7 months of age [27, 31]. During the training session, there were no significant differences in the level of exploratory preference between the two familiar objects and total exploratory time between the experimental and control groups, suggesting that both groups of mice have similar levels of motivation, curiosity, and interest in exploring familiar objects. In the retention session, however, the levels of exploratory preference for the novel objects were significantly higher in the control group than in the experimental group, indicating that tooth loss induced short-term memory impairment in AppNL-G-F mice (Fig. 1a). We further examined long-term memory by the passive avoidance test and found that the latency time to enter the dark compartment was shorter in the experimental group than in the control group (Fig. 1b), suggesting impaired long-term memory caused by tooth loss.
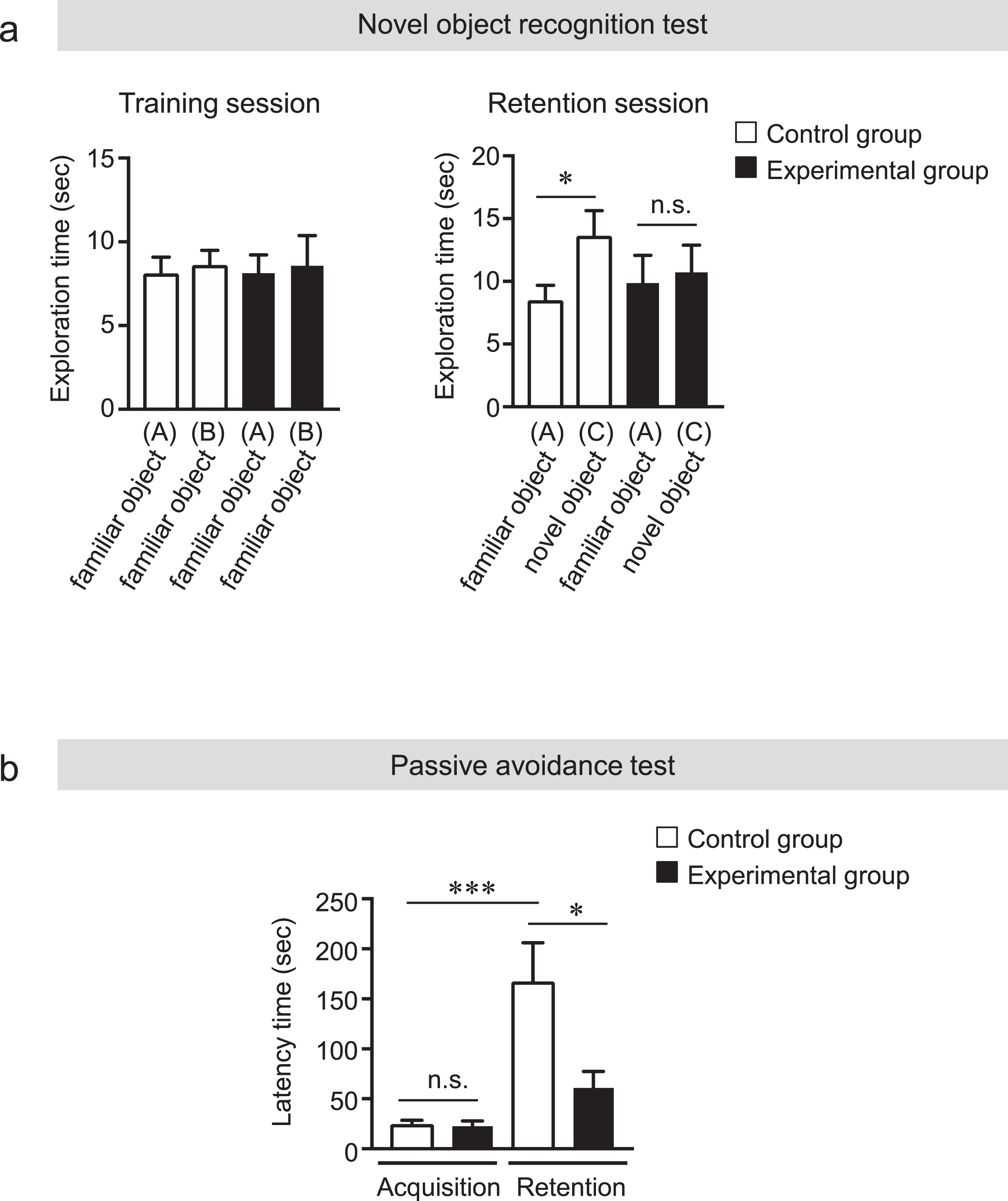
Tooth loss induces memory impairment. a) Novel objective recognition test. During the training session, the control and experimental mice showed the same exploratory time between the two familiar objects (A, B). During the retention session, the control mice spent a longer time exploring the novel object (C) than the familiar object (A). The experimental mice, however, spent the same exploratory time between the two familiar objects (A) and the novel object (C). All the values are presented as the mean±SEM, n = 7–8. *p < 0.05 versus control group, n.s., no significant difference, as determined by Student’s t-test. b) Passive avoidance test. The latency time of the mice entering the dark compartment was recorded 48 h after the training session. The latency time of the experimental group was significantly shorter than that of the control group. The cut-off latency time was set at 300 s. All the values are presented as the mean±SEM, n = 7–8. *p < 0.05, ***p < 0.001, n.s., no significant difference, as determined by one-way ANOVA.
Tooth loss did not change Aβ levels
In AppNL-G-F mice, Aβ plaque deposition starts at 2 months and saturates at around 7 months [20]. Since tooth loss was observed to induce memory impairment in the behavioral test, we determi-ned whether the impaired cognitive function of AppNL-G-F mice after tooth loss results from alt-ered Aβ deposition and insoluble Aβ levels. Sagittal brain sections were immunostained with the anti-Aβ antibody (82E1) that recognizes both Aβ40 and Aβ42 to detect human Aβ deposition. The percentages of Aβ-immunopositive areas in the hippocampus and cortex of mice were not significantly different between the control and experimental groups (Fig. 2a). Next, we measured insoluble Aβ40 and Aβ42 levels in the hippocampus and cortex of AppNL-G-F mice by ELISA. Supporting the histological findings, ELISA results showed no significant differences in insoluble Aβ40 and Aβ42 levels both in the hippocampus and cortex of the AppNL-G-F mouse brain between the two groups (Fig. 2b). These findings are consistent with our previous findings that tooth loss does not have any effect on Aβ deposition and insoluble Aβs levels [10].
