
Abstract
Alzheimer’s disease (AD) is the most common and devastating neurodegenerative condition worldwide, characterized by the aggregation of amyloid-β and phosphorylated tau protein, and is accompanied by a progressive loss of learning and memory. A healthy nervous system is endowed with synaptic plasticity, among others neural plasticity mechanisms, allowing structural and physiological adaptations to changes in the environment. This neural plasticity modification sustains learning and memory, and behavioral changes and is severely affected by pathological and aging conditions, leading to cognitive deterioration. This article reviews critical aspects of AD neurodegeneration as well as therapeutic approaches that restore neural plasticity to provide functional recoveries, including environmental enrichment, physical exercise, transcranial stimulation, neurotrophin involvement, and direct electrical stimulation of the amygdala. In addition, we report recent behavioral results in Octodon degus, a promising natural model for the study of AD that naturally reproduces the neuropathological alterations observed in AD patients during normal aging, including neuronal toxicity, deterioration of neural plasticity, and the decline of learning and memory.
Alzheimer’s disease (AD) is one of the most common and devastating neurodegenerative diseases that occur during aging and is characterized by a progre-ssive neurodegeneration process that produces learning and memory loss. Although there is no consensus yet on the origin of AD, we can mention some candidates: the amyloid-β protein (Aβ) cascade, i.e., Aβ accumulation (soluble or in plaques) [1, 2]; the accumulation of phosphorylated tau protein (tangles) [3]; and a neurovascular failure-inducing degeneration [4, 5]. AD is associated with the accumulation and deposition of Aβ, astrogliosis, oxidative injury, the formation of neurofibrillary tangles, cell death, and neurotransmission alterations that impair synaptic plasticity (SP) and cognition. One of the dominant working hypotheses involves the Aβ cascade, which is supported by research that makes use of transgenic mice expressing familial mutations of the human amyloid precursor protein (APP) and presenilin and results in the development of deficits in neural plasticity, learning, and memory. Nevertheless, these transgenic mice rarely develop neurofibrillary tangles and exhibit little synaptic and neuronal loss [6–13]. Instead of using mice, which are short-lived, another approach involves the use of long-lived animal models that naturally and progressively express hallmarks of AD as they age [13, 14] and constitute more realistic models of AD. These models are vital to test different etiologies, e.g., the amyloid cascade of the disease [15, 16]. In this regard, one promising model is the rodent, Octodon degus (degus), which develops brain changes during aging similar to those observed in patients with AD [13, 17–20]. Another critical issue that must be taken into account to advance in the treatments for AD is to include neural plasticity as part of the treatment since it contributes to sustaining memory and learning and the nervous system’s self-repair [21–24].
Here we review the advantages and disadvantages of animal models that naturally develop AD neuropathology, as well as some therapeutic procedures, such as environmental enrichment, physical exercise, transcranial stimulation, neurotrophins, and direct electrical stimulation of the amygdala, that improve neural plasticity and achieve a functional recovery of learning and memory [21–24].
NEURAL PLASTICITY AND NEURODEGENERATION
In contrast to what was previously believed, the central nervous system (CNS), unlike thought for many years, can dynamically modify its properties in response to changes in the environment. This view extends to neural plasticity’s mechanisms associated with learning and memory and the recovery of function after injury [21–24]. In its broadest sense, neural plasticity means the capacity for functional or morphological changes, of the CNS and its component elements (e.g., nerve cells and synapses), by external agents’ actions. This plasticity must be differentiated from genetically programmed modifications. External agents are usually sensory stimuli and traumatic injuries, which make each unique and different experience. Thus, neural plasticity stands out for its ad-aptive value, allowing the compensatory changes induced by experience to occur in the CNS continuou-sly [21, 25]. The mechanisms of neural plasticity are diverse. They can vary from extensive morphological modifications, such as those observed in the regeneration of axons and new synapse formation, to subtle molecular changes that alter the cellular response to neurotransmitters [26, 27]. In this sense, two neural plasticity forms can differentiate morphological or growth plasticity and functional plasticity [21, 28], where morphological plasticity includes neurogenesis, regeneration, axonal collateral formation, and reactive synaptogenesis. Santiago Ramón y Cajal was the first to propose plasticity in the number and strength of neural connections as the physical basis of learning and memory.
