
Abstract
Alzheimer’s disease (AD), the main cause of dementia worldwide, is characterized by a complex and multifactorial etiology. In large part, excitatory neurotransmission in the central nervous system is mediated by glutamate and its receptors are involved in synaptic plasticity. The N-methyl-D-aspartate (NMDA) receptors, which require the agonist glutamate and a coagonist such as glycine or the D-enantiomer of serine for activation, play a main role here. A second D-amino acid, D-aspartate, acts as agonist of NMDA receptors. D-amino acids, present in low amounts in nature and long considered to be of bacterial origin, have distinctive functions in mammals. In recent years, alterations in physiological levels of various D-amino acids have been linked to various pathological states, ranging from chronic kidney disease to neurological disorders. Actually, the level of NMDA receptor signaling must be balanced to promote neuronal survival and prevent neurodegeneration: this signaling in AD is affected mainly by glutamate availability and modulation of the receptor’s functions. Here, we report the experimental findings linking D-serine and D-aspartate, through NMDA receptor modulation, to AD and cognitive functions. Interestingly, AD progression has been also associated with the enzymes related to D-amino acid metabolism as well as with glucose and serine metabolism. Furthermore, the D-serine and D-/total serine ratio in serum have been recently proposed as biomarkers of AD progression. A greater understanding of the role of D-amino acids in excitotoxicity related to the pathogenesis of AD will facilitate novel therapeutic treatments to cure the disease and improve life expectancy.
INTRODUCTION
Alzheimer’s disease (AD) is the main cause of de-mentia: according to the World Health Organization it accounts for 60–70% of all cases. AD is characterized by a progressive decline in cognition, memory, and executive function, starting from early lack of memory to gradual worsening of language, orientation, and behavior and extending to severe loss of memory and some bodily functions. This chronic neurodegenerative disease is the result of synapse dysfunction, synapse loss, and, ultimately, neuronal death. The etiology of AD appears to be complex and multifactorial. Its pathophysiology includes both structural and functional abnormalities. Upstream initiators of AD are represented by the amyloid-β (Aβ) and tau proteins; as AD progresses, multiple brain lesions develop over time, such as senile plaques, consisting of Aβ and neurofibrillary tangles containing phosphorylated tau, and loss of synaptic profiles [1].
An open issue concerns the complex relationships between neurons, glia, microglia, and vasculature, which contribute to synapse and circuit dysfunction [2]. Acute brain insults (hypoglycemia, neurologic trauma, stroke, and epilepsy) result in disproportionate excitatory glutamatergic neurotransmission and trigger synapse dysfunction and massive cell death in the central nervous system. Cognitive impairment and dementia are commonly associated with recovery from such insults, suggesting shared pathological mechanisms among various forms of dementia, in-cluding AD. Aβ is thought to play a causal role in the pathogenesis of AD [3]. The action of Aβ on syn-apses engages glutamate receptors, especially the N-methyl-D-aspartate (NMDA) receptors, which act as common initiators of various forms of neuronal dys-function/damage. Several single nucleotide polym-orphisms in the genes encoding NMDA receptor subunits have been reported to be implicated in the pathophysiology of neurological disorders such as AD, schizophrenia, and depression.
To activate NMDA receptors, binding of the activator glutamate and a coagonist is required: this coa-gonist was originally proposed to be glycine (Gly) but in the last 15 years studies have established that D-serine (D-Ser) is frequently the preferred coagonist [4–6]. Owing to its critical role in the mammalian brain, the processes for D-Ser synthesis, transport, and degradation have been investigated in depth [7–9]. Furthermore, a second D-amino acid (D-AA), D-aspartate (D-Asp), acts as a NMDA receptor agonist [10]. Indeed, D-Asp stimulates mGlu5 receptors and presynaptic AMPA (α-amino-3-hydroxy-5-met-hyl–4-isoxazolepropionic acid) receptors: it is considered to be a signaling molecule involved in neural development, influencing brain morphology and behavior in adulthood [11, 12]. D-Asp is also involved in the synthesis of different hormones and of melatonin [11, 13].
D-AAs (Fig. 1) are only present in low amounts in nature and were long considered to be of bacterial origin as they are key components of peptidoglycan in the bacterial cell wall and of certain antibiotics [14]. Now we know that D-AAs are constituents of biologically active peptides secreted by amphibians and are present in the cellular fluids of marine worms and invertebrates [15] and in foodstuffs (both of natural origin or that develop during processing) [16]. D-AAs are also present in animals, originating from endogenous microbial flora, diet, or racemization of L-enantiomers. In 1992, Hashimoto and collaborators determined a relevant level of D-Asp and D-Ser in the central nervous system (CNS), making it then possible to ascribe a distinctive function to D-AAs in mammals [17, 18]. Here, we would like to note that D-Asp had already been detected in human brain about ten years previously, having been found in myelin-rich white matter: this was a relevant discovery as a correlation between the level of D-Asp in white matter and age was identified, although the presence of D-Asp was at first incorrectly attributed to the natural rate of racemization of L-Asp occurring in proteins with aging [19]. In recent years, alterations in physiological levels of different D-AAs have been related to various pathological states, e.g., chronic kidney disease [20] and neurological disorders such as AD, bipolar disorder, and schizophrenia [21–25].
The mirror structure of the most relevant D-amino acids related to neuromodulation. Figure generated with Biorender.com.
NMDA RECEPTORS AND ALZHEIMER’S DISEASE
In large part, excitatory neurotransmission in the mammalian CNS is mediated by glutamate: its receptors, mainly ligand-gated ionotropic glutamate receptors, are involved in synaptic plasticity, the mechanism underlying learning and memory [26]. On the one hand, the NMDA receptors, tetrameric channels constituted by two GluN1 subunits together with either two GluN2 subunits (selected among GluN2A-GluN2D isoforms) or a combination of GluN2 and GluN3 subunits (alternatively GluN3A or GluN3B isoforms), play a main role [27] (Fig. 2). For activation, NMDA receptors require the principal agonist glutamate at the GluN2 subunit and a coagonist at the so-called “glycine-binding site” on the GluN1 subunit. The coagonist, which could be represented by D-Ser or Gly, plays an important modulatory role in NMDA receptor function [4–6, 29]. At resting membrane potential (≈–70mV), Mg2 + blocks the Ca2 + channel of the NMDA receptor. Activated AMPA receptors remove this blockade by depolarization following the strong and prolonged release of glutamate from the presynaptic terminal during the induction of long-term potentiation (LTP): this promotes the influx of Ca2 + ion and, through the activation of Ca2+/calmodulin-dependent protein kinase II-mediated signaling cascade, enhances synaptic strength. On the other hand, a modest activation of NMDA receptors induces a small increase in postsynaptic Ca2 + levels and elicits phosphatase activity, yielding long-term depression (LTD) [30]. The interplay at the tripartite synapsis under physiological conditions is reported in Fig. 3.