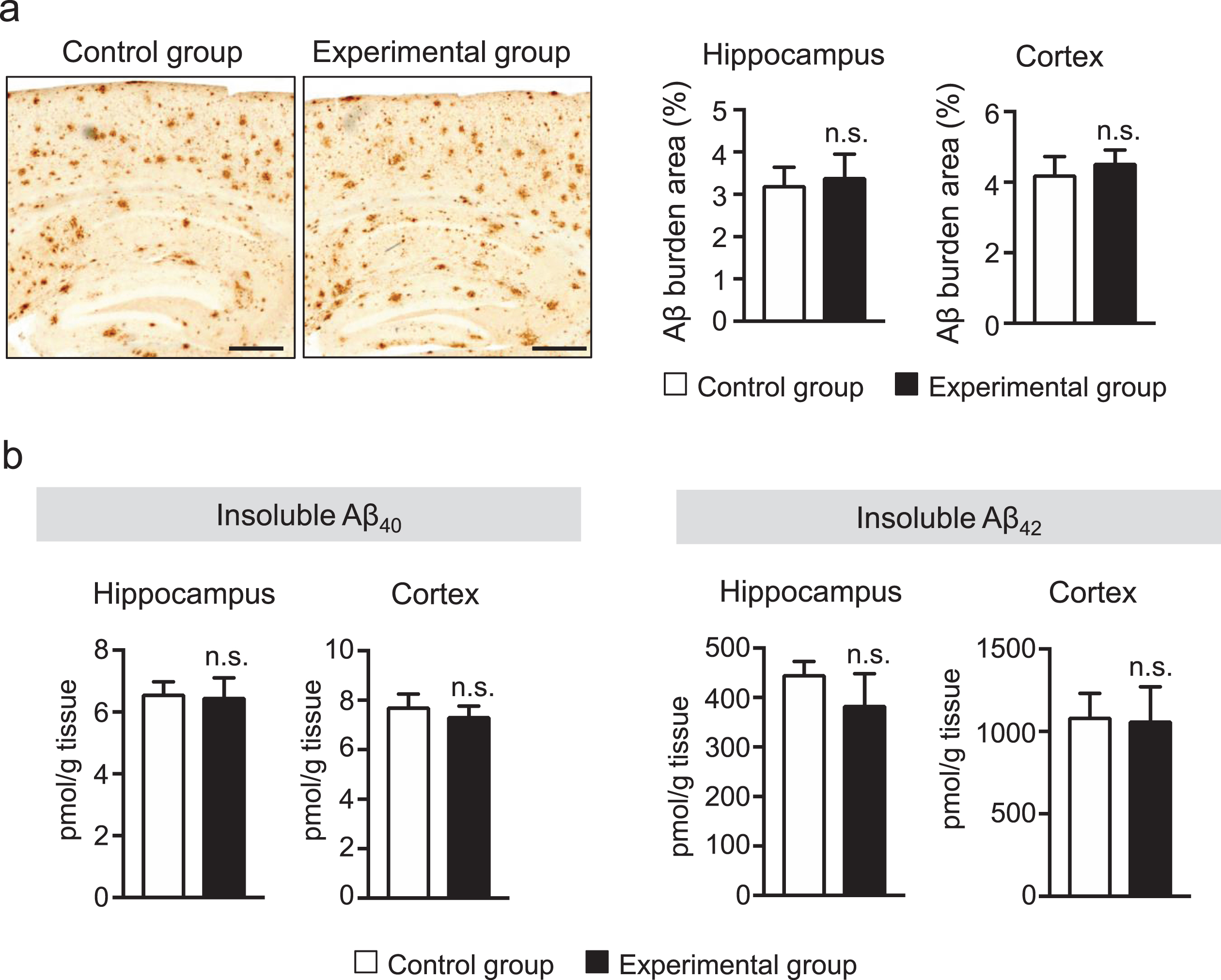
Tooth loss has no effect on Aβ deposition and insoluble Aβ40 and Aβ42 levels. a) Sagittal brain sections were stained with the anti-Aβ antibody (82E1), which recognizes both Aβ40 and Aβ42, to detect human Aβ deposition. Representative images are shown in the left panel. Aβ deposits were quantified as the percentage of immunostained area divided by the examined area (right panel). b) Insoluble Aβ40 and Aβ42 levels in the cortex and hippocampus of AppNL-G-F mice were measured by sandwich ELISA. Aβ levels were normalized to brain tissue weight. Aβ deposition and insoluble Aβ40 and Aβ42 levels in the cortex and hippocampus were not significantly different between the control and experimental groups. The data are expressed as the mean±SEM, n = 7–8, n.s., no significant difference, as determined by Student’s t-test. Scale bars: 500μm.
Tooth loss decreases the neuronal activity in the cortex and hippocampus
Afferent sensory input is highly dependent on masticatory function or hardness of the diet, and several regions of the CNS are activated during mastication. Previous studies have shown the link between reduced neuronal activity and synapse formation due to reduced mastication [9]. The immediate early c-Fos gene is transcribed in neurons within minutes after stimulation by depolarization and subsequent neurotropic intercellular signals [32]. Therefore, we measured the number of activated neurons in the hippocampus and cortex of AppNL-G-F mice by immunostaining the sagittal brain sections with using the anti-c-Fos antibody. The number of c-Fos-positive neurons was significantly lower in the hippocampal cornu ammonis (CA) 1 and CA3 regions in the experimental group than in the control group (Fig. 3a). Interestingly, there was also a significant decrease in the number of c-Fos-positive neurons in the cortex of the experimental group compared with that of the control group (Fig. 3b). It has been reported that masticatory dysfunction contributes to a decrease in the density of synapses caused by the reduced neuronal activity [33]. Therefore, we also evaluated the level of synapses in the cortex and hippocampus using the anti-synaptophysin (SYP, presynaptic protein) and anti-PSD95 (postsynaptic protein) antibodies by western blot analysis. The results of this analysis showed that the protein levels of synaptophysin and PSD95 were significantly decreased in both the hippocampus and cortex in the experiment group compared with the control group (Fig. 4a, b). Previous reports suggested that synaptic numbers and presynaptic protein expression may be mediated via ERK-and cAMP-response element binding protein (CREB) [34, 35]. Therefore, we measured the protein levels of phosphorylated (p)-CREB and pERK in the hippocampus and cortex of mice brains. We found that pCREB level was significantly decreased in both the hippocampus and cortex of the experimental group compared with those of the control group (Fig. 4a, b). Interestingly, however, the pERK level was significantly increased in the hippocampus and cortex of the experimental group compared with those of the control group suggesting that tooth-loss-induced ERK activation may not be involved in presynaptic protein expression. (Fig. 4a, b). Taken together, tooth loss was shown to result in synaptic dysfunction probably due to the reduction in neuronal activity caused by tooth loss.