Years later, the psychologist Donald Hebb [29] proposed plasticity as the mechanism by which the coincidence of pre- and postsynaptic activity could modify the neural connections in specific structures of the brain. In 1973, the first experimental evidence was found that supported the hypothesis of Cajal and Hebb, that is, that synapses can change as a result of their activity [30–33]. This phenomenon has been known as long-term potentiation (LTP) and consists of a sustained increase in synaptic transmission efficiency after stimulating an afferent pathway with high-frequency stimuli. The entire transmission process occurs faster and to a greater extent [34, 35]. Such changes happen immediately and have a variable duration, depending on the protocol used for th-eir induction, ranging from a few hours and days to weeks. Since its discovery, LTP is proposed to be the cellular basis of the processes that underlie learning and memory [34–36]. In particular, when LTP’s efficacy decreases during, e.g., neurodegeneration and or aging, there is also a decline in subjects’ cognitive capacity. The presence of amyloid plaques, neurofibrillary tangles, Lewy bodies, synaptic dystr-ophy, synaptic loss, loss of dendritic extent, and neurons loss in the brain [37, 38] has been described as a normal process observed during human aging. Although these changes are more subtle and selective than in AD patients, a critical consequence is a decay in neural plasticity (e.g., LTP) during aging [39]. Hence, failing neural plasticity mechanisms could accelerate transit from normal aging to neurodegeneration [40]. Numerous studies have shown that AD patients’ memory failures do not correlate well with the amyloid plaque burden in recent years. Instead, the loss of synaptic markers in the human cortex and hippocampus is a better predictor of clinical symptoms and disease progression [41–44]; however, there is no biomarker to measure the synaptic integrity dir-ectly over time in AD patients. The methods used most frequently to assess synaptic integrity are electrophysiological and neuroimaging [44]. More direct methods include neuroanatomical studies [45, 46] and measuring mRNA or synaptic proteins [47, 48]. Of particular interest are soluble oligomeric species that may play an essential role in synaptic dysfunction and neuronal loss in AD since current evidence indicates that neuronal and a rise of Dickkopf-1 may cause synapse loss, an antagonist of the endogenous intracellular wnt pathway [49, 50], rather than by Aβ plaque deposition per se [51, 52].
Tau hyperphosphorylation seems to play a more critical role in synaptic dysfunction and cognitive decline, affecting organelles’ axonal transport, including the mitochondria [53, 54] and impair AMPA receptor clustering [55]. The colocalization of Aβ and tau [56], observed in AD patients, suggests potentiation of these adverse effects because tau could become hyperphosphorylated in the amyloid presence [57].
We hypothesize neurodegeneration and cognitive decay during aging could, at least in part, be related to a failure of neural mechanisms to process information and to accommodate new learning and memory, practically a loss of neural plasticity. Therefore, the modulation of neural plasticity mechanisms could potentiate the recovery of lost functions in AD pat-ients. Following this idea, we will expose some evidence that strongly supports our hypothesis.
ENVIRONMENTAL ENRICHMENT AND NEURAL PLASTICITY
Early evidence showed that environmental enrichment produces changes in cortical weight and thickness [58], and an increase of dendritic branching and length, the number of dendritic spines, and the size of synapses of some neuronal populations [59, 60] and dental gyrus neurogenesis [61]. Additionally, at the molecular level, environmental enrichment induces the expression of brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) [62, 63], synaptic proteins [64, 65], and NMDA (N-methyl-d-aspartate) and AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid) receptor subunits [66]. As a result, environmental enrichment increases SP, such as LTP [67]. More importantly, environmental enrichment improves learning and memory at the behavioral level in young and old animals [68, 69]. Exciting results have shown that environmental enrichment enhances learning and memory in rodent models of neurodegenerative diseases such as AD. At the molecular level, there is an increase in synaptophysin, NGF, and neprilysin expression. Also, Adlard et al. [70, 71] using mice expressing a double mutant form of APP, showed an increased learning rate in the water maze test. They decreased the expression of Aβ plaques combining environmental enrichment with running wheels for five months.