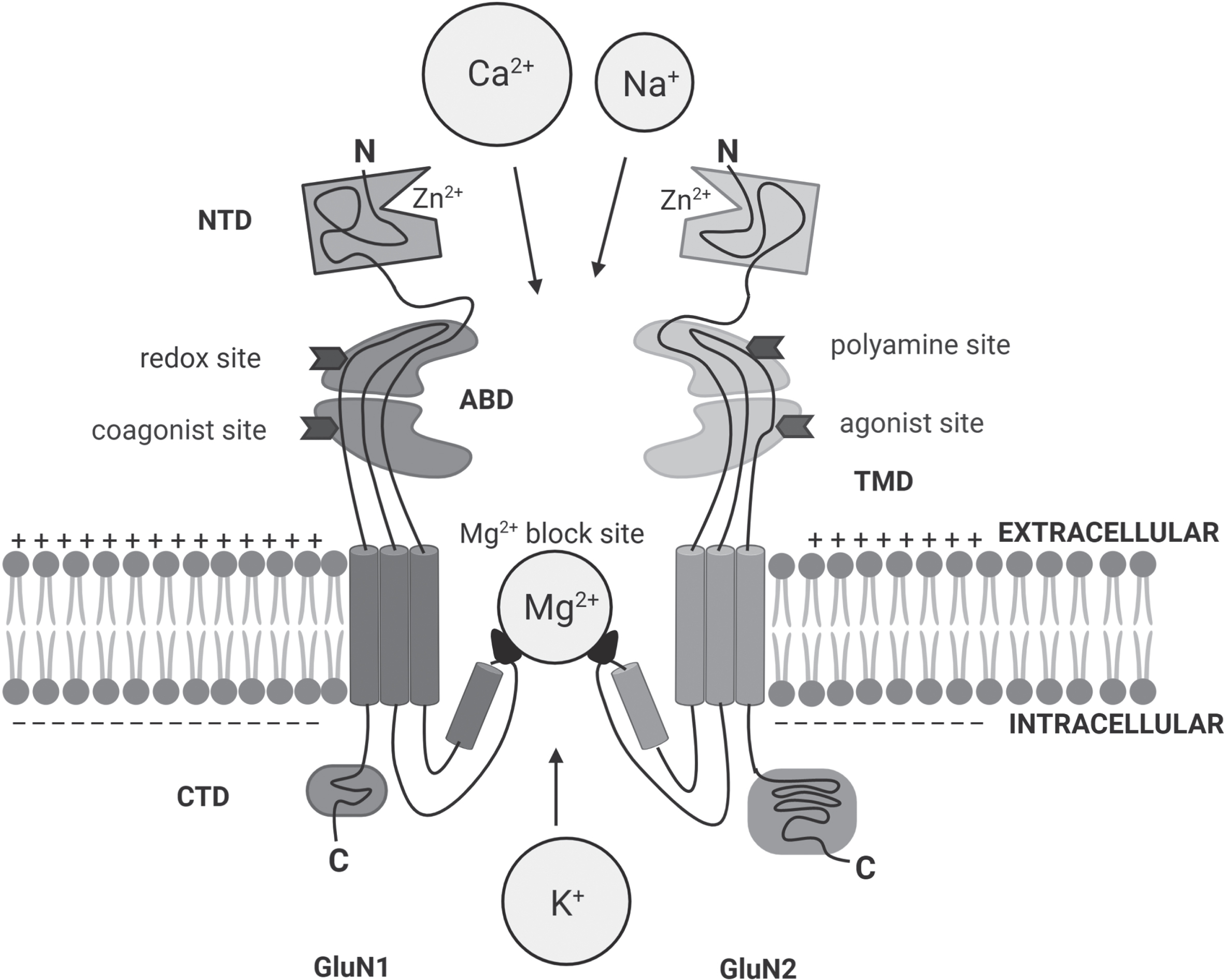
Scheme of the modular organization of NMDA receptors: heterotetramers are formed by two GluN1 subunits associated with the combination of two other subunits including GluN2(A-D) or a mixture of GluN2 and GluN3(A or B). The extracellular portion includes the NTD (N-terminal domain) and the ABD (agonist binding domain): the latter harbors the binding sites for the agonist glutamate and the co-agonist Gly/D-Ser (the glycine-binding site), as well as the redox and polyamine regulatory sites. The TMD (transmembrane domain) forms the ion channel that contains the site for Mg2 + blockade. The intracellular region is made by the CTD (C-terminal domain). Figure generated with Biorender.com.
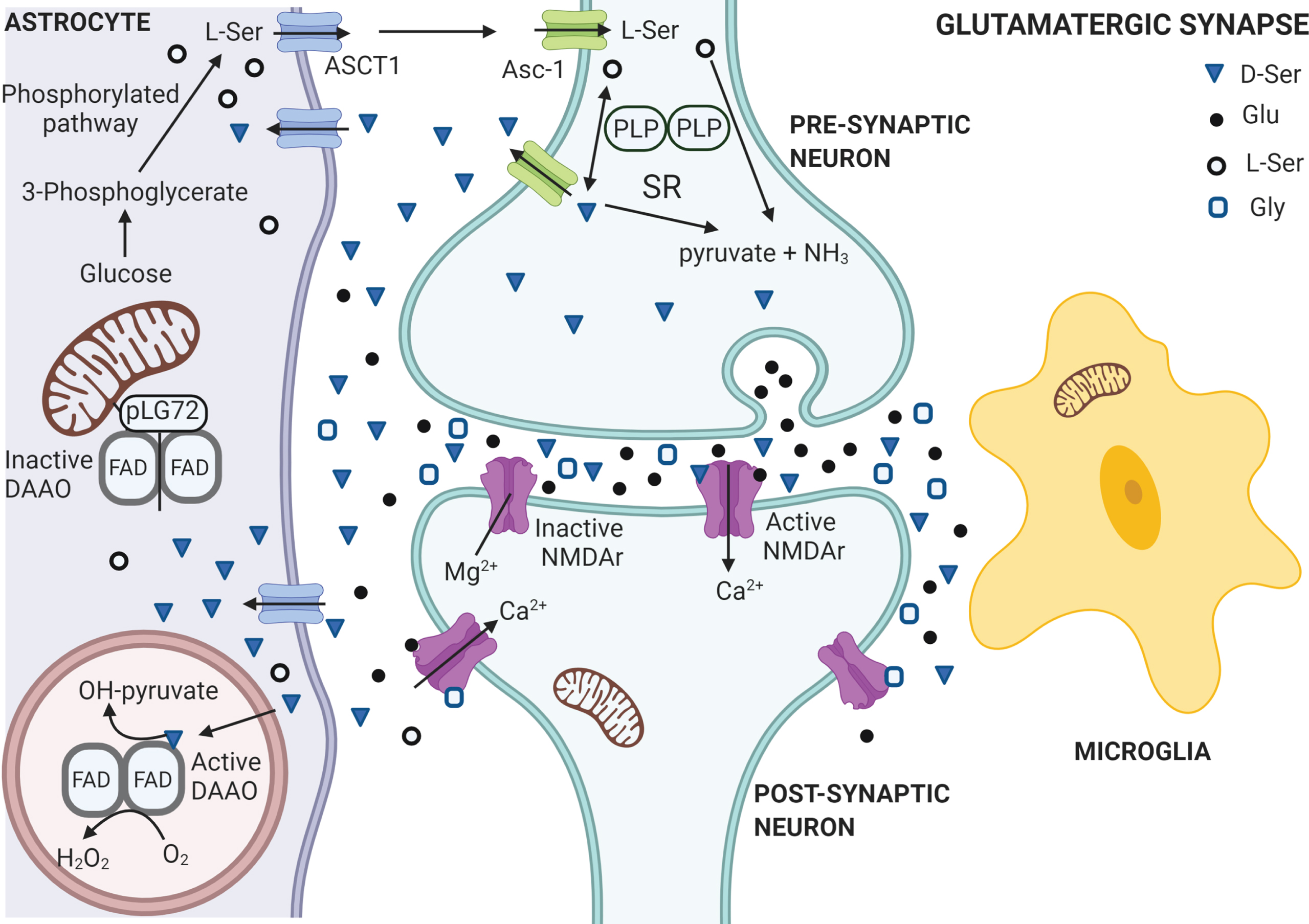
Schematic representation of the serine shuttle at the tripartite synapsis under physiological conditions. L-Ser is synthesized from glucose in the astrocytes through the phosphorylated pathway from the glycolytic intermediate 3-phosphoglycerate. L-Ser is released in external medium through the ASCT1 subtype of neutral amino acid transporters and then it is taken up by neurons through the Asc-1 subtype transporters. In neurons L-Ser is racemized into D-Ser by serine racemase (SR), which is delivered back to extracellular space through Asc-1 hetero-exchange with L-Ser. Extracellular D-Ser binds to NMDA receptors concomitantly with glutamate, activating the receptors at the membrane of postsynaptic neurons, thus promoting functional plasticity at synapses. Gly acts mainly on extrasynaptic NMDA receptors (NMDAr) while the synaptic ones are preferentially targeted by D-Ser as co-agonist. In healthy individuals, in absence of a presynaptic glutamatergic signal, the Mg2 + ion occupies the calcium channel in postsynaptic NMDA receptors. In neurons, both D- and L-Ser can be degraded by SR into pyruvate and NH3 by the water elimination reaction. D-Ser is picked up by astrocytes from the synaptic cleft through ASCT1 hetero-exchange with the L-enantiomer, where it is degraded by the peroxisomal enzyme D-amino acid oxidase (DAAO), which activity is controlled by pLG72 (located on the cytosolic side of outer mitochondrial membrane) to prevent an excessive degradation of D-Ser. Figure generated with Biorender.com.
A classical view of AD pathogenesis proposes that an excessive stimulation of glutamate signaling results in excitotoxicity, which is mediated by incr-eased calcium influx in post-synaptic neurons [31]. The prolonged calcium overload leads to gradual loss of synaptic functions followed by synaptotoxicity and neuronal cell death, which, in turn, correlates with the progressive decline in memory and cognition in AD [32, 33]. Astrocytes also play a significant role in this process owing to their ability to control the synaptic level of glutamate. Excitotoxicity is triggered by alterations in glutamate and calcium metabolism, dy-sfunction of glutamate transporters, and glutamate receptors, such as NMDA receptors, as well as a con-sequence of mitochondrial dysfunction, physical neuronal damage, and oxidative stress, see [34] for a recent review. To give a complete picture, we would mention that recent literature suggests that AD results from presynaptic failures before postsynaptic dysfunction ensues [35–38].
