
Abstract
Background:
The early cellular stresses leading to Alzheimer’s disease (AD) remain poorly understood because we cannot access living, asymptomatic human AD brains for detailed molecular analyses. Sortilin-related receptor 1 (SORL1) encodes a multi-domain receptor protein genetically associated with both rare, early-onset familial AD (EOfAD) and common, sporadic, late-onset AD (LOAD). SORL1 protein has been shown to act in the trafficking of the amyloid β A4 precursor protein (AβPP) that is proteolysed to form one of the pathological hallmarks of AD, amyloid-β (Aβ) peptide. However, other functions of SORL1 in AD are less well understood.
Objective:
To investigate the effects of heterozygosity for an EOfAD-like mutation in SORL1 on the brain transcriptome of young-adult mutation carriers using zebrafish as a model organism.
Methods:
We performed targeted mutagenesis to generate an EOfAD-like mutation in the zebrafish orthologue of SORL1 and performed RNA-sequencing on mRNA isolated from the young adult brains of siblings in a family of fish either wild type (non-mutant) or heterozygous for the EOfAD-like mutation.
Results:
We identified subtle differences in gene expression indicating changes in mitochondrial and ribosomal function in the mutant fish. These changes appear to be independent of changes in mitochondrial content or the expression of AβPP-related proteins in zebrafish.
Conclusion:
These findings provided evidence supporting that EOfAD mutations in SORL1 affect mitochondrial and ribosomal function and provide the basis for future investigation elucidating the nature of these effects.
INTRODUCTION
To reduce the prevalence and, therefore, the socioeconomic impacts of Alzheimer’s disease (AD), we must understand its molecular basis. Analysis of postmortem, human AD brains can give some insight into the disease mechanism. However, by the end stages of the disease, damage to the brain is considerable and little therapeutic intervention is possible. An understanding of the molecular changes that occur decades before symptoms arise is necessary for development of effective preventative or ameliorative treatments. However, we cannot access living, pre-symptomatic AD patient brain tissue for detailed molecular analysis. Therefore, analysis of animal models of AD is required.
A small number of genes are known to influence strongly the development of AD. Mutations in the presenilins (PSEN1 and PSEN2), and the gene encoding amyloid β A4 precursor protein (AβPP), are causative for the early-onset (<65 years of age), familial form of AD (EOfAD). EOfAD is an autosomal dominant disorder, and accounts for only a small proportion of all AD cases. The vast majority of AD cases arise sporadically and have an age of onset later than 65 years (LOAD). Variation in at least 20 genes is associated with increased risk of developing LOAD, with the ɛ4 allele of the apolipoprotein E gene (APOE) contributing the greatest risk [1, 2]. Since EOfAD and LOAD show similar disease progression and pathology (reviewed in [3]), analysis of EOfAD animal models may give insight into both subtypes of AD. Intriguingly, the gene sortilin-related receptor 1 (SORL1) appears to be associated with both EOfAD and LOAD [1 , 4–10] and may provide a mechanistic link between these two AD subtypes. However, SORL1 is the least studied of the EOfAD genes and its role in AD is unclear.
The majority of research investigating the role of SORL1 in AD is based around the role of its protein product in the intracellular trafficking of AβPP. It is well accepted that the binding of AβPP to SORL1 protein determines whether AβPP is directed through a recycling pathway, or is steered through the endolysosomal system to generate amyloid-β (Aβ, the primary component of the senile neuritic plaques found in AD brains) (reviewed in [11]). SORL1 also binds Aβ itself, guides it to the lysosome for degradation [12], and can act as a receptor for APOE [13]. These findings have been made mostly using cell lines in which SORL1 is overexpressed or removed. However, these manipulations do not closely reflect the pathophysiological state of SORL1 expression in AD.
We recently published an investigation of the effects on young adult zebrafish brain transcriptome state of heterozygosity for an EOfAD-like mutation and/or a putatively null mutation of the zebrafish orthologue of SORL1 [14]. Heterozygosity for the EOfAD-like mutation resulted in subtle effects on the young adult brain transcriptome, with only one gene detected as differentially expressed. Analysis at the level of genetic pathways (gene sets) found evidence for changes in cellular processes previously unknown to require sorl1, such as energy metabolism, protein translation and degradation. These effects were also observed in the brains of fish heterozygous for the putatively loss-of-function mutation in sorl1, suggesting they are due to haploinsufficiency. Transheterozygosity for the EOfAD-like and null mutations gave apparent effects on iron homeostasis and other cellular processes distinct from those detected in the heterozygous fish brains [14].
Here, we aimed to further our understanding of the effects of EOfAD mutations in SORL1 by generation and analysis of an additional zebrafish sorl1 mutation model. We performed targeted mutagenesis to generate a line of zebrafish carrying a W1818* mutation in sorl1, equivalent to the human SORL1 EOfAD mutation W1821* [4]. We then compared the brain transcriptomes of young adult sorl1 W1818* heterozygous and wild type sibling fish to identify, in an unbiased and objective manner, the changes in gene expression caused by this mutation. Consistent with our previous work, heterozygosity for the W1818* mutation results in subtle changes to brain gene expression. Genetic pathways involved in energy production and protein translation were altered. The W1818* mutation of sorl1 did not appear to affect Appa/Appb protein levels, cellular mitochondrial content, or iron homeostasis.
MATERIALS AND METHODS
Zebrafish husbandry and animal ethics
All zebrafish used in this study were maintained in a recirculating water system on a 14 h light/10 h dark cycle, fed dry food in the morning and live brine shrimp in the afternoon. All zebrafish work was conducted under the auspices of the Animal Ethics Committee (permit numbers S-2017–089 and S-2017–073) and the Institutional Biosafety Committee of the University of Adelaide.