OCTODON DEGUS, A NATURAL MODEL OF AGING AND NEURODEGENERATION
Although transgenic models have been instrumental in understanding familial forms of AD (5%of cases), these models, by not reproducing the full spectrum of neurodegeneration, are ineffective for clinical trials for sporadic AD (95%of cases) [12]. Aging is a critical factor that allows the gradual manifestation of the pathological mechanisms that accompany neurodegeneration and dementia in patients with AD. In general, the use of transgenic animals, although useful, is limited to detailed comparisons and related to the overexpression of specific proteins, such as the amyloid-β protein precursor. Another drawback in mice is that their lifespan is relatively short, 18–24 months, which is not sufficient to study the slow pro-cess that accompanies aging. In this respect, a limitation of currently validated animal models is that few allow for studying the real impact of natural aging on neurodegeneration development.
In the last few years, we have introduced the rodent, Octodon degus (degus), a natural model candidate to study aging and neurodegeneration since their brain reproduces changes observed in AD patients [13, 17–20]. Degus are mainly diurnal, medium-sized rodents and live in groups with high social interaction in the wild and under laboratory conditions [72]. Degus Aβ peptide shows a high 97.5%amino acid homology with humans, differing in only one amino acid, unlike rat and mouse, which differ from the human sequence in 3 amino acids [16]. Perhaps be-cause of this, some aged degus naturally develop AD-like pathologies, including the brain expression of the neuronal AβPP (β-APP695), display both intracellular and extracellular deposits of Aβ, intracellular accumulations of tau-protein and ubiquitin, a strong astrocytic response, and acetylcholinesterase-rich pyramidal neurons. Moreover, during aging, degus present symptoms associated with neurodegeneration and develop cognitive impairments including object recognition, spatial memory, and SP associated with the NMDAR-dependent process, which declines in an age-dependent manner (LTP and LTD). In degus, these impairments correspond to a form of sporadic AD with an increase of Aβ and soluble Aβ*56 (12-mer) oligomer, suggesting a critical factor for neural toxicity [51, 73], and tau deposition [74]. Consequently, SP in degus is affected during aging, especially at the postsynaptic level, with a decrease in LTP, protein expression (PSD-95, GluR2, NR2B), and cognitive performance (object recognition, T-maze), as we have described earlier [17, 51]. Here we present behavioral results based on a natural burrowing test for degus [75], that promise is an excellent biomarker for AD neurodegenerative disorder.
ACTIVITIES OF DAILY LIVING
A wide variety of behavioral tests are designed to assess animals’ cognitive states and defined brain structures. We have used a behavioral test based on rodents’ natural and spontaneous affinity to burrow, observed in rugged environments [76]. Importantly, designing behavioral tests based on natural or spontaneous behaviors provides a clear advantage in moti-vating animals for testing and reducing stress levels [77]. For example, natural or spontaneous behaviors have shown a good association with Activities of Daily Living (ADLs). Thus, in humans, ADLs are among the first activities affected in neurodegenerative diseases and are defined as necessary personal care activities (dressing, grooming, bathing, toileting, eating, and ambulation) or complex activities (meal preparation, shopping, telephone use, among others) [78]. The latter was one reason why we wanted to study the task of burrowing or digging to characterize the natural cognitive state of our subjects [79]. Burrowing is a natural behavior expressed by many rodent species, as they take advantage of their natural environment to protect themselves from predators, adverse weather conditions or store food and build a shelter for their offspring [80].
A burrowing task (BT) corresponds to the ADL type’s spontaneous activity and requires hippocampus function, as the induction of a cytotoxic injury decreased the burrowing performance [81]. Importantly, this test is fast, economical, and easy to impl-ement in the laboratories in its practical part. In a preliminary study (see methods in the Supplementary Material), we tested twenty-five degus aged between 40–75 months. The results show that 44%(n = 11) of degus exceeded the 10%threshold (grams of burrowed pellets) and were classified as Good Burrowers (GB), while 56%(14 degus) were below the threshold and were classified as Bad Burrowers (BB) (p < 0.05) (Fig. 1B). No statistical differences were found either by sex (p > 0.05) (Fig. 1C) or age (40–55 versus 56–75 months old, p > 0.05) (Fig. 1D). Figure 1E shows the GB number that changed from 58.3%to 50%, from 40–55 versus 56–75 months old, respectively. ADL-type behaviors are one of the first tasks that humans lose with aging and neurodegeneration [79]. To check if degus BT classification is related to their motor performance, we carried an open field (OF) test in which the degu is free to explore for 5 min. The results shown in Fig. 1F show that BB traveled a distance of 28.5 + 5 m, while GB traveled 32.4±3 m, with no significant differences (p > 0.05). The differential exploration of the center versus periphery of an OF is used to determine the animal’s anxiety level [82]. Figure 1G indicates individual results, where -1 indicates more time spent exploring the center, and +1 indicates a preference for the periphery. We did not see a particular difference between GB and BB (Fig. 1G) (BB: –0.39±0.13, GB: –0.27±0.09 m, p = 0.49), indicating that degus are not influenced by anxiety. Previous related results, according to burrowing test performance in degus, have established a good correlation with AD biomarkers of neuroinflammation [75].

