
Abstract
The worldwide prevalence of sporadic (late-onset) Alzheimer’s disease (sAD) is dramatically increasing. Aging and genetics are important risk factors, but systemic and environmental factors contribute to this risk in a still poorly understood way. Within the frame of BioMed21, the Adverse Outcome Pathway (AOP) concept for toxicology was recommended as a tool for enhancing human disease research and accelerating translation of data into human applications. Its potential to capture biological knowledge and to increase mechanistic understanding about human diseases has been substantiated since. In pursuit of the tau-cascade hypothesis, a tau-driven AOP blueprint toward the adverse outcome of memory loss is proposed. Sequences of key events and plausible key event relationships, triggered by the bidirectional relationship between brain cholesterol and glucose dysmetabolism, and contributing to memory loss are captured. To portray how environmental factors may contribute to sAD progression, information on chemicals and drugs, that experimentally or epidemiologically associate with the risk of AD and mechanistically link to sAD progression, are mapped on this AOP. The evidence suggests that chemicals may accelerate disease progression by plugging into sAD relevant processes. The proposed AOP is a simplified framework of key events and plausible key event relationships representing one specific aspect of sAD pathology, and an attempt to portray chemical interference. Other sAD-related AOPs (e.g., Aβ-driven AOP) and a better understanding of the impact of aging and genetic polymorphism are needed to further expand our mechanistic understanding of early AD pathology and the potential impact of environmental and systemic risk factors.
Keywords
BACKGROUND
Dementia is currently the 5th leading cause of death worldwide [1]. While < 5%of the Alzheimer’s disease (AD) cases are hereditary, the vast majority results from a complex interplay between aging, genetics, systemic and environmental factors, and lifestyle [2, 3]. While clinical symptoms are similar, clinical hereditary AD develops early in life, while clinical sporadic (late-onset) AD (sAD) becomes more prevalent at 65 + years of age. However, subjective indications of memory loss may already appear long before the diagnosis of sAD [4, 5] and correlate with changes in hippocampus and entorhinal cortex, both responsible for information processing and memory formation [6].
Being a high impact pathology, AD has triggered intense research efforts to understand disease pathology and to facilitate drug development. Yet, this research did not translate in any significant impact on the quality of life of patients other than alleviation of symptoms. Important reasons for a 99%failure rate may include disease complexity, reliance on animal models representing hereditary AD pathology, and focus on the symptomatic late phase of the disease [7–9].
In sAD, the mechanistic networks responding to systemic and environmental factors, and lifestyle, and deregulating amyloid-β protein precursor (AβPP) and tau processing are still poorly understood [10]. While Aβ has long been considered as the main villain in the widely pursued Aβ cascade hypothesis, its cytotoxic potential [11] and its capacity to induce tau accumulation [12] has been challenged by data showing that tau, rather than Aβ drives disease [13–15]. For this alternative hypothesis, tau must be capable of impairing Aβ and tau metabolism [16] directly or by affecting downstream processes (e.g., Ca2 + homeostasis [17], oxidative stress [18], neuroinflammation [19], cell cycle regulation [20], apoptosis [21], autophagy [22], synaptic function [23], glucose [24], and lipid metabolism [25]) on a background of continuous deterioration driven by accumulation of aging-related oxidative stress-induced epigenetic alterations [26–32].
Since tau oligomers and aberrant phosphorylation of tau protein are linked to tau pathology, processes known to induce, or to be induced by, tau variants may hide clues regarding initiation and progression of sAD [33]. Evidence suggests that Aβ oligomers may contribute to these tau-mediated processes by selectively impairing fast transport of mitochondria and by causing mitochondrial fragmentation. Both effects are abolished by inhibition of glycogen synthase kinase 3β (GSK3β), known to be involved in tau phosphorylation, while they appear to be independent from each other [34].
Gene clustering and ontology have revealed susceptibility genes primarily involved in cholesterol and lipid metabolism, neuroinflammation, and endosomal vesicle cycling, but genes contributing to cytoskeleton function, axonal transport, cell migration, and synaptic function emerged as well [35]. Age-related cholesterol and glucose dysmetabolism, both preceding the onset of memory deficit and contributing to disease progression, have been proposed as main triggers for sAD initiation, dysregulation of age-sensitive downstream processes, and production of pathogenic Aβ and tau [36].
The variability in longevity between individuals in different communities suggests that not all risk for acquiring sAD is accounted for by aging and genetic factors. Various microbial, nutritional, and environmental factors may cause transcriptional dysregulation of processes involved in sAD progression [37]. How aging, genetics, and external risk factors co-trigger pathology remains unclear.