The electrophysiological function of NMDA rec-eptors is directly modulated by Aβ (Fig. 4) since: 1) it interacts with the receptors and, in cultured neurons, leads to internalization of synaptic NMDA receptors and the depression of related currents [39]; 2) it induces glutamate release from astrocytes, which, in turn, activates the pathological, extrasynaptic NMDA receptors on neurons and the ensuing decrease in miniature excitatory postsynaptic currents [40]; 3) its oligomers change the activation of the NMDA receptor-dependent signaling pathway by impairing LTP and inducing LTD; and 4) NMDA receptor antagonists prevent the structural effects induced by Aβ. For reviews, see [31, 41].
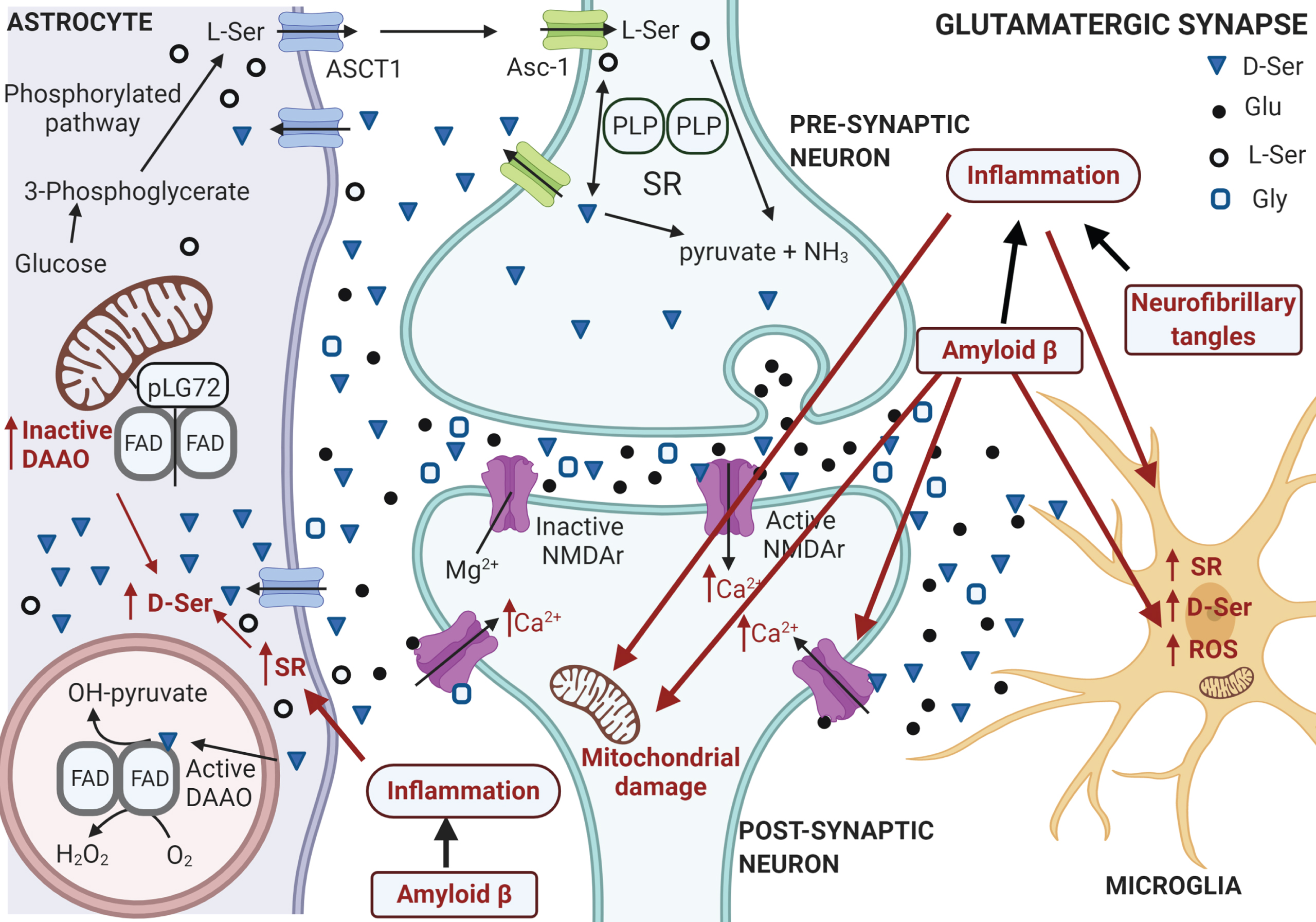
Schematic representation of the serine shuttle at the tripartite synapsis under AD. Aβ plaques and neurofibrillary tangles induce brain inflammation (see words and arrows depicted in red): both inflammation and Aβ damage neuronal mitochondria. Under these conditions the neuron is unable to maintain the correct resting potential: Mg2 + vacates the calcium channel at postsynaptic NMDA receptors allowing the influx of excessive levels of calcium ion. Aβ may further exacerbate the alteration due to excessive Ca2 + level by enhancing its entry interacting with postsynaptic NMDA receptors and activating D-Ser synthesis in microglia and astrocytes. The final result is a pathological neuronal overactivation. Neuronal cell survival can be also compromised by insufficient synaptic NMDA receptor signaling: the block of NMDA receptor function leads to neuronal apoptosis and degeneration. Lower glucose metabolism is associated to early stage AD since lack in ATP production affects ionic and neurotransmitter gradients. Indeed, a decrease in the astrocytic glucose metabolism could affect L-Ser production from the glycolytic intermediate 3-phosphoglycerate and, in turn, the D-Ser level. Figure generated with Biorender.com.
NMDA receptors also play a major role in neuronal survival by activating the neuronal survival pathway [42, 43]: blocking NMDA receptor function causes neuronal apoptosis and degeneration [44, 45]. Neuronal cell survival is compromised both by insufficient synaptic NMDA receptor signaling and excessive stimulation of glutamatergic signaling, res-ulting in excitotoxicity by excessive Ca2 + entry, primarily through these receptors. The level of NMDA receptor signaling must be balanced to promote neuronal survival and prevent neurodegeneration: this signaling in AD is affected mainly by glutamate availability and modulation of the receptor’s functions [31].
While the synaptic NMDA receptors are prefere-ntially activated by the coagonist D-Ser, the extrasynaptic ones interact better with the coagonist Gly [46] and are preferentially targeted by the antagonist memantine [47]. Extrasynaptic NMDA receptor-in-duced responses seem to be tightly related to the phy-siological changes occurring in AD: Aβ specifically activated extrasynaptic NMDA receptors, which cau-sed synaptic loss, an outcome that was antagonized by memantine [40, 48].