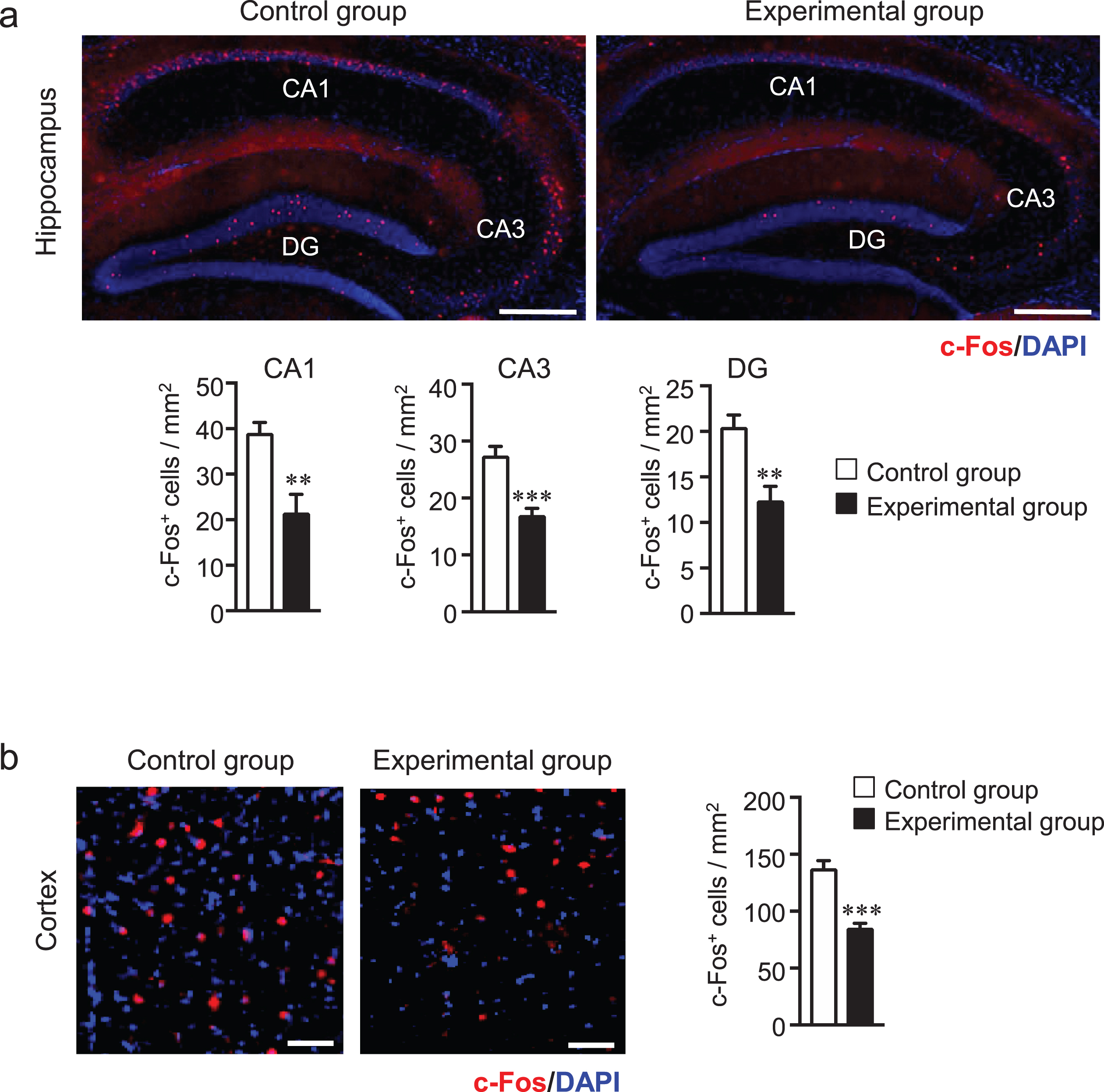
Tooth loss decreases neuronal activity. c-Fos-positive neurons in the hippocampus and dentate gyrus (a) and cortex (b). Sagittal brain sections were stained with the anti-c-Fos (red) antibody and cell nuclei were stained with DAPI (blue). Quantification of c-Fos-positive cells in CA1, CA3, and DG in (a) and in the cortex in (b). CA, cornu ammonisi; DG, dentate gyrus. All the values are presented as the mean±SEM, n = 6. *p < 0.05, **p < 0.01, ***p < 0.001 versus control group, as determined by Student’s t-test. Scale bars in (a): 500μm and in (b): 100μm.
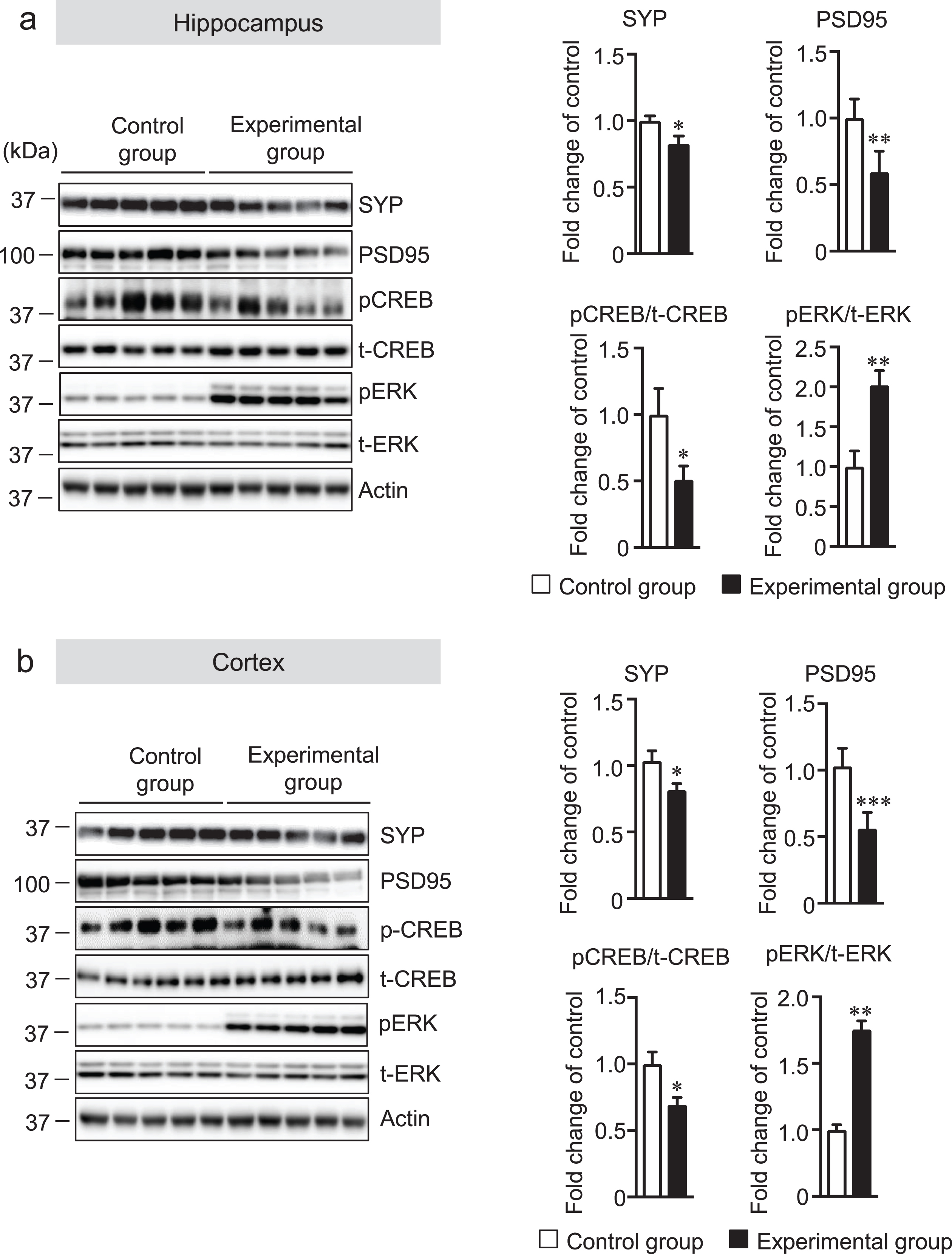
Tooth loss decreases the protein levels of synaptophysin (SYP, presynaptic protein) and PSD95 (postsynaptic protein), and pCREB, and increases pERK protein level. Protein levels in the hippocampus (a) and cortex (b) of mouse brains were determined by western blot analysis, quantified by densitometry, then normalized to actin level. The data are expressed as the mean±SEM, n = 7, *p < 0.05, **p < 0.01, ***p < 0.001 versus control group, n.s., no significant difference, as determined by Student’s t-test.