The burrowing task in aged degus. A) Burrowing setup. Left: Burrowing apparatus (gray plastic tube with two screws at the entrance for support) filled with rabbit food pellets (1,300 g). Center: localization of the setup against the wall of a circular OF (diameter 180 cm). Right: degus put in the maze for free exploration; the burrowing content is measured as grams of pellets displaced out of the tube, corresponding to the BT performance. B) Burrowing classification according to degus performance in terms of the weight of pellet burrowed (n = 25). A threshold value of 10%of the total pellet burrow was determined (130 g) to separate Good Burrowers (GB) from Bad Burrowers (BB). C) Burrowing performance, according to sex. D) Burrowing performance in animals aged 40–55 months and 56–75 months. The red line represents the threshold value. E) Percentage of GB as a function of age. F) Distance traveled in an OF. G) Time in center versus periphery in the OF to measure anxiety level. The black line corresponds to the ratio of exploration time in center versus periphery, which were the same. Data are mean ± SD. Statistical analysis using the T-test. ****p < 0.0001.
PHYSICAL EXERCISE AND NEURAL PLASTICITY
Previous studies have demonstrated that physical exercise can improve neural plasticity mechanisms, increase BDNF, vascular endothelial growth factor (VEGF) and insulin-like growth factor 1 (IGF-1) [83–88]; enhance LTP and LTD in the dentate gyrus, increase spine density, dendritic branching, and neurogenesis [89–91], suggesting that all these factors contribute to the cognitive and neural plasticity improvement observed in physically trained model animals. On the other hand, it has been observed that exercise increases capillarization [92], decreases oxidative stress [93], and reduces Aβ load and the levels of hyperphosphorylated tau proteins [70, 94–97]. More importantly, physical exercise improves physical and executive function and spatial memory of patients with mild, moderate, or severe AD [88, 98]. Several studies have suggested that the activity carried out by free access to a wheel can prevent or delay cognitive deterioration occurring during a neurodegenerative process.
In a preliminary experiment in our laboratory, we have studied in degus of different ages the effect of voluntary long-term physical exercise on their cognitive capacities. Specifically, we have tested the loco-motor activity (open field), object recognition, and spatial (8-arm-maze) memory in young and old degus. Our results (unpublished) suggest that both young and old exercised degus reach a better cognitive performance than degus without (wheel) activity. Moreover, after four months without access to the freewheel, both degu groups show an increased cog-nitive deterioration [99]. Therefore, voluntary exercise may be an effective therapeutic strategy to reduce AD’s cognitive impairment. In another study (unpublished), we conducted a pilot study to determine the hippocampus’s neurogenesis level during aging, which usually decreases in rodents and primates, including humans. For this, we studied the morphology of the hippocampus (gyrus dentate) during aging, finding a dramatic decrease in neurogenesis between 7 to 96 months in degus, which contrasts with the number of cells in CA1, which do not change with age [100].
TRANSCRANIAL STIMULATION AND NEURAL PLASTICITY
Over the last few years, transcranial stimulation has been shown to promote neural plasticity mechanisms and cognitive improvement in neurodegenerative disorders. Early research in humans showed that repetitive transcranial magnetic stimulation (rTMS) produces a neural potentiation measured at EEG electrodes located bilaterally over the premotor cortex [101]. Interestingly, high-frequency rTMS induces LTP-like cortical plasticity within the precuneus in AD patients [102]. However, some results are contradictory, perhaps due to different protocols utilized in each study. For example, a study accomplished by Chen et al. [103] in an animal model of AD using rTMS showed an enhancement of cognitive function, a reduction of neuronal apoptosis, and an increase in the levels of BDNF, NGF, and doublecortin.