AD has been also linked to the level of NMDA receptor coagonist D-Ser: in AD hippocampus or Aβ-treated cultured microglia, D-Ser levels increased due to a parallel increase in expression of serine racemase, the enzyme assigned to synthesize D-Ser (Fig.4) [49]. In addition, a decrease in forebrain D-Ser levels, obtained by knocking out serine racemase, ameliorated both NMDA- or Aβ-caused neurotoxicity [50]. Notably, six modulators acting at the glycine-binding site of NMDA receptors and showing procognitive and antidepressant properties were identified: three of them are D-AAs, namely, D-Ser, D-cyclo-serine, and D-alanine, reviewed in [51].
D-Asp levels have been also related to the modulation of excitatory neurotransmission. Knockout mice for the enzyme assigned to catabolize D-Asp, namely, D-aspartate oxidase [52], demonstrated that the persistently high levels of this amino acid caused age-dependent effects: spatial memory and cognitive ability improved in young individuals, followed by rapid deterioration of learning and memory abilities in the same animals, leading to precocious brain aging [53, 54]. Interestingly, the high D-Asp levels in these knockout mice resulted in changes in tau phosphorylation [55]. Degradation of D-Asp by D-aspartate oxidase prevented NMDA receptor hyperstimulation, while an increase in D-Asp level rescued the age-related cognitive impairment [54]. In a murine model of neuropathic pain, D-Asp treatment reduced abnormal behaviors and normalized Aβ expression, probably by increasing steroid levels in the prefrontal cortex and in the hippocampus [56].
D-AMINO ACID LEVELS IN ALZHEIMER’S DISEASE PATIENTS
During the last 20 years, several studies aimed to quantify D-Asp and D-Ser in CNS and compare hea-lthy subjects (HS) and AD patients. It should be noted, however, that most of this work, in particu-lar that conducted before the 2000s, presents some issues. Firstly, different analytical methods were employed to determine amino acid levels (these included gas chromatography, enzymatic determinations, and diverse HPLC methodologies) and, in some cases, the analytical procedures employed were not described in detail. Secondly, all these studies were carried out on a limited number of subjects (often less than ten). These concerns resulted in contrasting and somewhat confusing results, also ascribable to the lack of standardized and accurate procedures and appropriate controls, as those proposed in [57]. Results obtained in these studies are summarized in Tables1–3 and Supplementary Tables 1–3.
Change in D- and L-Ser levels and D/(D+L)-Ser ratio between AD and HS samples expressed as percentage. Δ% =100*([AD] - [HS] / [HS])
Change in D- and L-Asp levels and D/(D+L)-Asp ratio levels between AD and HS samples expressed as percentage. Δ% =100*([AD] - [HS] / [HS])
N.D., not determined.
Change in D- and L- levels and D/(D+L) ratios of different amino acids between AD and HS samples expressed as percentage. Δ% =100*([AD] - [HS] / [HS])
N.D., not determined.