Genome editing
To introduce mutations at the W1818 site in zebrafish sorl1, we used a TALEN pair designed and purchased from Zgenebio Biotech Inc. (Taipei City, Taiwan). The target genomic DNA sites (5’ to 3’) were ATGAGGTGGCGGTGTG (left) and GTAGGACTGTCTCCAT (right) (Supplementary Figure 1). The DNAs encoding the TALEN pairs were supplied in the pZGB2 vector, which were linearized with NotI (NEB, Ipswich, USA) before transcription of mRNA in vitro using the mMESSAGE mMACHINE T7 in vitro transcription kit following the manufacturer’s protocol (Invitrogen, Carlsbad, USA). The mRNAs encoding the TALEN pairs were diluted to a final concentration of 200 ng/μL each, and approximately 2–5 nL of the TALEN pair mRNA solution was injected into zebrafish embryos at the one cell stage. We eventually isolated a family of fish carrying an 11 nucleotide deletion in sorl1, W1818*, following the strategy described in [14]. Briefly, injected embryos were raised until approximately three months of age, then were outcrossed to Tübingen strain fish of approximately the same age to generate F1 families. Since the injected fish are mosaic for any mutations at or near the W1818 codon, we performed a T7 endonuclease I (T7EI) assay (NEB) on ten F1 embryos from a clutch to confirm whether any mutations had occurred in the germline of the injected parent. The PCR primers used to amplify the W1818 region for the T7EI assay have sequences (5’ –3’) F: TTAGGACCTCCTGTCAGCATTTCT and R: ACAAAATAAAAGTGTATGTGC. If the T7EI assay indicated that mutations were indeed in the germline, the remaining F1 embryos of the clutch were raised to adulthood. Sanger sequencing of the W1818 site was performed on genomic DNA extracted from fin biopsies of F1 adult fish to characterize any mutations (sequencing was performed by the Australian Genome Research Facility (AGRF, Adelaide, AUS)). Single heterozygous mutant fish (W1818*/+) from the F1 family were mated with single wild type (+/+) fish to generate F2 families. Heterozygous mutants from F2 families were in-crossed to produce F3 families containing homozygous mutant individuals.
Hypoxia treatment and allele specific digital quantitative PCRs
Adult zebrafish (6 months of age) were subjected to a hypoxic environment by placement in oxygen-depleted water for 3 h (oxygen concentration of 6±0.5 mg/L in normoxia and 1±0.5 mg/L in hypoxia). After treatment, fish were immediately sacrificed in a loose ice slurry and their brains removed. Total RNA was then extracted from whole zebrafish brains using the QIAGEN RNeasy® Mini Kit (Qiagen, Venlo, Netherlands) according to the manufacturer’s protocol. Recovered RNA concentrations were estimated using a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific Inc, Waltham, MA, USA). cDNA was synthesized using random hexamers in the Superscript III First Strand Synthesis System (Invitrogen) according to the manufacturer’s instructions. 50 ng of each resulting cDNA sample was then used in allele-specific digital quantitative PCR (dqPCR) as described in [15]. To detect the two transcripts of sorl1, we used a W1818* mutation-specific forward primer with sequence 5’ GTGGCGGTGTGATGATGG 3’, and a wild type-specific primer with sequence 5’ TGGCGGTGTGGGCTCAC 3’. A common reverse primer sequence spanning the junction between exons 41 and 42 of sorl1 (to reduce amplification from any genomic DNA carried over during RNA extraction) was 5’ GTAGAACACAGCGTACATCTCTGC 3’. Comparisons of mean transcript abundances per 50 ng of brain-derived cDNA were made using a two-way ANOVA with Tukey’s post hoc test for multiple comparisons.
Western blot analysis
Whole zebrafish brains were placed in 100μL of 1X Complete Protease Inhibitor Cocktail (Roche Holding AG, Basel, Switzerland) in RIPA buffer (Sigma-Aldrich Corp. St. Louis, MO, USA), immediately homogenized using a handheld motorized pestle for 1 min on ice, then incubated at 4°C for 2 h with gentle rocking. Cellular debris was sedimented by centrifugation with a relative centrifugal force of 16,100 g for 10 min. 25μL of 4X NuPAGE™ LDS Sample Buffer (Thermo Fisher Scientific) was added to the supernatant (75μL) containing total protein, then samples were heated at 80°C for 20 min. Total protein concentrations were determined using the EZQ® Protein Quantitation Kit (Molecular Probes, Inc. Eugene, OR, USA) following the manufacturers protocol. Samples were prepared for SDS-PAGE by adding 2.5μL of 10X NuPAGE™ Sample Reducing Agent (Invitrogen) to 75μg of total protein. The volume was increased to 25μL with 1X LDS buffer in RIPA buffer. Samples were heated at 80°C for 10 min and centrifuged briefly to sediment insoluble material. Supernatants containing the soluble proteins were subjected to SDS-PAGE on NuPAGE™ 4–12%Bis-Tris Protein Gels (Invitrogen) in the Mini Gel Tank and Blot Module Set (Thermo Fisher Scientific), using 1X NuPAGE™ MOPS SDS Running Buffer (Invitrogen). Resolved proteins were then transferred to PVDF membrane in 1X tris glycine SDS, 20%methanol. The PVDF membranes were blocked in 5%Western Blocking Reagent (Roche) then probed with primary and secondary antibodies. Antibody incubations were performed at either 1 hour at room temperature, or overnight at 4°C with gentle rocking. Antibody dilutions can be found in Table 1. Horse-radish peroxidase (HRP) signals were developed using Pierce™ ECL Western Blotting Substrate (Thermo Fisher Scientific) and imaged with a ChemiDoc™ MP Imaging System (Bio-Rad Laboratories, Hercules, CA, USA). The intensities of the signals were measured using the Volume tool in Image Lab 5.1 (Bio-Rad Laboratories). The relative intensities of the signals were normalized to β-tubulin and compared across genotypes by one-way ANOVA tests. Original blot images can be found in Supplementary Figure 7.
Antibodies used in this study
RNA-seq analysis
We used a total of 12 fish in the RNA-seq analysis (i.e., n = 6 per genotype, with mostly equal numbers of males and females), based on a previous power calculation indicating that n = 6 would provide approximately 70%power to detect the majority of expressed transcripts in a zebrafish brain transcriptome at a fold- change >2 and at a false discovery rate of 0.05 (data not shown). Whole heads were removed and preserved in approximately 600μL of RNAlater (Thermo Fisher Scientific). Total RNA was extracted from the brains using the mirVana miRNA isolation kit (Thermo Fisher Scientific) following the manufacturer’s protocol. Then, any genomic DNA carried over during the total RNA extraction was removed using the DNA-free™ Kit (Ambion, Waltham, USA). RNA was stabilized using RNAstable® (Biomatrica, San Diego, CA, USA) following the manufacturer’s protocol to minimize RNA degradation. Briefly, RNA was resuspended in DEPC-treated water, applied directly to a RNAstable® tube and dried with a vacuum concentrator. Dried RNA was sent to Novogene (Hong King, China) for cDNA library synthesis and RNA-sequencing. The cDNA libraries were generated using NEBNext Ultra RNA Library Prep Kit for Illumina (NEB) and RNA was sequenced using the Illumina Novaseq PE150 platform.