On the other hand, repetitive transcranial direct current stimulation (tDCS) produced spatial memory recovery in an AD rat model [104]. These authors suggest that the improvement is due to tDCS modulating synaptic plasticity through calcium or sodium channel regulation and increasing cell proliferation in the subventricular zone. However, Gondard et al. [105], using the same technique, did not find positive effects on learning and memory. Moreover, few randomized controlled trials using rTMS or tDCS in patients with mild to moderate cognitive impairment and AD demonstrated an improvement in cognitive functions in a different cognitive test such as the Mini-Mental State Examination (MMSE) or the AD Assessment Scale-Cognitive Subsection (ADAS-cog) [106–115]. In addition, it has been demonstrated by Zhang et al. [116] that rTMS combined with cognitive training improves cognitive function, which suggests that combined therapies could lead to better results in AD patients.
NEUROTROPHINS AND NEURAL PLASTICITY
Neurotrophins are growth factors that are essential in neuronal development, function, survival, and plasticity in the developing and adult CNS. They consist of four structure-related proteins: nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and neurotrophin-4 (NT-4/5) [117]. Neurotrophins exert their effects through membrane receptors that connect with different intracellular molecular cascades, such as MAP-kinase, PKC, and phosphatidylinositol 3-kinase (PI3-K), modifying gene expression and causing the synthesis of proteins [118]. The latter enables them to induce and modulate growth and functional neuroplasticity [119, 120]. On the other hand, neurotrophins can also indirectly support SP processes and reinforce the influence of non-glutamatergic afferents modulating LTP [121, 122]. BDNF in the adult brain increases synaptic transmission, facilitates SP, and promotes synaptogenesis [123]. Previous studies in healthy animals have shown that BDNF is crucial for forming and retaining hippocampal-dependent memory, fear memory extinction, and motor learning [124]. Moreover, previous AD animal models have demonstrated that BDNF administration decreases cognitive impairment and synapse loss, and neuronal abnormalities without causing Aβ and tau pathology [125–131]. Other authors have found that BDNF in neuronal cultures decreases production, and its removal contributes to an increase of Aβ [132, 133]. Likewise, Murer et al. [134] showed that neurons expressing BDNF did not present NFT, and by contrast, neurons with NFT did not express BDNF. In addition, Wang et al. [135], report that TrkB, an agonist antibody AS86 induces neurite outgrowth and enhanced spine growth with decreased cell death in cultured neurons. Furthermore, in this study, the use of AS86 rescued the cognitive impairments in APP/PS1 mice. On the other hand, the expression of BDNF in the hippocampus, temporal cortex, and parietal cortex is reduced in AD patients [136–138]. Also, in patients with sporadic and autosomal dominant AD, the BDNF Val66Met polymorphism impairs episodic memory and hippocampal activity when measured by Fluorodeoxyglucose-positron emission tomography (FDG-PET) [139, 140].
AMYGDALA STIMULATION AND NEURAL PLASTICITY
The amygdala is a subcortical structure critical for emotional and motivational reactions [141, 142]. It also contributes to memory consolidation occurring in other brain areas [143]. Moreover, electrical stimulation of the basolateral amygdala (BLA) can reinforce memory-related synaptic mechanisms like LTP [144] via cholinergic afferents to the locus coeru-leus and noradrenergic afferents to the medial septum [145]. Interestingly, natural emotional and motiva-tional stimuli, like drinking water after two hours of deprivation, prolong LTP if temporally related to LTP induction [146]. This phenomenon, recognized as LTP-behavioral reinforcement, is mediated by noradrenergic receptors [147], dependent on the synthesis of new plasticity-related proteins [148], and the amygdala is an essential part of the neural circuit involved [149]. Following this idea, we have shown that post-training BLA electrical stimulation in healthy animals accelerates the acquisition of a motor skill in the staircase task [150] and improves spatial memory in fimbria-fornix (FF) lesioned animals [151]. It also increased BDNF protein expression and arc gene expression in the hippocampus [152, 153], an increase of the synaptogenesis related proteins MAP-2 and GAP-43 in the hippocampus and prefrontal cortex [154]. Furthermore, the amygdala’s stimulation produces an increase of c-Fos protein, an early expression transcription factor related to neural plasticity and memory, in brain regions like the hippocampus and prefrontal cortex [155]. Interestingly, Inman et al. [156] demonstrated that the amygdala’s direct electrical stimulation enhances humans’ declarative memory. BLA electrical stimulation shortly after the performance of the behavioral task produces a functional recovery by directly promoting plastic changes in the brain structure involved in the task or by activating other modulatory regions like the locus coeruleus or the septal area, which, in turn, modulate the neural plasticity mechanisms involved in memory in relevant areas, especially in the hippocampus and the prefrontal cortex. Interpreting these and previous results [152–155], we propose that BLA stimulation promotes norepinephrine and dopamine release in the prefrontal cortex. In contrast, norepinephrine and acetylcholine are released in the hippocampus, which via G protein activates CREB and c-fos, BDNF, and arc gene expression.