D-Ser
Similar levels of D-Ser in the frontal cortex of AD patients and HS were found by [58], whereas Madeira et al. [24] reported that D-Ser levels were significantly higher in hippocampus and parietal cortex of neuropathologically confirmed AD patients but were the same as in HS in the occipital cortex. Concerning cerebrospinal fluid (CSF), a 5-fold higher mean level of D-Ser and a 2.4-fold higher D-/(D+L)-Ser ratio were determined in the ventricular CSF of AD patients than in HS [59]. A 5-fold increase in D-Ser levels in probable AD patients as compared to HS was also observed in the lumbar CSF [24]. These authors reported a positive correlation between D-Ser levels and the progression of cognitive decline/impairment as assessed both by the Clinical Dementia Rating, (CDR) score, a scale used to characterize five domains of cognitive and functional performance in AD dementia [60], and the Mini-Mental Scale Examination (MMSE) scale [61]. However, Biemans et al. [62] did not confirm these findings in their report: by employing an ultra-HPLC-tandem MS method, they found a 13% D-Ser increase in AD patients compared to HS only and no correlation with MMSE. Moreover, comparison of the mean value of D-Ser levels observed in the lumbar CSF of AD patients revealed that the concentration in the study of [62] was about 8-fold lower than the value reported by [24]. The D-/(D+L)-Ser ratio determined in the latter study in control samples (≈8%) was then confirmed by [63], which, however, did not report the mentioned increase in the AD cohort. A possible explanation for this discrepancy could be related to the different analytical methods employed, the different storage times of the samples, and the differences in MMSE values, in disease duration, and even in ethnic and genetic background of the two cohorts analyzed.
A slight decrease (–11%) in levels of L-Ser in serum of AD patients compared to HS, together with a moderate (–20%) and statistically significant decrease in D-/(D+L)-Ser ratio were reported in [21]. Nuzzo et al. did not report any changes in the D-Ser and D-/(D+L)-Ser ratios [63] but this conclusion is hampered by the lack of an enzymatic treatment to evaluate D-Ser concentration: it is conceivable that D-Ser levels were overestimated, as made apparent by the extraordinarily high D-/(D+L)-Ser ratio (≈20%) reported for postmortem cortical samples. On the other hand, different results were recently reported by [64] using a validated analytical method, where a moderately (+20%) and statistically significant increase both in D-Ser and in D-/(D+L)-Ser ratio were observed. Detailed analysis of these latter results showed a slight gender effect on serum D-Ser levels and a positive correlation between serum D-Ser levels and age of AD patients, which was not observed in HS. Most interestingly, a clearly increasing trend with disease progression (assessed by CDR score) for both D-Ser and D-/(D+L)-Ser ratio was apparent. A statistically significant increase was found for serum D-Ser levels between HS and AD patients with a CDR score of 2 and for serum D-/(D+L)-Ser ratio between HS and AD patients with a CDR score of both 1 and 2. This latter result is in line with previous observations by [24].
A very recent meta-analysis of serum and CSF levels of D-Ser in AD patients has been published [65]. The analysis of seven trials corresponding to 1186 participants showed that D-Ser levels were significantly higher in AD patients compared to controls. Moreover, a significant negative association was apparent between effect size and mean MMSE score in AD group.
In sum, we consider the increase in brain and blood D-Ser levels (observed in a large proportion of the available investigations, see Table 1) to be a hallmark of AD onset/progression that can be used as useful biomarker when sensitive analytical methods are combined with suitable controls.
D-Asp
The first paper reporting on D-Asp levels and AD stated that the D-/L-Asp ratio in white matter of human brain from HS increased during aging, from 1 to about 35 years, and that in the gray matter the average value of D-/L-Asp ratio was halved with respect to the one found in the white matter of the same subjects [66]. Moreover, no gender differences were observed. The D-/L-Asp ratio was the same in both white and gray matter of AD patients and in an age-matched cohort of HS [66]. In contrast, a different study reported that the free D-Asp level was more than twice as high in the white matter of AD patients than in HS, while a similar value was determined in gray matter between the two groups [67]. Notably, the same group, one year later, reported a significantly higher D-/(D+L)-Asp level in both gray and white matter by using an enzymatic assay on total protein hydrolyzed [68]. Later on, D’Aniello’s group conducted a detailed analysis in different brain regions. In this work, D-Asp levels in brain samples from AD patients were significantly lower than those observed in corresponding samples from HS in frontal, parietal, and temporal cortices and hippocampus and amygdala (43, 38, 35, 47, and 41%, respectively). In contrast, no significant differences were found in cerebellum, an area lacking the neuropathological changes of AD [69]. In addition to various brain tissues, D-Asp levels have been also assayed in lumbar and ventricular CSF. The mean values of D-Asp and D/(D+L)-Asp levels were 1.5- and 1.4-fold higher, respectively, in lumbar CSF of AD patients than in HS, and about 2.7-fold higher for both values in ventricular CSF [70]. In more recent studies, however, no D-Asp was detected [63] in lumbar CSF.
Very recently, different results were reported based on HPLC analyses in which the identity and concentration of D-Asp was evaluated through selective enzymatic degradation. No difference in D-Asp levels and D-/(D+L)-Asp ratio were reported in samples from the superior frontal gyrus of AD patients and HS [55]. Similarly, and independently of each other, two different groups found no significant modification in D-Asp concentration and D-/(D+L)-Asp ratio in serum from HS and AD patients [63, 64]. Analyzing the latter results based on the CDR score, a moderately decreasing trend from HS (CDR 0) to patients with a CDR score of 2 (moderate dementia) was observed for the D-/(D+L)-Asp ratio [64]. Interestingly, a decreased mean value of D-/(D+L)-Asp ratio in AD patients was reported by [55], although the difference from HS was not statistically significant. The lack of statistical significance in the latter studies might be ascribed to the very low concentration of D-Asp, close to the limit of detection for the analytical method used.
In conclusion, no correlation between D-Asp levels and AD progression is apparent (see Table 2). This correlates with the proposed role of D-Asp in the regulation of early neurodevelopmental processes that link this neuromodulator to disorders such as schizophrenia, autism spectrum disorders, and intellectual disability [63].
Other D-amino acids
D-alanine (D-Ala) in gray and white matter was studied by [67]: comparison of data from HS and AD patients showed that D-Ala levels were similar in white matter, whereas doubled D-Ala levels were apparent in the gray matter of AD patients as compared to HS. No difference was found in the D-/(D+L)-Ala ratio. In ventricular CSF, both D-Ala and D-/(D+L)-Ala ratio were similar in AD pati-ents and HS [59, 71]. The same paper reported the presence of D-arginine (D-Arg) in ventricular CSF: in this case, D-Arg and D-/(D+L)-Arg ratios were lower in samples from AD patients, but the difference was not statistically significant. Concerning D-Ala, Lane’s group concluded that serum levels of the D-enantiomer of alanine in AD patients at different cognitive decline stage were unrelated to cognitive deficits when assessed by the CDR scale, while a positive correlation was evident when the Alzheimer’s Disease Assessment Scale-Cognitive Subscale (ADAS-cog) was used, and relatively to the behavioral score only [72, 73]. Recently, a further study reported the absence of any significant difference in D-Ala levels in blood from patients with mild cognitive impairment (MCI) and dementia compared to HS [74].