Demultiplexed paired-end fastq files (with adapter sequences removed) were supplied by Novogene. We first inspected the quality of the raw data by fastQC and ngsReports. We then performed a pseudo alignment using kallisto [17] version 0.43.1, in paired end mode specifying 50 bootstraps. The index file for kallisto was generated according to the zebrafish transcriptome (primary assembly of GRCz11, Ensembl release 96), with the sequences for all unspliced transcripts additionally included. We imported the transcript counts estimated by kallisto for analysis using R [18], using the catchKallisto function of the package edgeR [19]. To obtain gene level counts, we summed the counts of all transcripts arising from a single gene. Normalization factors were calculated using the trimmed mean of M-values (TMM) method [20]. Lowly expressed genes are statistically uninformative as they provide little evidence for differential expression, and we considered genes to be detectably expressed if they contained at least contained a logCPM of 0.75 in at least 6 of the 12 RNA-seq libraries. After selecting detectable genes, library sizes ranged from 13,050,237 to 50,560,749. Although these library sizes varied considerably, a correlation between library size and the first two principal components was not observed, supporting that variation due to library size does not contribute to the two largest sources of variation in this dataset (Supplementary Figure 6).
We performed an initial differential expression analysis using the exact test function of edgeR [19]. A design matrix was specified with an intercept for each brain sample, and sorl1 genotype (W1818*/+) as the common difference. Only one differentially expressed gene was identified in this analysis (data not shown). Therefore, to assist with identification of dysregulated genes due to sorl1 genotype, we removed one factor of unwanted variation using the RUVg method of the package RUVSeq [21]. For RUVg, we set k = 1, and the negative control genes as the least 5000 differentially expressed genes (i.e., largest p-value) in the initial differential expression test. The W_1 offset term generated was then included in the design matrix for an additional differential expression test using a generalized linear model and likelihood ratio tests with edgeR [19, 22]. We considered genes differentially expressed if their FDR adjusted p-value was less than 0.05.
Gene set enrichment testing was performed using three different algorithms: fry [23], camera [24], and GSEA [25, 26]. Since each method gave different levels of significance, we combined the raw p-values from each method by calculating the harmonic mean p-value [27]. We considered a gene set to be significantly altered if the FDR adjusted harmonic mean p-value was less than 0.05. The gene sets we tested in this study were the KEGG [28], HALLMARK [29], and the zebrafish iron-responsive element [30] gene sets. The KEGG and HALLMARK gene sets for zebrafish were obtained from MSigDB [29] using msigdbR [31]. To perform promoter motif enrichment analysis, we used homer [32] as described in [33] on the top 100 most statistically significant differentially expressed genes due to sorl1 genotype when including the W_1 covariate in the design matrix.
Data visualization was performed using the packages ggplot2 [34], pheatmap [35], and upsetR [36].
The raw fastq files and the output from catchKallisto (i.e., the raw transcript abundances) have been deposited in Gene Expression Omnibus database (GEO) under Accession Number GSE156167. All code to reproduce the RNA-seq analysis can be found at https://github.com/karissa-b/sorl1_w1818x_6m.
RESULTS
Generation and characterization of the EOfAD-like mutation W1818* in zebrafish sorl1
In 2012, Pottier et al. [4] identified mutations in SORL1 segregating with EOfAD in families with autosomal dominant inheritance patterns. We previously analyzed the effects of a coding sequence-truncating mutation from this study (human C1478*) on the young adult brain transcriptome in a zebrafish model (zebrafish V1482Afs) [14]. To further our understanding of the effects of coding sequence-truncating mutations in SORL1, we generated an additional zebrafish model of a mutation from the Pottier et al. study, W1821* [4]. The W1821 codon is conserved in zebrafish (W1818). We edited this site in the zebrafish genome using targeted mutagenesis (Supplementary Figure 1) and isolated a line of zebrafish carrying an 11 nucleotide deletion, resulting in a protein coding sequence-level change equivalent to that caused by the human mutation (Fig. 1A).

The W1818* mutation in sorl1 results in a transcript subject to nonsense mediated mRNA decay. A) Alignment of zebrafish sorl1 and human SORL1 wild type and mutant exon 41 sequences. B) Schematic of Sorl1 protein with protein domains and the site of the W1818* mutation indicated. VPS10, vacuolar protein sorting 10 domain; LDLR, low density lipoprotein receptor; EGF, epidermal growth factor; FN, fibronectin; TMD, transmembrane domain; ICD, intracellular domain. C) Number of copies of the sorl1 wild type (WT) and mutant (W1818*) transcripts in 6-month-old wild type (+/+) and heterozygous mutant (W1818*/+) sibling brains under normoxia and acute hypoxia. p-values were determined by two-way ANOVA with Tukey’s post hoc test for multiple comparisons.
Protein coding sequence-truncating mutations in SORL1 have been shown to be subject to nonsense mediated mRNA decay (NMD) in lymphoblasts from human carriers [4]. Additionally, SORL1 expression is upregulated under hypoxia in vitro [37]. Therefore, we asked whether the W1818* transcript of sorl1 is also subject to NMD, and whether its expression is altered in vivo by hypoxia treatment. We performed allele-specific, digital quantitative PCRs (dqPCRs) on brain-derived cDNA from fish exposed to normoxia or acute hypoxia at 6 months of age. We found that the W1818* transcript is less abundant in W1818*/+ brains relative to the wild type transcript under both normoxia (p < 0.0001) and acute hypoxia (p = 0.0001), supporting that the W1818* transcript is subject to NMD. We also observed that the wild type transcript in W1818*/+ brains is less abundant than in +/+ brains under normoxia (p = 0.004) and acute hypoxia (p = 0.02). We did not observe any significant change of sorl1 transcript levels between normoxia and acute hypoxia (Fig. 1C), despite that these fish were likely showing a transcriptional response to hypoxia, as indicated by upregulation of pdk1 transcript levels (Supplementary Figure 2).