In turn, c-Fos and BDNF could induce synapto-genesis-related proteins like MAP-2 (post-synapti-cally) and GAP-43 (pre-synaptically), contributing to the observed behavioral recovery. In the lesioned hippocampus, since the fimbria fornix lesion el-iminates most subcortical afferents, similar plastic mechanisms could be initiated via entorhinal cortex afferents and be triggered by glutamate activation of NMDA and metabotropic receptors. The latter could improve the spatial memory storage but might also explain why the recovery is only partial. In a phar-macological study, we have demonstrated that noradrenergic agonists applied 10 min after the induction of an early-LTP could mimic BLA stimulation’s reinforcing effect. In contrast, cholinergic agonists were not able to do so [145]. Catecholaminergic afferents appear to be relevant to LTP’s early maintenance, while cholinergic afferents are required later. According to the model proposed in Fig. 2, the stimulation of the BLA in FF-lesioned and trained rats can partially activate the molecular mechanisms leading to neural plasticity and trace formation, producing a recovery of spatial memory. Altogether, BLA stimulation can improve or modulate the neuroplastic process implicated in recovering lost functions due to CNS injury. We expect that modulating neural plasticity mechanisms through BLA stimulation can also rescue lost functions, such as memory, in AD models.
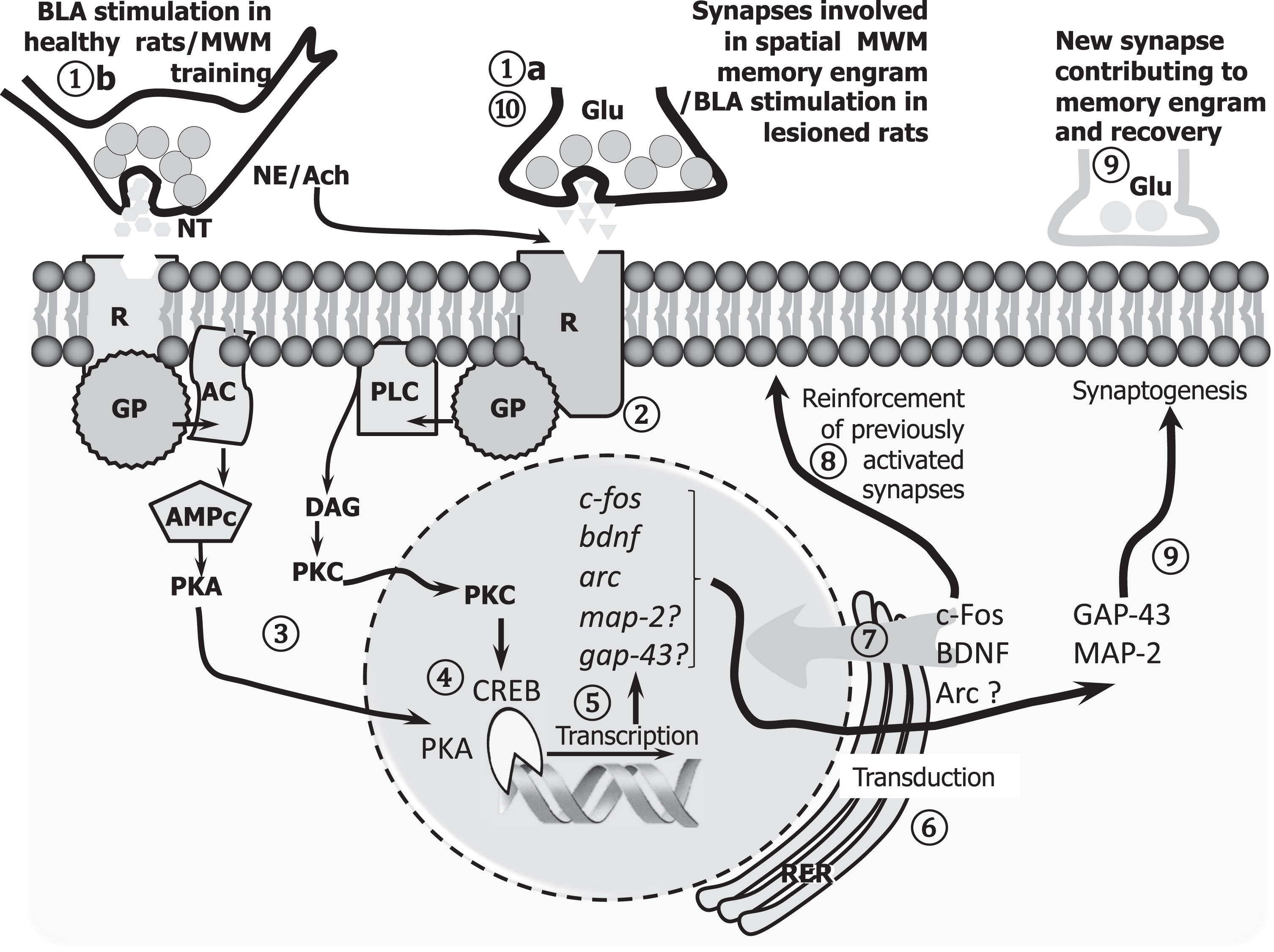
A cell model interpreting the synaptic plasticity mechanisms triggered by BLA stimulation on engram cells, promoting spatial memory in healthy and lesioned animals. In normal animals, the glutamatergic afferents (1a) to the dentate gyrus (DG) carry the information to be stored within the hippocampal memory system, probably as a long-lasting increase in the efficacy of those activated synapses (LTP). The activation of the amygdala contributes to reinforcing the LTP in the DG via the activation of norepinephrine (NE) afferents from the locus coeruleus, which also activates the septal cholinergic input required mainly by late phases of LTP (1b). Both transmitters activate intracellular second messenger cascades (2,3) that modify pre-existing proteins and regulate the expression of plasticity-related genes (4,5) like BDNF, Arc (functional plasticity), MAP-2, and GAP-43 (structural