Gly was assayed in different brain areas by [24]. In parietal cortex Gly levels were significantly higher in AD patients than in HS (about 1.5-fold), whereas they were the same in occipital cortex and in hippocampus. The same paper also reported slightly higher Gly levels in lumbar CSF of probable AD patients than in HS (+15%), but this increment was not statistically significant.
According to the available results, however, we cannot reach conclusions concerning a relationship between D-Ala, D-Arg, or Gly levels and AD pathophysiology (Table 3).
D-AMINO ACID LEVELS AND COGNITION
NMDA receptors play a pivotal role in cognitive functions, particularly in learning and memory formation, development, behavior, and social interaction: they convert specific patterns of neuronal activity into long-term changes in synaptic structure and activity, processes considered to underlie higher cognitive functions [75, 76]. Their hypofunction or reduced activation can result in profound deficits in synaptic plasticity and cognitive processes. For example, Grin1D481N mice, in which behavioral phenotypes are relevant to schizophrenia and which pos-sess a point mutation in the NR1 glycine binding site resulting in a 5-fold decrease in NMDA receptor affinity for the coagonist, showed impairments in long-term spatial learning and memory, abnormally persistent latent inhibition (reflecting a deficit in selective attention), and reduced social approach behaviors [77]. Notably, all these deficits were rev-ersed by administering compounds that saturate the glycine-binding site of NMDA receptors, such as D-Ser [77]. A beneficial effect of a low dose of D-Ser (50mg/kg/day) on reversal learning was observed in APPswe/PS1 mutant mice harboring Aβ plaques in the brain [78]. Interestingly, the same low-dose D-Ser treatment boosted memory consolidation, object recognition, and working memory in normal mice, too [79]. Indeed, oral administration of D-Ser rescued the cognitive deficit observed in APP knockout mice and also restored the observed deficits in spine dynamics, adaptative plasticity, and morphology [80]. Here, mice lacking the enzyme responsible for D-Ser degradation (D-amino acid oxidase, DAAO, see below) underscore the crucial role of D-Ser in cognition and social behavior: in these animals, increased D-Ser levels were associated with better performance in both cognitive and behavioral tests [81–83]. Notably, increased DAAO levels were reported in peripheral blood of patients with AD or mild cognitive impairment, showing a positive correlation with the severity of the cognitive deficit and, in contrast with the known biological role of DAAO, with D-Ser levels [72]. This contradictory result requires further elucidation.
NMDA receptor hypofunction has been well documented in nonpathological brain aging and seems to arise from a reduction in D-Ser levels that should be mainly due to an impairment in the biosynthetic pathway [84, 85], while the affinity of D-Ser for the NMDA receptor-binding site should not be affected [84, 86]. Accordingly, exogenous D-Ser treatment enhanced synaptic plasticity in aged rats [86] and in a mouse model of accelerated senescence (SAMP8 mice) [87]. It is noteworthy that daily treatment with the well-known DAAO inhibitor sodium benzoate in individuals with amnestic mild cognitive impairment or mild AD produced significantly better improvement than placebo in several cognitive functions, such as processing speed and working memory [88]. The favorable effect of sodium benzoate in AD could be also ascribed to an antioxidant effect as DAAO inhibition prevents hydrogen peroxide from being generated during the enzymatic degradation of D-Ser [89]. The oxidative stress observed during aging can result in the oxidation of cysteines and in the alteration of the dimeric, active conformation of serine racemase: this effect can be prevented by long-term treatment with the reducing agent N-acetyl cysteine, resulting in potent NMDA receptor activation [90].
However, if increased D-Ser levels can improve cognitive performance by enhancing glutamatergic transmission, then excessive stimulation of NMDA receptors can promote excitotoxicity, which is associated with AD onset [91, 92]. A number of experimental results have linked NMDA receptors, D-Ser, and excitotoxicity: 1) the evidence that D-Ser mainly binds to synaptic NMDA receptors while glycine preferentially binds to extrasynaptic receptors [46]; 2) the known neuroprotective role for NMDA receptors located at synapse due to the enhancement of antioxidant defenses promoting the transcription of prosurvival factors and the stimulation of anti-apoptotic effects [48] and the neurotoxic role for extrasynaptic receptors [48, 93]. The activation of the latter receptors by glutamate or Aβ yields an increase in calcium levels in the postsynaptic cells that activates calpain and CDK5 pathways involved in tau phosphorylation and, finally, leads to an excessive neuronal oxidative stress that can destroy neuronal connections; 3) the evidence that activation of NMDA receptors containing GluN2B subunits (such as the extrasynaptic receptors) activate excitotoxic pathways directing neuronal death, while those containing GluN2A subunits stimulate signaling cascades associated with neuroprotection and regulate survival [94]. For the sake of completeness, we need to mention that the involvement of NMDA receptors in excitotoxicity is still under debate since these receptors seem to play a double role in excitotoxicity. While some studies suggest that NMDA receptor-induced excitotoxicity requires overactivation of both synaptic and extrasynaptic receptors [94, 95], other investigations suggest that activation of synaptic receptors counteracts excitotoxicity and activation of extrasynaptic receptors contributes to the excitotoxic cascade [96]; additional studies even propose that neurotoxicity might be dependent on the activity of synaptic NMDA receptors only [46]. Nevertheless, stimulation of extrasynaptic NMDA receptors is critical in AD. Actually, recent investigations reported that enhancement of synaptic NMDA receptor activity and inhibition of extrasynaptic receptors have protective effects against Aβ-induced neurotoxicity [97] and that stimulation of the latter receptors is associated with inactivation of extracellular signal-regulated kinase/mitogen-activated protein kinase signaling [98], which is essential for memory consolidation and synaptic plasticity. Furthermore, D-Ser released from reactive astrocytes can bind to extrasynaptic GluN2B-containing receptors, triggering the activation of excitotoxic signaling pathways and resulting in neuronal damage and death. At the same time, the use of an NMDA receptor coagonist resembling D-Ser and the selective blockade of extrasynaptic receptors significantly improved spatial and related forms of learning and memory under AD-like conditions [97]. A recent clinical study highlighted a positive correlation between D-Ser (and D-Ala) blood levels and decline in cognitive processes in AD patients [73]. This finding supports the reports of an increase in D-Ser levels in animal models of AD [24] and in blood/CSF of AD patients [24, 64], as well as the evidence that Aβ aggregates induced D-Ser release (Fig.4) [49]. However, the observed increase in D-Ser levels might also represent a protective mechanism to counteract Aβ signaling and prevent AD pathology, as recently suggested by [89]. D-Ser enhances neurogenesis and neuronal survival [99] and regulates apoptosis, inhibiting this process at early phases and stimulating it at later phases [100]. The beneficial effect due to enhancement or attenuation of NMDA receptor neurotransmission may depend on the phase of the disease: increasing D-Ser levels might be therapeutically beneficial in the early stages of AD.