To determine whether the W1818* mutation alters the expression of Sorl1 protein, we performed western immunoblot analysis on zebrafish brain lysates at 6 months of age, using an antibody raised against the C-terminus of human SORL1 (and that cross-reacts with zebrafish Sorl1 protein). A ∼250 kDa signal is observed in +/+ brains, and is absent in homozygous mutant brains, confirming that the W1818* mutation disrupts translation of Sorl1 protein (Supplementary Figure 3).
In summary, sorl1 does not appear to be upregulated by acute hypoxia in vivo, the W1818* results in a transcript which is likely subject to NMD, and the mutation disrupts wild type Sorl1 protein translation.
Transcriptome analysis of W1818*/+ and +/+ young adult zebrafish brains
Which genes are dysregulated as a result of heterozygosity for the EOfAD-like, W1818* mutation in sorl1 in young adult brains? To address this question, we performed mRNA sequencing on the brains from fish either heterozygous for the W1818* mutation and their wild type siblings at 6 months of age (n = 6).
The overall similarity between gene expression profiles can be explored using principal component analysis (PCA). Samples with similar gene expression profiles cluster together in a PCA plot. In our RNA-seq dataset, no clear separation of male and female brain samples was observed, supporting our previous observations that sex does not have a large effect on the zebrafish brain transcriptome [14, 38]. We observed partial separation of W1818*/+ and wild type samples across principal component 2 (PC2), accounting for approximately 16% of the total variation in this dataset (Fig. 2A). Therefore, heterozygosity for the W1818* mutation likely does not have widespread effects on the transcriptome of young adult zebrafish brains.
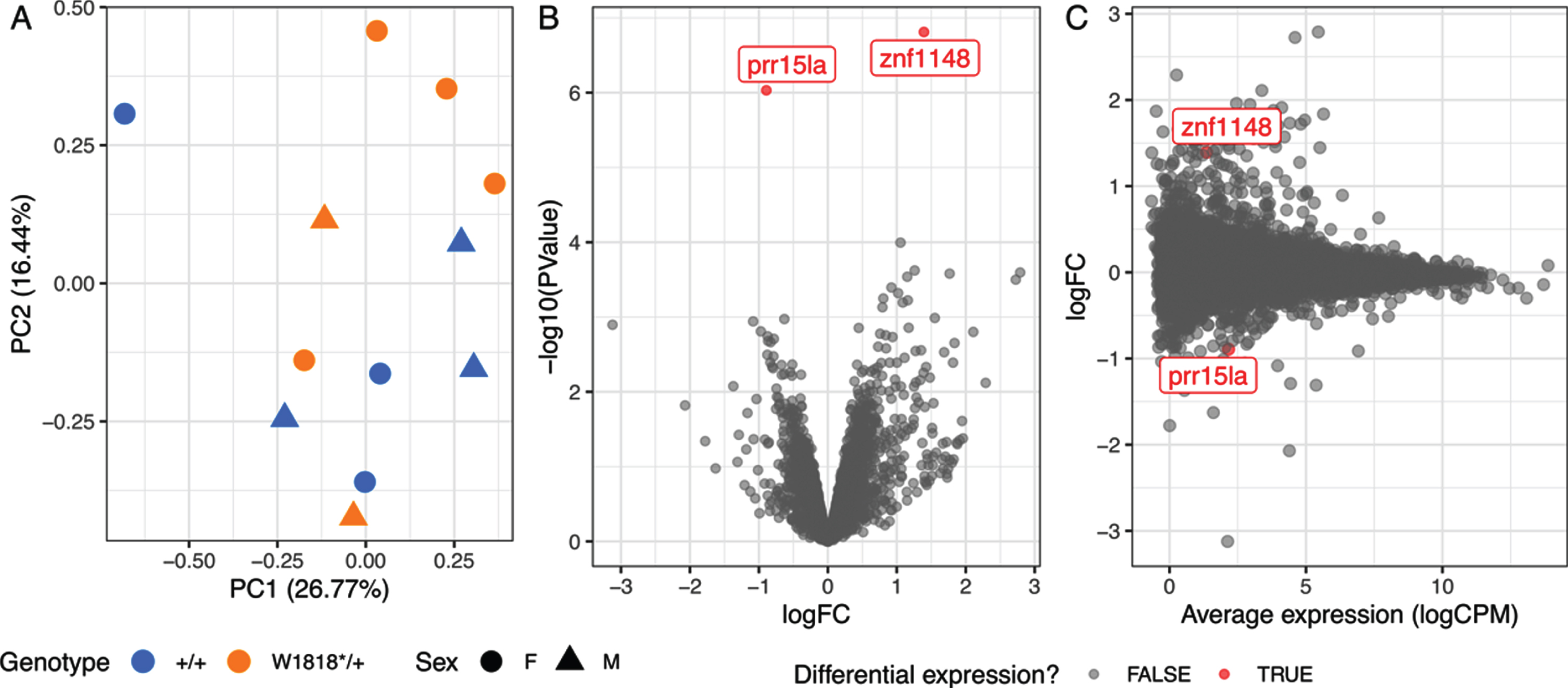
Heterozygosity for the W1818* mutation in sorl1 has subtle effects on young-adult brain transcriptomes. A) Plot of principal component 1 (PC1) against PC2 from a principal component analysis. Circles indicate female samples (F) and triangles indicate male (M) samples. B) Volcano plot showing the significance (-log10(P-Value)) and log2 fold change (logFC) of genes in W1818*/+ brains relative to +/+ sibling brains. The genes identified as significantly differentially expressed are colored red. C) Mean-difference (MD) plot of gene expression in W1818*/+ brains relative to +/+ sibling brains.
Differential gene expression analysis supported our conclusions from the PCA. Only two genes were identified as differentially expressed to a statistically significant degree due to the W1818*/+ genotype: zinc finger protein 1148 (znf1148) and proline rich 15 like a (prr15la) (Fig. 2B, C and Supplementary Table 1). The functions of these genes are not known. BLAST searches identified ZNF99 and PRR15L as candidate human orthologues of znf1148 and prr15la respectively. However, broad-scale analysis of conservation of synteny between the genomic regions occupied by the zebrafish and human genes was unable to support these orthologies (Supplementary Figure 4).