Tooth loss induces glial activation in the hippocampus
Microglia are considered the innate immune cells of the CNS. Upon activation, microglia release neuroinflammatory cytokines and cytotoxic compounds that disrupt homeostatic processes and neuronal functions [36, 37]. Reactive astrocytes also play an essential role in neuroinflammation. Recent data show that reactive microglia produce inflammatory cytokines that can activate astrocytes [38]. Although glial activation is a prominent feature in AD, whether tooth loss can activate the glial cells has not been explored. Thus, we investigated the effect of tooth loss on glial activation in the brain. We first measured the protein levels of Iba1 (a microglia marker) and GFAP (an astrocytic marker) in the hippocampus and cortex by western blot analysis. We found that both Iba1 and GFAP protein levels were increased in the hippocampus, but not in the cortex, of the experimental group compared with that of the control group (Fig. 5a, b). We further investigated the mRNA expression levels of the neuroinflammatory cytokines (TNF-α, IL-6, and IL-1β) and anti-inflammatory cytokines (IL-10 and TGF-β) in hippocampal and cortical tissues by qRT-PCR analysis. As shown in Fig. 5c, the mRNA expression levels of TNF-α, IL-6, and IL-1β significantly increased in the hippocampus, but not in the cortex, of the experimental group compared with those of the control group. Although the mRNA expression levels of IL-10 and TGFβ were not statistically significant, they tended to be decreased in the hippocampus, but not in the cortex, of the experimental group compared with those of the control group. These results further confirmed the results of immunohistochemical staining of the brain sections with the anti-Iba1 and -GFAP antibodies. We observed that the number of Iba1-positive cells was significantly higher in the hippocampus of the experimental group than in that of the control group (Fig. 6a). In the cortex, however, the number of Iba1-positive cells was not significantly different between two groups (Fig. 6b). In addition to Iba1-positive cells, the number of GFAP-positive cells was also significantly higher in the hippocampus of the experimental group than in that of the control group (Fig. 7a) but not in the cortex (Fig. 7b). These findings suggest that tooth loss induces glial activation in the hippocampus, which may induce neuronal death in the hippocampus.
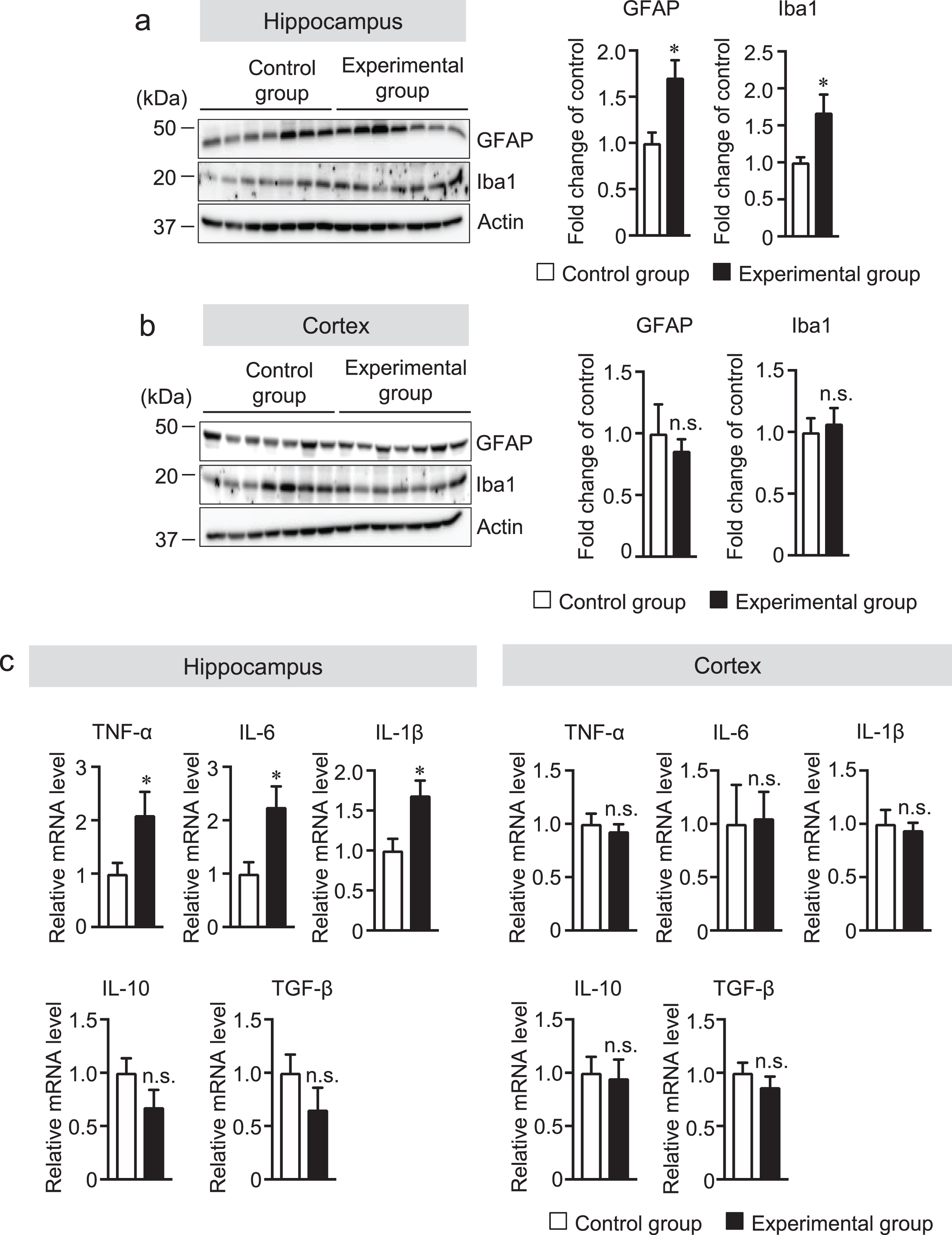
Tooth loss increases the protein levels of GFAP and Iba1. Protein levels in the hippocampus (a) and cortex (b) of mouse brains were determined by western blot analysis, quantified by densitometry, then normalized to actin level. The data are expressed as the mean±SEM, n = 7, *p < 0.05 versus control group, n.s., no significant difference, as determined by Student’s t-test. (c) The mRNA expression levels of TNF-α, Il-6, IL-1β, IL-10, and TGF-β were determined by qRT-PCR analysis. The mRNA expression levels of TNF-α, Il-6, IL-1β, IL-10, and TGF-β were normalized to the corresponding amount of GAPDH mRNA. All the values are presented as the mean±SEM, n = 7, *p < 0.05 versus control group, n.s., no significant difference, as determined by Student’s t-test.