In agreement with its role as an endogenous ago-nist, D-Asp was also shown to affect NMDA receptor-dependent transmission, synaptic plasticity, dendritic morphology, and cognition. Studies performed on animal models with nonphysiologically high D-Asp levels (i.e., mice lacking the D-Asp-degrading enzyme D-aspartate oxidase or mice treated with D-Asp) showed an increase in NMDA receptor-dependent early- and late-phase hippocampal LTP [53, 102] associated with structural synaptic variations: increased dendritic length and spine density and a greater dendritic complexity of hippocampal and cortical neurons [101]. Behavioral studies showed a significant improvement in spatial learning and memory formation in these animals [11, 103]. However, subsequent studies have highlighted that the persistent upregulation of D-Asp levels caused a precocious and progressive decay of synaptic transmission and hippocampal memory in 13/14-month-old mice [54], which was paralleled by a loss of excitatory glutamatergic synapses and reduction in synaptic GluN1 and GluN2B subunits [104]. Moreover, the deregulated D-Asp concentration in these mice was associated with precocious oxidative stress and caspase-3 activation, and ultimately led to cell death, thus suggesting that D-aspartate oxidase, by strictly regulating D-Asp levels, plays a neuroprotective role and prevents early neurodegenerative processes triggered by excessive NMDA receptor stimulation [12].
A further D-amino acid, the partial agonist D-cycloserine was shown to improve cognitive functions both in animal studies [105–107] and in patients with dementia [108, 109], although conflicting results were also reported [110, 111]. However, as several cognitive beneficial effects were observed in healthy individuals [112–114], it has been speculated that this molecule may have a different effect on mood and learning, depending on the stage of dementia [115].
Recently, a negative correlation between D-glutamate (D-Glu) blood levels and some cognitive functions (i.e., comprehension, naming objects, and following commands) was reported in AD patients [73], further confirming previous findings from the same group [72]. In contrast, D-Ala levels did not correlate with any of the cognitive functions analyzed, but higher D-Ala and lower D-Glu levels were associated with an increase in the ADAS-cog behavior scores, which reflect the severity of behavioral and psychological symptoms of dementia [73]. Very recently, a new chiral tandem liquid chromatography-MS/MS system was used to analyze the amount of chiral amino acids in blood from HS and individuals with MCI and dementia (diagnosis of MCI was assessed by MMSE score) [74]. An increase in blood D-Pro levels (1.5-fold when evaluated as D-/(D+L) ratio) was reported in MCI patients versus a matched cohort, a parameter that was strongly associated with the early stage of cognitive decline when examined in combination with D-Ser levels. Accordingly, the authors proposed blood D-Pro levels as a biomarker for the diagnosis of dementia and as a novel brain health index correlated to cognitive functions. This study suffers from some limitations, however: the enrollment of female individuals only, the low number of individuals in the dementia group, and the sole use of the MMSE score to assess cognitive function. Furthermore, it is noteworthy that D-Pro is the only D-amino acid that is catabolized by both DAAO and D-aspartate oxidase [52, 116].
METABOLISM OF D-AMINO ACIDS AND ALZHEIMER’S DISEASE
As stated above, dysfunctional glutamatergic transmission mediated by altered NMDA receptor activity has been proposed to contribute to the pathogenesis of AD [41, 117]. About 25 years ago, it was reported that D-cycloserine and D-Ser enhance memory performance in individuals with AD [108, 109] and restore retrograde memory in rats after perirhinal cortex injury [118], suggesting that D-Ser plays a complex and important role in the onset of AD. Thus, in the last two decades, the contribution of D-Ser in AD pathogenesis has been extensively investigated, also focusing on the enzymes involved in its metabolism.
In the brain, D-Ser is synthesized from the L-enantiomer by the PLP-dependent enzyme serine racemase (SR, EC 5.1.1.18) and is degraded either by SR itself or DAAO (EC 1.4.3.3) [7]. SR is mainly expressed in neurons and to a lesser extent in astrocytes and microglia [119–122], while DAAO is expressed in astrocytes [123–126]. SR is functionally regulated by several mechanisms, including cofactor and ligand binding (Mg2 +, Ca2 + and ATP), protein interactors, oligomerization state, subcellular localization, and post-translational modifications [127–132]. DAAO activity is influenced by cofactor and ligand binding, protein mistargeting, post-translational modifications, and protein interactors (pLG72, Basson) [7, 133–136].
In 2004, Wu and collaborators reported that primary cultures of microglia treated with Aβ and the secreted forms of AβPP (sAPP) induced the release of an increased amount of D-Ser, which was coupled with an increase in transcription and protein level expression of the dimeric (active) form of SR [49, 137]. Moreover, it was reported that primary cultures of hippocampal neurons responded to the conditioned medium from treated microglia with a rapid increase in Ca2 + levels, while the addition of DAAO reduced the ability of microglia-treated medium to induce the Ca2 + response, supporting the hypothesis that glial D-Ser is a cause of excitotoxicity (see Fig.4) [49, 136]. These conclusions were further confirmed as injecting Aβ in the hippocampus of SR knockout mice (showing a 90% decrease in D-Ser content in the brain compared to control mice) resulted in less degeneration than in wild-type animals [50]. Moreover, a strong upregulation of SR in reactive astrocytes in an AD rat model (TgF344-AD rats heterozygous for an APPsw/PS1ΔE9 transgene) and in human postmortem hippocampus and cortex samples from AD patients has been reported, supporting the notion that the increase in glial D-Ser may be associated with extrasynaptic NMDA receptor activation, thus playing an important role in excitotoxicity and neuronal damage (see Fig.4) [25]. Two studies conducted on postmortem AD brains, animal and cellular models of AD, and blood and CSF from probable to severe AD patients reported a significant increase in D-Ser levels [24, 73] and increased mRNA and protein levels of SR in homogenates from hippocampus or primary neuronal cultures treated with Aβ [24].
Similarly, DAAO has been related to AD progression, too. DAAO levels have been found to rise in the peripheral blood of patients with AD, with an increasing trend for severe AD [72]. Over the years, several studies focused on identifying and using DAAO inhibitors in different diseases [138], e.g., the inhibitor sodium benzoate was used for pain relief [139] and to treat early psychosis [140]. In randomized, double-blind, placebo-controlled trials, sodium benzoate (250–750mg/day for 24 weeks) improved the AD assessment scale ADAS-cog score, increasing neurocognitive function in patients with mild AD who did not show behavioral and psychological symptoms of dementia (DPSD) [88], while it did not show any efficacy in AD patients with DPSD treated with 622mg benzoate/day for 6 weeks only [141].