To obtain a more complete view of changes to brain gene expression due to heterozygosity for the W1818* mutation in sorl1 we performed enrichment testing on particular gene sets (below) using the entire list of detectable genes in the RNA-seq experiment. For insight into the cellular processes represented by the gene expression changes we used the KEGG and HALLMARK gene sets from the Molecular Signatures Database (MSigDB, [26, 29]). To test for possible iron dyshomeostasis, we used our recently defined gene sets representing the genes containing iron-responsive elements (IRE) in the untranslated regions (UTRs) of their mRNAs [30]. We applied the self-contained gene set testing methods fry [23] and GSEA [25, 26], and the competitive gene set testing method camera [24], and combined the resulting p-values by calculating the harmonic mean p-value, a method of combining dependent p-values [27]. We further protected against type I errors by performing an FDR adjustment on the resulting harmonic mean p-value.
In all, we identified six gene sets significantly altered as a group after FDR adjustment of the harmonic mean p-value (Fig. 3). However, the subsets of “leading edge” genes identified by the GSEA algorithm (which can be thought of as those genes driving the enrichment of the gene set) for four of these six gene sets (HALLMARK_ and KEGG_OXIDATIVE_PHOSPHORYLATION, KEGG_HUNTINGTONS_DISEASE, and KEGG_PARKINSONS_DISEASE) all share many genes, showing that the statistical significance of these gene sets is, essentially, being driven by the same signal (genes encoding components of the mitochondrial electron transport chain). No IRE gene sets were found to be significantly altered, consistent with our previous observations for heterozygosity for the V1482Afs EOfAD-like allele of sorl1 [14].
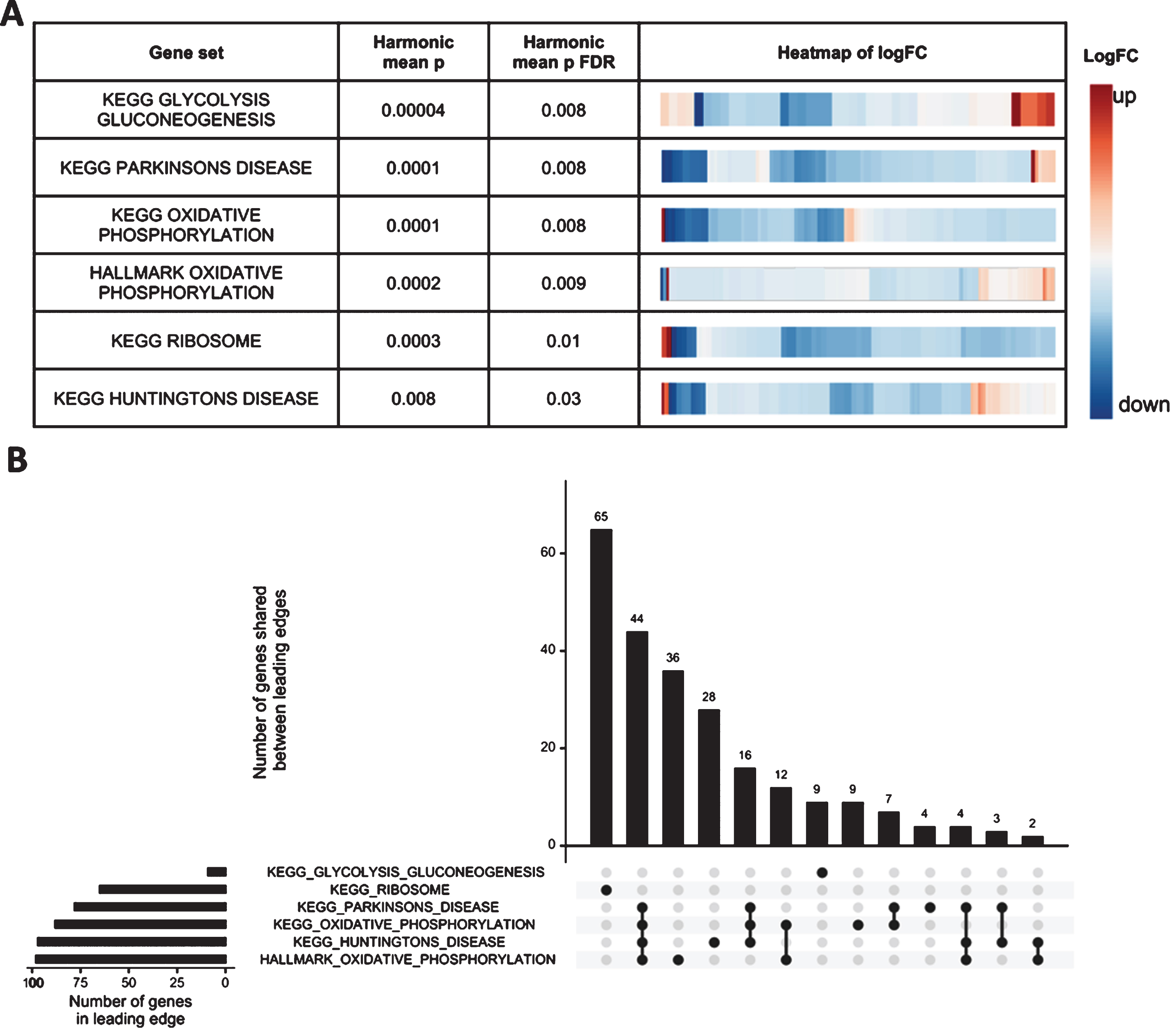
Mitochondrial and ribosomal function are predicted to be affected by heterozygosity for the W1818* mutation in sorl1. A) Table indicating the significantly altered KEGG and HALLMARK gene sets (FDR adjusted harmonic mean p-value < 0.05) in W1818*/+ brains relative to +/+ brains. Heatmaps indicate the log2 fold change (logFC) of all detectable genes within the gene sets, clustered by their Euclidean distance. B) Upset plot of the leading edge genes from the GSEA algorithm for the significant gene sets indicating the number of shared genes driving the enrichment of each gene set. See the online version for color.
Since our transcriptome analysis was of bulk mRNA isolated from entire zebrafish brains, differences in cell type proportions between W1818*/+ and +/+ brains could cause artefactual apparent changes in gene expression levels. To investigate this possibility, we visualized the expression levels of marker genes present in four broad cell types within zebrafish brains: neurons, astrocytes, oligodendrocytes [39] and microglia [40] (Fig. 4A). Since we did not observe any obvious differences in marker gene expression levels between genotypes, it is unlikely that changes in cell type proportions cause the differential expression of genes observed in the W1818* heterozygous fish brains.