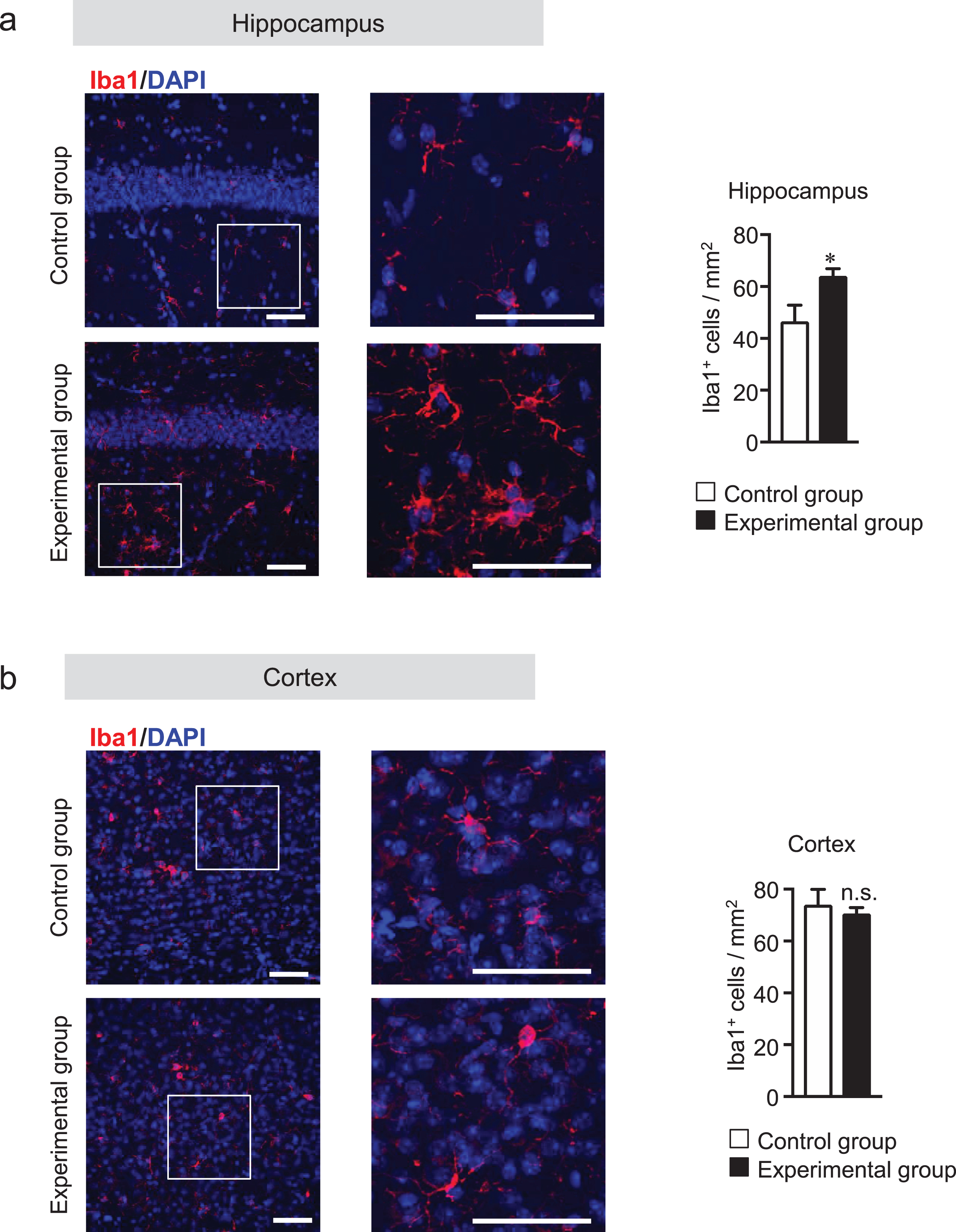
Tooth loss enhances microglial activation in the hippocampus. Sagittal brain sections were stained with the anti-Iba1 (red) antibody, and cell nuclei were stained with DAPI (blue). Representative images of hippocampus (a) and cortex (b). Numbers of Iba1-positive cells in hippocampus (a, right panel) and cortex (b, right panel). High magnified images of the squared region in the left panels are shown in the adjacent right panels. The data are expressed as the mean±SEM, n = 6, *p < 0.05 versus control group, n.s., no significant difference, as determined by Student’s t-test. Scale bars: 100μm.
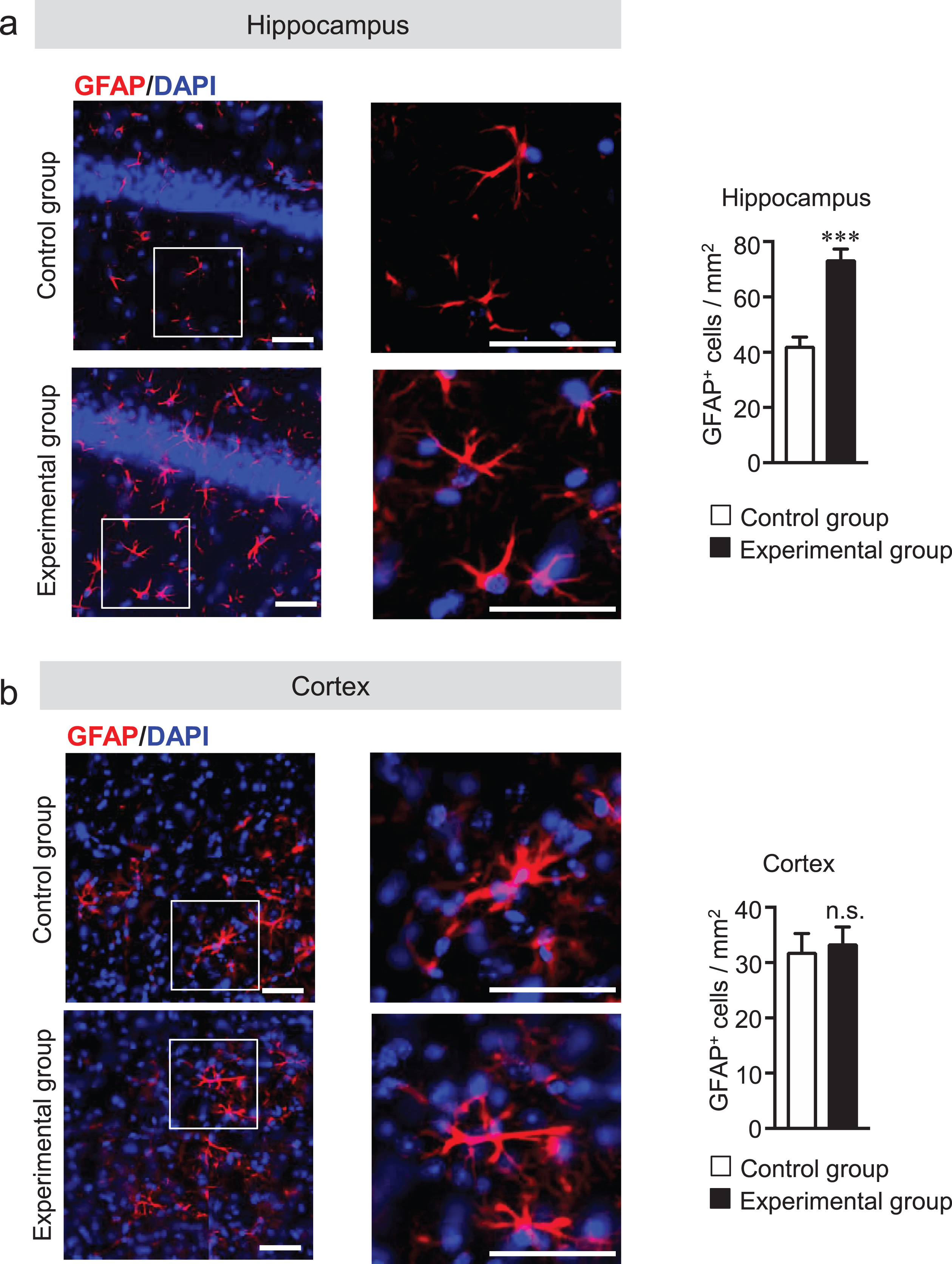
Tooth loss enhances astrocyte activation in the hippocampus. Sagittal brain sections were stained with the anti-GFAP (red) antibody, and cell nuclei were stained with DAPI (blue). Representative images of hippocampus (a) and cortex (b). Numbers of GFAP-positive cells in hippocampus (a, right panel) and cortex (b, right panel) are shown. High magnified images of the squared region in the left panels are shown in the adjacent right panels. The data are expressed as the mean±SEM, n = 6, ***p < 0.001 versus control group, n.s., no significant difference, as determined by Student’s t-test. Scale bars: 100μm.