plasticity) (6,7). Altogether, the potentiation of existing synapses (8) and the formation of new ones (9) are cellular mechanisms by which memory is stored. In FF lesioned animals, this sequence is affected by the interruption of both NE and ACh afferents; however, the stimulation of the BLA can still (at least in part) contribute to consolidation via the glutamatergic afferents from the entorhinal cortex (10), probably by the activation of metabotropic glutamate receptors, which share common postsynaptic molecular cascades with other transmitters (2). Such a partially restored function could explain the memory recovery achieved by BLA post-training stimulation, resulting in an amelioration, but not in a full recovery.
CONCLUSIONS
Neural plasticity is a fundamental property of a healthy CNS which supports functions like learning and memory and functional recovery based on synaptic efficacy modification, synaptogenesis, sprouting, axonal regeneration, and neurogenesis. In contrast, the cognitive decline that occurs during aging is accompanied by an increase and accumulation of Aβ protein in the brain, neurofibrillary tangles, synaptic dystrophy, loss of neurons, and reduction of brain volume. These changes could overcome a physiological threshold beyond which neural plasticity mechanisms fail, and thus neurodegeneration is triggered. Exciting results have shown that optogenetic induction of LTP in the perforant path synapses of dentate-gyrus cells or optogenetic reactivation of the dentate gyrus cells in double transgenic mouse models of AD restore long-term memory and spine density [157, 158]. Moreover, environmental enrichment, natural behavioral tests, physical exercise, transcranial stimulation, neurotrophins such as BDNF, and direct amygdala electrical stimulation all induce plastic changes, rescue damaged synapses, and improve memory. As dis-cussed here, the use of natural animal models, which recapitulate the main findings associated with the neurodegenerative diseases that occur during the slow, progressive, aging process seen in humans, is critical when evaluating neurorestauration alternatives. As illustrated in this work, the combined use of natural models with a relatively long lifespan combined with interventions that promote neural plasticity represents an effective way to screen for AD preclinical treatments.