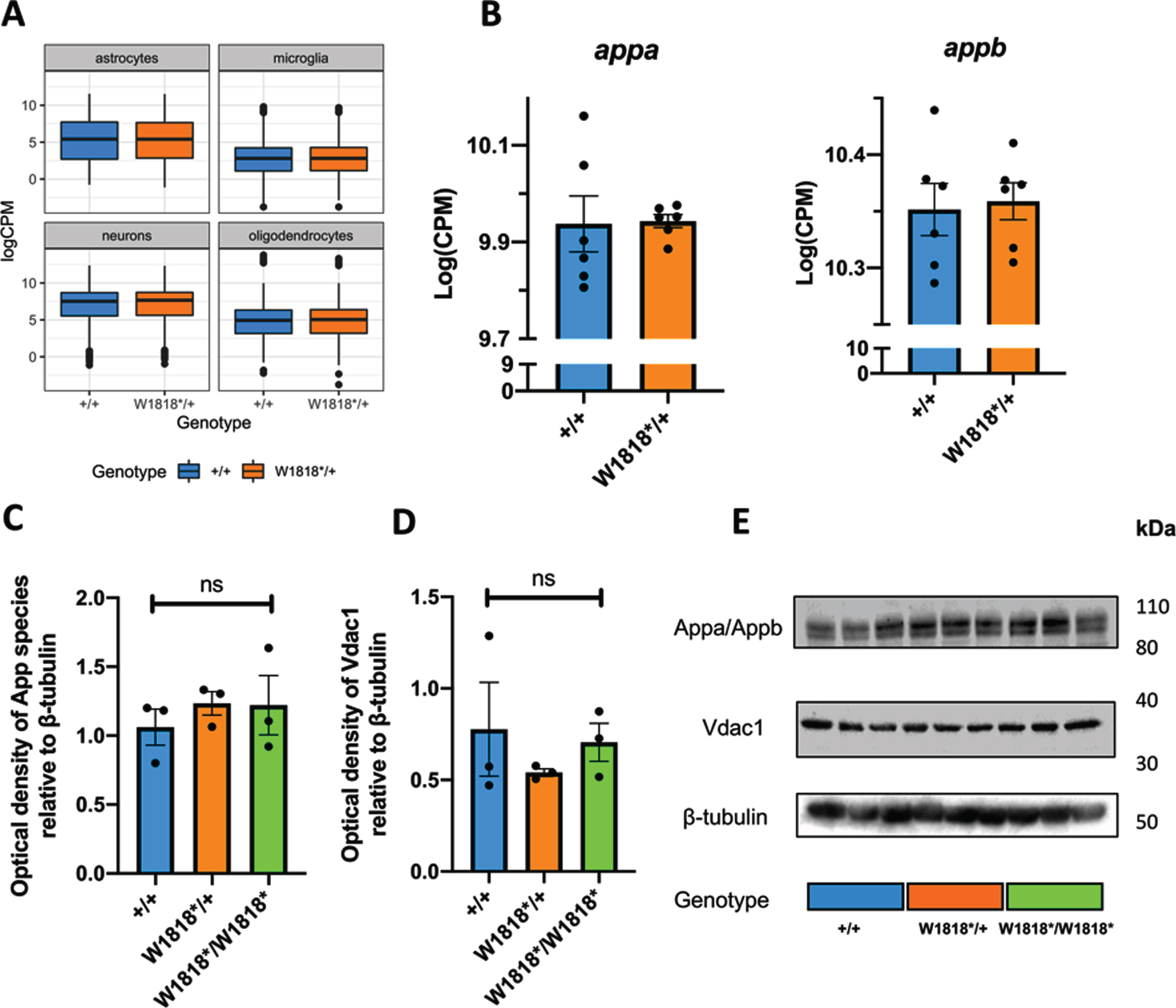
Brain transcriptome changes in W1818*/+ mutants are unlikely to be due to changes in cell type proportions, altered expression of appa/b, or mitochondrial content. A) Distribution of expression (logCPM) of marker genes of astrocytes, microglia, neurons and oligodendrocytes in wild type (+/+) and heterozygous mutant (W1818*/+) brains. B) Expression (logCPM) of AβPP orthologues appa and appb in wild type and heterozygous mutant brains. C) Quantification of western blot analysis of expression of App proteins (Appa and Appb) in 6-month-old wild type, heterozygous and homozygous mutant zebrafish brains. D) Quantification of western immunoblot analysis of expression of the mitochondrial marker Vdac1 in 6-month-old wild type, heterozygous and homozygous mutant zebrafish brains. E) Representative images of western immunoblots. Significance levels in C) and D) were determined by one-way ANOVA (ns, not significant).
Differences in transcription factor activity could also be driving the changes to gene expression observed in W1818*/+ brains. To explore this, we performed promoter motif enrichment analysis using homer [32] on the 100 most statistically significantly differentially expressed genes due to sorl1 genotype. We identified that the promoter motif for hepatocyte nuclear factor 4α (Hnf4α) appears significantly over-represented in the promoters of the top 100 most DE genes (Bonferroni adjusted p-value = 0.04). Expression of hnf4α itself was low in these zebrafish brains and was insufficient to be regarded as detectable in this RNA-seq experiment (<0.75 cpm in at least 6 of the 12 RNA-seq libraries). However, examination of the logCPM values for hnf4α before filtering found that one mutant brain displayed far higher expression of hnf4α than the others and is clearly an outlier. A PCA on the expression of all genes predicted to contain a Hnf4α-binding motif (HNF4ALPHA_Q6 gene set from the C3 category of MSigDB) revealed no obvious separation between W1818*/+ and +/+ samples, suggesting that sorl1 genotype does not result in distinct expression patterns for these genes (Supplementary Figure 5).
In summary, heterozygosity for the W1818* mutation appears to have subtle effects on the expression of genes involved in energy production and protein translation in young adult zebrafish brains. The subtle effects of this EOfAD-like mutation are consistent with the typically lesser pathogenicity of such mutations in SORL1 compared to, for example, EOfAD mutations in PSEN1 [6, 41] and may reflect the earliest cellular changes that, after decades, lead to overt Alzheimer’s disease.