Tooth loss induces neuronal cell loss in the hippocampus
Tooth loss and soft diet suppress neurogenesis leading to pyramidal neuronal loss in the hippocampus [9, 10]. In addition, the inhibition of microglia activation improves neurogenesis and cognitive performance [39, 40]. Therefore, we hypothesized that tooth-loss-induced glial activation may mediate neuronal cell loss in the hippocampus. We determined the number of pyramidal neurons in the CA1 and CA3 regions in the hippocampus by immunostaining the sagittal brain sections with anti-NeuN antibody, and then NeuN-positive cells were quantified in terms of fluorescence intensity. The fluorescence intensity of the CA1 and CA3 regions of the hippocampus was lower in the experimental group than in the control group (Fig. 8b, c). No such reduction in the number of NeuN-positive cells was observed in the cortex (data not shown) of the experimental group compared with the control group. These findings indicate that the glial activation due to tooth loss induces neuronal cell loss in the CA1 and CA3 regions of the hippocampus.
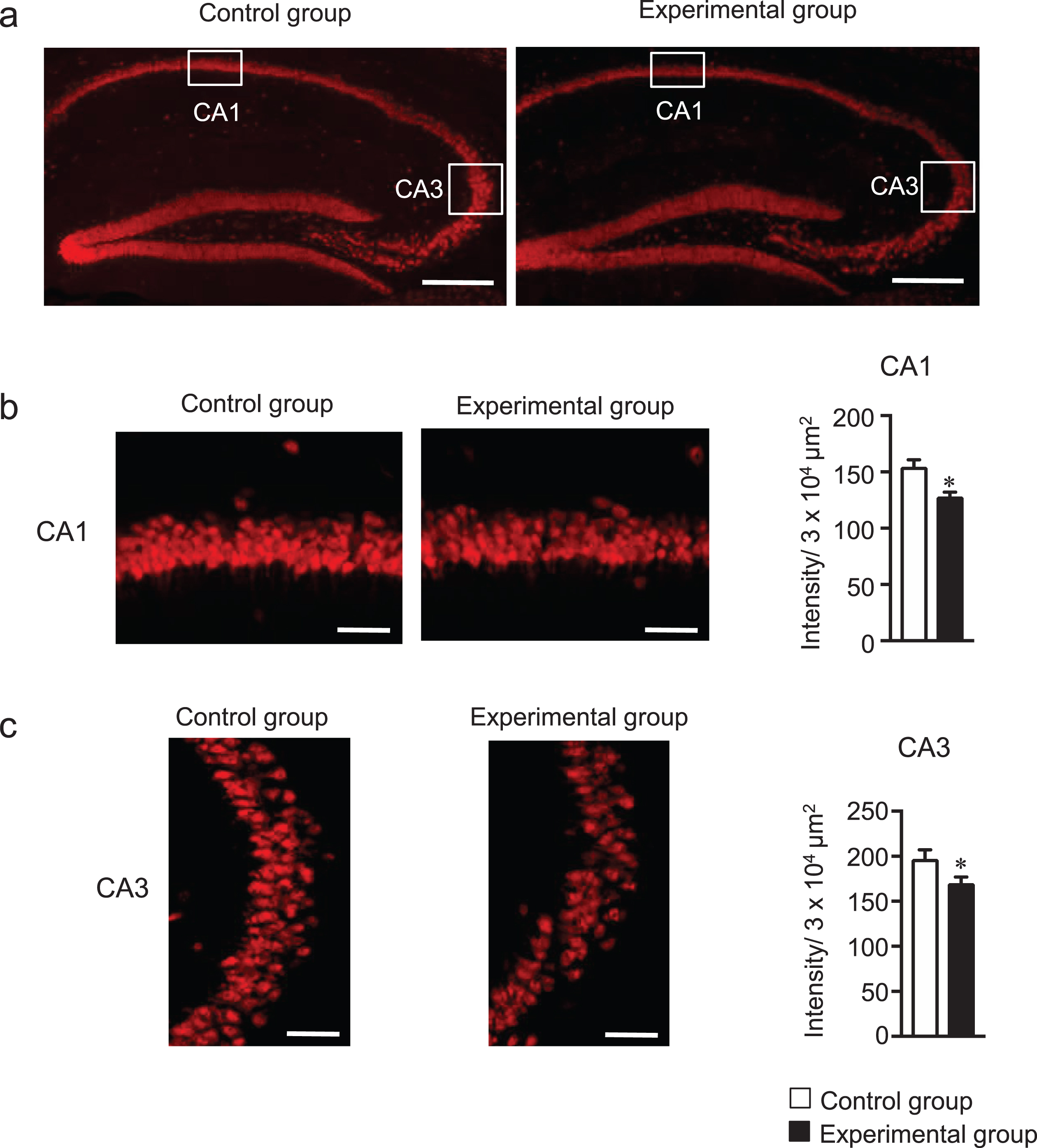
Tooth loss induces neuronal cell loss in the hippocampus. (a) Representative image of NeuN fluorescence staining in hippocampus. Sagittal brain sections were stained with the anti-NeuN (red) antibody. Fluorescence staining of NeuN in CA1 (b) and CA3 (c) regions of hippocampus and measurement of fluorescence intensity of NeuN-positive cells. The data are expressed as the mean±SEM, n = 7, *p < 0.05 versus control group, as determined by Student’s t-test. Scale bars in (a): 500μm and in (b and C): 50μm.