A large percentage of AD patients show psychotic symptoms, such as delusions and hallucinations: linkage and association studies supported the hypothesis that psychiatric symptoms in AD could be due to genetic factors [142–144]. Interestingly, the G72 gene, encoding a small protein (pLG72) that acts as a negative effector of DAAO [145–148], was previously linked to schizophrenia, bipolar disorder, and psychotic illness [145, 148–150]. With the aim to clarify the role of the G72 gene in the occurrence of psychosis in AD, 185 individuals with AD and depression in the early phase of disease were genotyped. This work identified an intronic mutation of the G72 gene (rs2153674), which is associated with frequent and severe delusions [151]. Moreover, some years later, another intronic genetic mutation (rs778296) was associated with the early onset of AD, while the rs3391191 mutation encoding for the pLG72 R30K variant (associated to schizophrenia onset) [145] was found to influence the age of onset of the disease in PSE1 E280A AD patients [152, 153]. To confirm the hypothesis that pLG72 contributes to the pathogenesis of AD, the peripheral blood of patients with dementia (in whom severity of cognitive deficit was evaluated based on the CDR scale) was analyzed: here, significantly higher pLG72 levels were observed in patients with MCI and mild, moderate, and severe AD than in patients with HS and the highest levels in patients with mild disease [154]. This work reported statistically significant differences in D-Ser levels and D-/(D+L)-Ser ratios among the five groups (values that increased with the dementia rating) and an association between pLG72 and L-Ser levels. These results support the view that pLG72 may contribute to AD onset by affecting DAAO activity [154].
A recent investigation highlighted a correlation between glucose metabolism, serine metabolism, and AD [155]. It is known that lower glucose metabolism is associated with early-stage AD and that mouse models of AD show reduced uptake and metabolism in the brain (probably depending on impaired vascular delivery) [156, 157]: a lack of ATP production affects ionic and neurotransmitter gradients. The astrocytic glucose metabolism is important for L-Ser production from the glycolytic intermediate 3-phosphoglycerate by phosphoglycerate dehydrogenase (PHGDH) belonging to the “phosphorylated pathway” (see Figs.3 and 4) [158]. Notably, a lower level of both L- and D-Ser was reported in early-stage AD mice, resulting in a lower level of occupancy at the NMDA receptor coagonist site in hippocampal slices and impaired LTP: such a deficit was rescued by administering exogenous D-serine [155]. The dietary supplementation of D-Ser rescued impaired spatial memory at the whole-animal level, even for PHGDH knockout mice. Indeed, L-Ser administration rescued the same deficits in the 3xTg-AD mice, which in any case did not show any alteration in PHGDH activity and serine metabolism. The lowered PHGDH level in late-stage AD (reactive) astrocytes may represent a further element affecting serine biosynthesis. This work proposed that oral L-Ser supplementation would be an effective therapy for AD by enhancing NMDA receptor activity [155]. However, such an activation might be potentially dangerous since NMDA receptor-dependent excitotoxic mechanisms have been proposed to contribute to synapse loss [159]. However, D-Ser, a coagonist for NMDA receptors, is frequently present at subsaturating levels, whose effects rely on the presence of the agonist glutamate: it has been proposed that increasing D-Ser levels “could enhance signal to noise within AD circuits by improving the efficiency of phasic/synaptic NMDA receptor signaling, which mediates synaptic plasticity as well as neuroprotective signalling” [160].
CONCLUSIONS
The tripartite glutamatergic synapses play an important role in the pathophysiology of AD [91]. Among the major factors affecting NMDA receptor-mediated neurotransmission in AD, availability of the agonist glutamate and modulation of the receptor’s functions are extremely important [40, 161]. NMDA receptor function is modulated by both D-Ser and D-Asp and the noncompetitive antagonist memantine has been approved for treatment of moderate to advanced AD [91]. Here, the distinct preference of the coagonist for synaptic and extrasynaptic NMDA receptors makes it possible to identify separate neuronal outcomes, inducing cell survival or cell death. The possibility to selectively inhibit NMDA receptor-mediated excitotoxicity alone may delay the progression of synaptic disruption observed in AD. However, it must be considered that, although memantine treatment has shown encouraging results in preclinical trials and in AD models, clinical studies did not report satisfactory effects. The benefits of memantine on cognition, global functioning, and “activities of daily living” were often absent. Nevertheless, this lack of efficacy could arise from late administration or from the need for combined use with other therapies. For a review, see [162]. The pioneering work of the Joseph Coyle laboratory about 20 years ago demonstrated that AD patients receiving D-cycloserine, a molecule exhibiting partial agonist activity at the glycine site of NMDA receptors, resulted in a significant improvement in cognitive parameter scores [109]. Most recently, aged humans with impaired memory capacities in the Groton maze computer test showed an improvement in performance after receiving a D-Ser-enriched drink [163]. A greater understanding of the role of D-AAs in excitotoxicity related to the pathogenesis of AD will facilitate novel therapeutic treatments for AD as well as for other acute and chronic diseases in which excitotoxicity is a core feature. Here, it is feasible to control D-Ser levels by acting directly on the two enzymes related to its metabolism, namely SR and DAAO. The previous PLP-containing enzyme is modulated by Mg2 + and ATP [12], while the activity of the flavooxidase is robustly stimulated by the FAD cofactor [116, 133–135]. Indeed, D-Ser level is also affected by the phosphorylated pathway that generates the precursor L-Ser from the glycolytic intermediate 3-phosphoglycerate [129].
As current therapeutics do not cure the disease and do not improve life expectancy when administered in late-stage AD, biomarkers to identify early-stage disease are needed, especially those related to NMDA receptors, which seem to be involved in synaptic dysfunction in early stages of AD. The potential use of both D-Ser and D-/(D+L)-Ser ratio in serum as biomarkers of AD was recently proposed by [64, 164] following the observation of a clearly increasing trend with progression of the disease (assessed by CDR score).
Three main limitations of the available investigations related to D-amino acids levels and AD onset are apparent. Firstly, various analytical methods have been used over the years, frequently not paying sufficient attention to appropriate controls, as stated in [57]. Secondly, investigations were carried out on a limited number of patients and with a short follow-up, as also stated by [65]. Finally, disease progression was evaluated according to different scales and frequently information about relevant characteristics of individual subjects was omitted, such as blood levels of Aβ42 and (phosphorylated) tau and medications used. These findings suggest that further investigations are needed. In addition to enrolling a larger cohort of patients (with a full characterization of peripheral biomarkers of AD and treatments used), an investigation in patients showing amnestic mild cognitive impairment (i.e., CDR=0.5) and evaluated by imaging analysis (i.e., PET) for the presence of a neurodegenerative process would seem necessary to validate the use of D-Ser level as a biomarker of AD onset.
Footnotes
ACKNOWLEDGMENTS
This work was supported by a grant from Ministero Universitá e Ricerca Scientifica PRIN 2017 (Grant 2017H4J3AS) to Loredano Pollegioni, and from Fondo di Ateneo per la Ricerca to Luciano Piubelli and Loredano Pollegioni. Valentina Rabattoni is a PhD student of the Life Sciences and Biotechnology course of University of Insubria.