Heterozygosity or homozygosity for W1818* does not appear to affect the abundance of App species, or mitochondrial mass
The most characterized cellular role of SORL1 protein is the sorting of AβPP throughout the endolysosomal system (reviewed in [11]). Therefore, we sought to investigate whether the W1818* mutation of sorl1 causes changes in expression of the zebrafish forms of AβPP. In zebrafish, two paralogous genes show orthology with human AβPP: appa and appb. Inspection of our RNA-seq data did not reveal any differences in the expression levels of appa or appb between heterozygous mutant and wild type brains (Fig. 4B). We also used an antibody against human AβPP that cross-reacts with both zebrafish Appa and Appb proteins for immunoblot examination of App species in the brains of wild type, heterozygous and homozygous mutant zebrafish at 6 months of age (Fig. 4C). We did not observe any significant differences between genotypes, supporting that the changes to sorl1 gene function/expression are likely independent of changes to expression of appa or appb.
Finally, we hypothesized that, in W1818*/+ brains, changes to expression of genes involved in oxidative phosphorylation could cause, or be due to, changes in cellular mitochondrial content. To explore this, we performed western immunoblotting against Vdac1, a protein highly abundant in the outer mitochondrial membrane, in lysates from entire zebrafish brains. We did not observe any significant effect of sorl1 genotype on the levels of Vdac1, supporting that cellular mitochondrial content is unchanged between genotypes (Fig. 4D).
DISCUSSION
We cannot access tissue from the living brains of SORL1 mutation carriers for detailed molecular analysis. This restricts our ability to elucidate the early changes which eventually lead to AD and requires that we examine animal models instead. Our particular approach involves generating genetic models of EOfAD mutations in zebrafish which closely mimic the genetic state of human EOfAD (i.e., here we analyzed the effects of a single, heterozygous mutation in the endogenous sorl1 gene of zebrafish). This approach avoids potentially confounding assumptions such as that homozygous animals simply show more extreme phenotypes than heterozygotes, or that transgenic animals overexpressing mutant genes cause effects similar to endogenous human mutations (assumptions commonly made in AD genetics research). We also performed our analyses when zebrafish are 6 months old and recently sexually mature. We regard this as the equivalent of early adulthood in humans. At this age changes to gene expression should reflect pathological processes (or the responses to these) occurring long before any cognitive deficits become apparent. Finally, analysis of large families of sibling fish raised in an identical environment (the same tank) reduces both genetic and environmental variation between samples (i.e., noise) and increases our ability to detect subtle changes in gene expression due to heterozygosity for the EOfAD-like mutations.
In this study, we built on our previous analysis [14] investigating the effects of protein coding sequence-truncating mutations in sorl1 implicated in EOfAD. We generated a zebrafish model of the W1821* mutation in human SORL1 (zebrafish sorl1 W1818*). We showed that transcripts of the W1818* allele are likely subject to NMD (consistent with observations of coding sequence-truncating mutations in human SORL1 [4]), and that W1818* transcripts cannot generate full-length Sorl1 protein.
We did not observe any significant differences between the expression of sorl1 transcripts in zebrafish brains under normoxia and hypoxia in vivo. This contrasts with the observations of Nishii et al. [37], using a hematopoietic stem cell line. These researchers saw increased SORL1 protein levels under hypoxia. However, a recent meta-analysis of RNA-seq datasets investigating transcriptional responses to hypoxia in both humans (128 datasets) and mice (52 datasets) showed that SORL1 was seldom found to be differentially expressed under hypoxia. When SORL1 was identified as DE, either up- or downregulation was observed [42]. Therefore, whether SORL1 is regulated by hypoxia, and in which cell types this may occur, requires further investigation.
Heterozygosity for EOfAD-like mutations in sorl1 causes subtle changes in young adult brains
Transcriptome analysis of young adult, W1818*/+ mutant zebrafish brains relative to their +/+ siblings identified statistical evidence for two DE genes: znf1148 and prr15la. Little or no information on the function of these genes can be found in the scientific literature. However, the candidate orthologue of prr15la in humans, PRR15L, was seen as associated with human intelligence in a meta-analysis of genome-wide association studies totaling 78,308 individuals [43]. That only a small number of genes was identified as DE is to be expected from the apparently semi-penetrant effects of EOfAD mutations in SORL1 (reviewed in [11, 44] and see below) and the young age of the brains examined (long before any overt pathology would be expected. It is also consistent with our previous analysis of another EOfAD-like mutation in sorl1 (zebrafish sorl1 V1482Afs modelling human SORL1 C1478* ), for which heterozygosity gave rise to only one significantly DE (downregulated) gene: cytochrome c oxidase subunit 7A1 (cox7a1) [14]. This gene appeared as slightly, but non-significantly, upregulated in W1818*/+ brains (logFC = 0.5, pFDR = 1) (Supplementary Table 1) so the differences in the genes identified as DE between the W1818* and V1482Afs heterozygous mutant brains is likely an effect of statistical noise. In contrast, analysis of an EOfAD-like mutation in the zebrafish orthologue of PSEN1, the gene most commonly mutated in EOfAD [45], detected 251 DE genes in the brains of young adult heterozygous zebrafish [46]. Thus, it appears that EOfAD mutations in SORL1 cause less severe effects on cellular state compared to EOfAD mutations in PSEN1. This is more consistent with SORL1’s action as a LOAD genetic risk locus than as an EOfAD-causative locus. Campion and colleagues [44], noted that the ages of onset of AD in patients with mutations in SORL1 are proximal to the arbitrary LOAD age threshold of 65 years of age (between 56 and 80 years, compared with PSEN1 mutation carriers, which have an overall median age of disease onset of approximately 42 years [47]). Also, some SORL1 mutation carriers with early onset AD carry additional LOAD genetic risk variants in APOE, TREM2, and ABCA7, and/or have aged (>66 years), unaffected siblings who carry the same SORL1 mutation. Together, these observations, along with our in vivo transcriptomic studies, support that SORL1 is more likely a genetic risk locus for LOAD than an EOfAD locus.