Tooth loss increased pJNK, pERK, and HSP90 protein levels in the hippocampus
Chronic stress due to decreased mastication resulting from tooth loss impairs learning and memory [16, 41] and alters the density and morphology of microglia [42]. In addition, chronic stress also activates protein kinase JNK and ERK, and heat shock protein 90 (HSP90) [43, 44]. Therefore, we hypothesized that tooth loss may induce chronic stress in the brain of mice, which leads to glial activation. We measured the protein levels of pJNK and HSP90 in the hippocampus and cortex of mouse brains. We found that pJNK and HSP90 levels were significantly upregulated in the hippocampus, but not in the cortex, of the experimental group compared with that of the control group (Fig. 9a, b). These findings indicate that tooth loss induces chronic stress in the hippocampus, which may induce the activation of both microglia and astrocytes in the hippocampus.
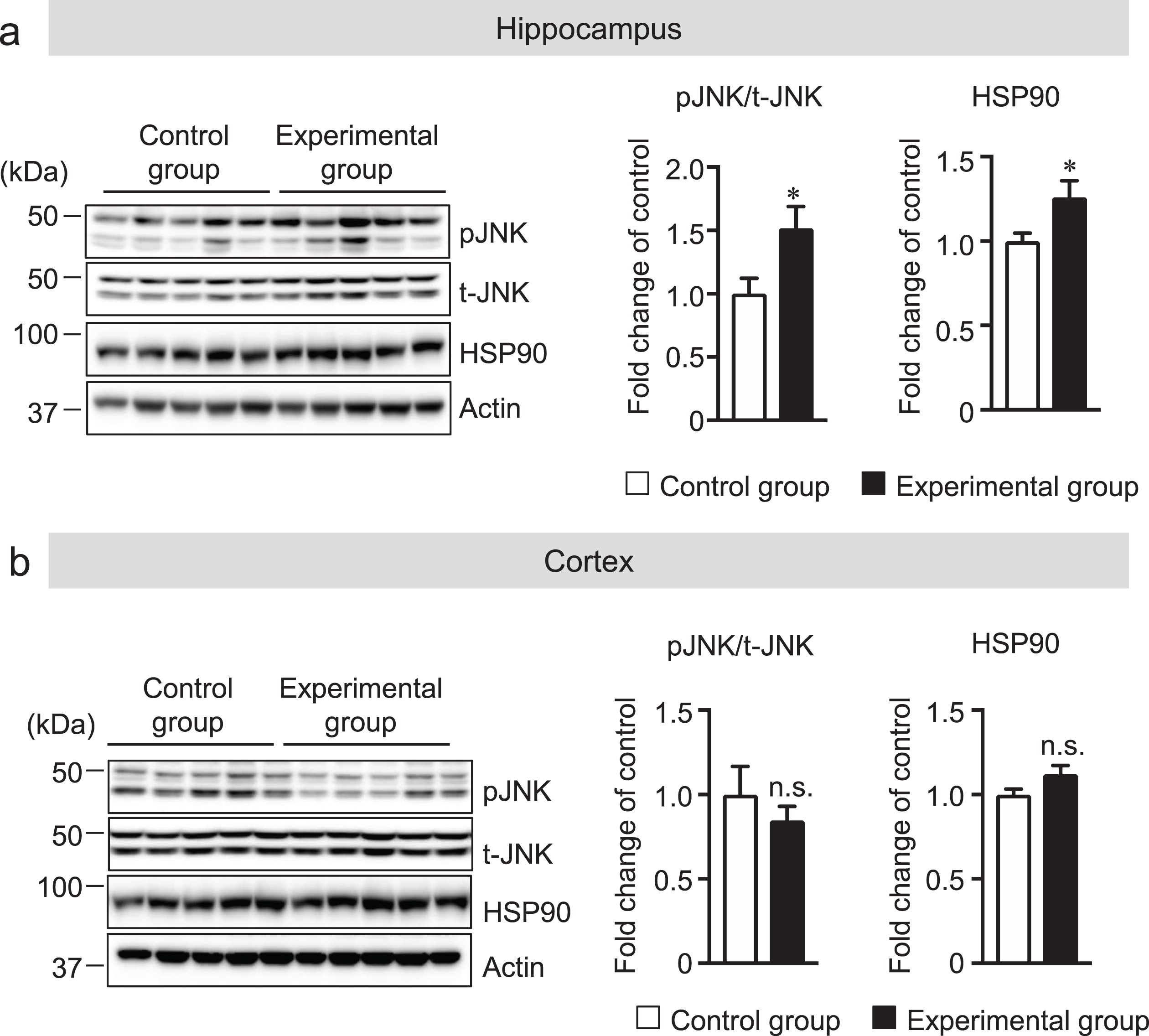
Tooth loss increases the protein levels of pJNK and HSP90. The protein levels in the hippocampus (a) and cortex (b) of mouse brains were determined by western blot analysis, quantified by densitometry, then normalized to actin. The data are expressed as the mean±SEM, n = 7, *p < 0.05 versus control group, n.s., no significant difference, as determined by Student’s t-test.
DISCUSSION
Several lines of evidence suggest the association of tooth loss with both memory impairment and AD pathogenesis, as determined using senescence-accelerated prone 8 (SAMP8) mice, AβPP transgenic mice as the AD model, and wild-type mice [14–16, 41]. However, the mechanisms by which tooth loss induces memory impairment and AD pathogenesis remain unclear. In this study, we investigated the effects of tooth loss on cognitive dysfunction and AD pathogenesis including Aβ levels, neuronal activity, neuronal cell loss, glial activation (microgliosis and astrocytosis), and stress-activated protein levels (p-JNK and HSP90) using AppNL-G-F mice.
AppNL-G-F mice exhibit memory and learning deficits starting at 7 months of age, and Aβ plaque deposition occurs at 2 months of age and saturates at around 7 months of age [20]. In agreement with this study, the molar-intact AppNL-G-F mice used in our experiment did not show memory impairment at 4 months of age. However, these mice whose molar teeth were extracted at 2 month of age showed memory impairment after 2 months. On the basis of the amyloid cascade hypothesis, it is well known that Aβ deposition in the brain plays a crucial role in AD pathogenesis [45]. Therefore, we determined whether tooth loss can affect Aβ levels in the brains and found that tooth loss did not affect both Aβ deposition and insoluble Aβ levels in the cortex and hippocampus, which is consistent with the finding of a previous study that tooth loss induces memory impairment in AβPP transgenic mice (J20) via an amyloid-cascade-independent pathway [10].