A promotor enrichment analysis of the 100 genes showing the strongest signal (smallest p-values) for differential expression due to sorl1 genotype identified the DNA-binding motif for Hnf4α as significantly enriched. HNF4α, a gene associated with maturity-onset diabetes of the young (MODY, a form of non-insulin-dependent diabetes mellitus [48]), is primarily expressed in the liver (reviewed in [49]) and has been reported to play roles in gluconeogenesis [50] and cholesterol homeostasis [51]. Yamanishi and colleagues [52] reported that HNF4α is also expressed in the brains of mice where it appears to drive transcriptome changes in mice housed under stressful conditions. Expression of the hnf4α gene in the brains of our zebrafish was very low so that its transcripts were excluded during the pre-processing preceding our differential gene expression analysis. However, inspection of the logCPM values from before exclusion of genes with low expression identified one W1818*/+ male mutant sample with a relatively higher expression of hnf4α (likely an outlier). Nevertheless, a PCA on the expression of all detectable genes predicted to contain the Hnf4α DNA-binding motif (250 genes) did not reveal any convincing evidence of global dysregulation of Hnf4α-regulated genes in W1818*/+ brains.
Changes to mitochondrial and ribosomal function are observed in young-adult, sorl1 mutant zebrafish, and in human AD
Enrichment testing of all the detectable genes in the RNA-seq experiment identified subtle changes to expression of genes involved in energy production and protein translation. Reassuringly, we previously identified these processes as altered due to heterozygosity for the EOfAD-like V1482Afs mutation of sorl1, and for a putatively null mutation in sorl1 in young adult zebrafish brains [14], suggesting that changes to mitochondrial and ribosomal function are an effect-in-common of EOfAD mutations in SORL1, and arise through a haploinsufficiency mechanism (i.e., due to decreased sorl1 function). Whether these mitochondrial and ribosomal functions are directly reliant on Sorl1 protein expression, or change as part of a homeostatic response to a deficiency of another, unknown, Sorl1-dependent function, is unclear. To our knowledge, the role of SORL1 in the context of mitochondrial and ribosomal function has not been investigated previously. However, it is well accepted that these processes are affected early in AD. Studies exploiting positron emission tomography with 2-[18F] fluoro-2-deoxy-d-glucose (FDG-PET) (measuring glucose metabolism in the brains of living subjects) have shown that glucose metabolism gradually declines during the conversion from mild cognitive impairment (MCI) to AD, particularly in the parieto-temporal and posterior cingulate cortices [53 –57]. In our zebrafish model, genes encoding the components of the mitochondrial electron transport chain are mostly downregulated. Interestingly, the overall direction of change for genes involved in glycolysis and gluconeogenesis is up, supporting that a metabolic shift occurs from oxidative phosphorylation towards glycolysis as the primary source of energy for the brain.
In W1818*/+ brains, we observed downregulation of genes encoding ribosomal proteins. Ribosomes isolated from postmortem MCI and AD patients show less capacity for protein synthesis [58 –60], increased levels of oxidized ribosomal RNA (rRNA), and bound ferrous iron (Fe2+) relative to control ribosomes [61]. Oxidative stress within cells (such as due to increased redox active metals like Fe2+, or free radicals generated during normal mitochondrial respiration) oxidises rRNA and decreases ribosomal activity [62]. Further investigation is needed to determine whether protein synthesis is actually affected in W1818*/+ brains.
We recently developed an enrichment analysis-based method to detect signs of iron dyshomeostasis in RNA-seq data. The method exploits gene sets encompassing genes containing iron-responsive elements (IREs) in the 5’ or 3’ UTRs of their mRNAs [30]. We did not observe any evidence for changes to expression of these gene sets, suggesting that intracellular iron (Fe2+) levels are not significantly altered in W1818*/+ brains relative to their +/+ siblings. This is consistent with our previous analysis, where only complete loss of wild type sorl1 resulted in significant changes to expression of genes which IREs in the 3’ UTRs of their mRNAs [14]. Nevertheless, we cannot exclude that iron homeostasis is affected in the heterozygous mutant brains. Due to the nature of bulk RNA-seq, we may have failed to observe opposite directions of change in gene expression in different cell types.
No evidence that mutation of sorl1 significantly changes expression of genes affecting the endo-lysosomal system in young adult zebrafish brains
We did not observe any gene sets involved in endolysosomal system function to be significantly altered in W1818*/+ brains, despite that SORL1 protein is thought to act, primarily, within the endolysosomal system (reviewed in [11]). Knupp and colleagues [63] showed that complete loss of SORL1 in neurons (and not microglia) derived from human induced pluripotent stem cells (hiPSCs) resulted in early endosome enlargement, a phenomenon previously observed in post mortem EOfAD and LOAD brains [64 –66]. Our previous four-genotype analysis also did not reveal any direct evidence for dysregulation of genes in the endolysosomal system [14] although iron homeostasis appeared disturbed in brains lacking any wild type sorl1 expression, and that might be due to a disturbance of cellular iron importation in which the endolysosomal system plays an important role (e.g., [67]). The results of our two independent zebrafish studies do not support that heterozygosity for mutations in sorl1 affects the endolysosomal system at the level of gene regulation. Nevertheless, changes may be occurring in our mutant fish at the protein level without affecting the transcriptome. Also, as mentioned previously, humans heterozygous for SORL1 mutations generally show ages of AD onset (if affected) close to the arbitrary late onset threshold of 65 years of age [4, 44]. Therefore, it is possible that any endolysosomal defects in heterozygous W1818* zebrafish brains are not be observable until later ages.
In conclusion, we have provided evidence supporting that mutation of sorl1 affects mitochondrial and ribosomal function. Our bioinformatic analysis provides the basis for future experiments to elucidate the nature of these effects.
Footnotes
ACKNOWLEDGMENTS
The authors would like to acknowledge Dr. Seyyed Hani Moussavi Nik for obtaining part of the funding for this work, and his assistance in the genome editing to generate the W1818* mutant line of zebrafish, Ms. Nhi Hin for providing the zebrafish iron-responsive element (IRE) gene sets and her valuable advice and discussions on the bioinformatic analysis, and Dr. Zijing Zhou for his assistance in performing the western blots. This work was supported with supercomputing resources provided by the Phoenix HPC service at the University of Adelaide.
This work was funded partially by an Alzheimer’s Australia Dementia Research Foundation Project Grant titled “Identifying early molecular changes underlying familial Alzheimer’s disease” awarded on 1 March 2017 from Alzheimer’s Australia Dementia Research Foundation (now named Dementia Australia). KB is supported by an Australian Government Research Training Program Scholarship. ML and MN were both supported by grants GNT1061006 and GNT1126422 from the National Health and Medical Research Council of Australia (NHMRC). ML and SP are employees of the University of Adelaide.