Several clinical and animal studies have demonstrated that the sensory part of the trigeminal nerve conveys sensory signals from the natural teeth and their adjacent periodontal mechanoreceptors to many areas in the CNS and, thus, affects hippocampal function [13]. For example, molarless rats show reduced sensory stimulation from periodontal ligaments, which in turn affects the morphology and function of neurons in the hippocampus [14, 15]. In addition, the trigeminal information input that is linked to masticatory muscles and periodontal ligaments has been recognized as having a facilitatory effect on synaptic transmission in the cerebral cortex [46]. In this study, we found that tooth loss significantly decreased neuronal activity (as indicated by the number of c-Fos-positive neurons) in the hippocampal CA1 and CA3, and DG regions. The neuronal activity also significantly decreased in the cortex of experimental mice. This is possibly due to the changes in the activity of motor neurons that regulate the masticatory muscles [7] and the reduction of sensory input from sensory receptors caused by the impaired masticatory function, which reduced the synaptic density in the cerebral cortex [47]. In addition, the reduction of the number of c-Fos-positive neurons may attribute to neuronal loss found in decreased NeuN intensity in CA1 and CA3 regions of the hippocampus. Numerous studies have shown that neuronal activity plays an important role in the regulation of synaptic strength and neuronal circuit refinement [48, 49]. In this study, tooth loss markedly decreased both presynaptic (SYP, synaptophysin) and postsynaptic density protein 95 (PSD95) protein levels in both the hippocampus and cortex. There is considerable evidence that the activation of the ERK-CREB signaling pathway mediated synaptic number and presynaptic protein expression. CREB, an important transcription factor, modulated the presynaptic protein synaptophysin, by enrichment of phosphorylated CREB on promoters of this gene, which may contribute to long-term synaptic plasticity. In this study, although tooth loss attenuated CREB activation, the ERK was activated in the hippocampus indicating that the ERK activation caused by tooth loss could not contribute to CREB activation and other pathway(s) may involve the attenuation of CREB activation. Although ERK is known to play a critical role in hippocampus synaptic plasticity, abnormal ERK activation in the hippocampus may impair hippocampal function and contributes to memory deficits in AD patients [50]. ERK activation is also known to be involved in AD and neuroinflammation in which the ERK activation induced neuroinflammation [51]. In this study, we found tooth loss increased the levels of neuroinflammatory cytokines, which may be involved in ERK activation induced by tooth loss. Taken together, our finding indicates that the reduced synaptic formation in the hippocampus and cortex of molarless mice is likely to be due to the decreased neuronal activity and neuronal cell loss in the CA1 and CA3 regions (see the discussion below).
Neuronal cell death and synapse loss are among most common, and earliest, pathophysiological features of neurodegenerative diseases including AD. Previously, it was thought that this early synaptic loss was mainly due to the direct effects of Aβ oligomers and fibrils on neuronal Aβ receptors or secondary neuroinflammation. However, recent studies have revealed that reactive glial cells are actually the ones that drive the initial neuronal cell loss and synaptic loss in AD [52–54]. The activated microglia can release neuroinflammatory cytokines such as TNF-α and IL-6, which can indeed induce synaptic loss [55, 56]. Reactive astrocytes also play an essential role in neuroinflammation. Recent data show that inflammation-induced reactive microglia produce neuroinflammatory cytokines, such as TNF-α and IL-6, that can activate astrocytes, and in turn can rapidly induce synaptic loss [38]. However, glial activation by tooth loss has not been fully explored. Thus, we also investigated the effect of tooth loss on glial activation. Here, we found that tooth loss significantly increased the numbers of microglia and astrocytes in the hippocampus, but not in the cortex. This was accompanied by increased mRNA expression levels of TNF-α, IL-6, and IL-1β in the hippocampus. Taken together, tooth loss may cause both microgliosis and astrocytosis resulting in neuronal cell loss in the CA1 and CA3 regions of the hippocampus. However, whether tooth loss is directly involved in glial activation is unclear; one possible mechanism is that neuronal inactivation caused by reduced afferent sensory input may, at least in part, cause glial activation [33, 57]. Further studies are required to fully elucidate the potential mechanisms associated with tooth-loss-induced glial activation. In addition, we have previously reported that tooth loss in 3 x Tg-AD model mice showed neuronal death in the mesencephalic trigeminal nucleus (Vmes), resulting in the release of Aβ42. Released Aβ42 damaged the locus coeruleus (LC), which in turn induces a significant reduction in the hippocampal neurons in the CA1 and CA3 regions receiving projections from LC [58], which is another possibility that hippocampal neuronal cell death caused by tooth loss is involved in the neurodegeneration of Vmes and LC.
It has been reported that the reduced mastication due to tooth loss inhibits hippocampal neurogenesis caused by reduced levels of brain-derived neurotrophic factor (BDNF) in the hippocampus [59, 60]. However, it is as yet unclarified how tooth loss inhibits the neurogenesis and decreases BDNF levels in the hippocampus. A large number of studies have shown that inhibition of microglial activation has a profound neuroprotective effect associated with the improvement of spatial memory and neurogenesis under neuroinflammatory conditions [39, 40]. Furthermore, the enhanced pro-inflammatory signaling leads to a decrease in the mRNA expression levels of BDNF [61]. Here, we found that tooth loss induced glial activation and neuronal cell loss in the hippocampus. These findings indicate the tooth-loss-induced glial activation may inhibit neurogenesis via the decrease in BDNF levels. Although we did not examine the effect of tooth loss on neurogenesis in the hippocampus because of limited samples, recent studies showed that tooth loss and reduced mastication inhibited in BDNF expression and neurogenesis in the hippocampus [9, 10].
As mentioned above, tooth-loss-induced glial activation in the hippocampus may be dependent on neuronal inactivation. In this study, the molarless mice, however, did not show glial activation in the cortex where the neuronal activity was reduced, indicating that other factors could be involved in glial activation in the hippocampus. The hippocampus is sensitive to stress, as well as the aging process, and it is one of the first brain regions to be structurally and functionally modified by stress [62]. Chronic stress due to reduced mastication resulting from tooth loss impairs learning and memory [16, 41] and alters the density and morphology of microglia [42]. In addition, chronic stress also activates the protein kinase JNK and increases heat shock protein 90 (HSP90) levels [43, 44]. In this study, we found that tooth loss activated JNK and increased HSP90 levels in the hippocampus but not in the cortex, indicating that tooth loss may induce chronic stress in the hippocampus, which may in turn induce glial activation. Stress activates the hypothalamic–pituitary–adrenal axis, leading to glucocorticoid secretion from the adrenal cortex [63]. It has been reported that occlusal disharmony increases plasma glucocorticoid levels leading to reduction in neuronal activity in the hippocampus [64]. Therefore, tooth-loss-induced glial activation in the hippocampus may be involved in the increase in plasma glucocorticoid levels. To support this hypothesis, further investigation of increased plasma glucocorticoid levels in relation to tooth loss is necessary.
Human studies showed that tooth loss may affect to several physical conditions including feeding activity, which could reduce their body weight, and induce depression and anxiety [65]. In this study, however, body weight did not significantly differ between the experiment and control group (data not shown), which is consistent with our and other previous findings [66, 67]. Limitation of current study, however, is that it is difficult to separate the influence of depression and other psychological symptoms from surgical stress on the development of cognitive deficit in the molarless mice.
In summary, we have shown that tooth loss in AppNL-G-F mice induces memory impairment via the amyloid-cascade-independent pathway and the decrease in neuronal activity and synaptic density. Furthermore, tooth loss increased pJNK and HSP90 levels in the hippocampus, which may in turn lead to glial activation resulting in the neuronal cell loss in the CA1 and CA3 regions of the hippocampus. Our findings suggest that taking care of teeth is very important to preserve a healthy oral environment, which may reduce the risk of cognitive dysfunction.
Footnotes
ACKNOWLEDGMENTS
This work was supported by a Grant-in-Aid for Scientific Research B (16H05559) and a Grant-in-Aid for challenging Exploratory Research (15K15712) (to M.M.) from the Ministry of Education, Culture, Sports, Science and Technology, Japan. This work was also supported by the Project of translational and clinical research seed A from Japan Agency for Medical Research and Development (AMED, A-128) (to M.M.). We acknowledge the assistance of the Research Equipment Sharing Center at the Nagoya City University.