During the BioMed21 workshop series [38, 39], federal agencies, funding agencies, regulators, and academics explored existing approaches and potential improvements in development, access, storage, and evaluation of human-relevant information with the goal of enhancing human disease research. One recommendation is the adaptation of the Adverse Outcome Pathway (AOP) concept for mapping perturbations of normal human physiology during disease development. The AOP concept, currently applied in toxicology [40], has since been being applied to capture complex biological knowledge about non-communicable and communicable human diseases [41] (https://ec.europa.eu/jrc/en/event/webinar/intro-webinar-ciao-project). It is believed that this approach can enable new target discovery as exemplified by the AOP for developmental vascular toxicity (AOP43) providing mechanistic insight into unique modes of actions of two angiogenesis inhibitors with therapeutic potential [42].
In compliance with the OECD guidance on the development of AOPs, the evaluation of weight of evidence (WoE) approach was applied [40]. Existing lines of evidence were evaluated against hypotheses based on qualitative and/or quantitative methods to confirm the strength of the causal relationships between key events (KEs) and the quality of supportive data. A reduced modified set of Bradford Hill criteria, including biological plausibility, essentiality, and empirical support, have been proposed by AOP-Wiki User’s Guide for weighing the available evidence [41, 43]. With respect to biological plausibility of each key event relationship (KER), existing mechanistic links between upstream and downstream events based on biological knowledge are evaluated for mechanistic relevance. Essentiality implies the causality of the KEs in the AOP. Empirical support involves experimental data which strengthen the relationships between the KEs. In line with this BioMed21 recommendation and guided by a human-based integrated framework for AD research, KEs and KERs driving tau-mediated pathology resulting in memory loss were captured in an AOP, and potential plug-ins for environmental risk factors were mapped on this AOP to portray the potential of these risk factors on sAD progression [8]. Plotting neurotoxicity data on a neurodegeneration AOP is not free of challenges. Genetic polymorphism and various chemical exposure regimes may trigger multiple pathways making it difficult to identify molecular initiating events (MIEs) that are causally responsible for triggering downstream KEs resulting in an adverse outcome (AO). Human neurological disorders may display diverse pathologies with similar clinical phenotypes; however, the understanding of molecular and cellular mechanisms behind adult neurodegenerative disorders is incomplete [37].
Since the complexity of AD initiation and progression can only be captured by a network of AOPs each covering adverse pathways that contribute to dementia, the proposed tau-driven AOP should be considered a first proof-of-concept for the application of the AOP to acquire better understanding of the age-related dysfunctional biological processes of sAD and the potential impact of environmental factors on these processes. It may also be a first step toward a case study demonstrating applications and benefits of predictive, mechanism-based approaches in the context of translation and human disease biology, a major recommendation emerging from the BioMed21 workshop series [39].
LITERATURE SEARCH METHODOLOGY WITH FOCUS ON HUMAN INFORMATION
The applied information emerged from systematic reviews and pivotal primary research manuscripts. If required, additional literature searches were performed to identify secondary research studies. The search strategy included strings of keywords linked by the three basic Boolean operators (AND, OR, NOT) covering cellular and molecular mechanisms driving processes involved in AD and neurotoxicity: tau and Aβ processing [16], calcium homeostasis [15, 17], oxidative stress [44], neuroinflammation [19], cell cycle regulation [20], apoptosis [21], autophagy [22], synaptic function [23], glucose metabolism [24], and cholesterol metabolism [25]. The biological tools and readouts overlap with the human-based integrated framework for AD research proposed by Pistollato et al. [8]. Bibliographic databases such as PubMed, MEDLINE, EMBAS,E and the Alzheimer’s Science Hub were visited. Search filters were designed to prioritize systematic reviews and manuscripts including pathway analysis published later than 2000. All searches were limited to English language studies only. Human in vivo, in vitro, and ex vivo information was prioritized, while non-human data were considered only to fill out information gaps. Information gaps were defined as molecular and cellular events, or relationships between events, for which only in vivo, in vitro, or ex vivo animal data were found related to established KEs for AD. Such data were applied only when there was a plausible relevance for human pathology, i.e., to the extent that the knowledge provided by the animal data could fit into the general understanding of the targeted human pathological processes. Irrespective of the species providing the data, only validated, or plausible information, defined as information confirmed by subsequent studies from the same group or similar studies by other groups, was retained.
DEFINING A STARTING POINT AND MINIMIZING COMPLEXITY
To address the complex interplay among the various processes in a conceivable way several assumptions were introduced before developing the AOP blueprint: Aging is broadly accepted as the main driving force, while MIEs with subsequent focused molecular interactions triggering downstream pathological processes have not been specified. Here, the bidirectional relationship between brain glucose and cholesterol dysmetabolism was adopted as hypothetical starting point because both increase with age, affect production and clearance of tau and Aβ [45–47], as well as downstream KEs preceding memory deficit and disease progression [36]. Arguments against assumption (1) include the complexity of both phenomena and evidence substantiating a negative impact of aging not only on glucose and cholesterol metabolism, but also on mitochondrial function and autophagy, among others [26–32]. Their intimate interconnection makes it difficult to discriminate between cause and consequence. For the sake of simplicity, changes in adenosine triphosphatase (ATP)-dependent processes were considered as consequences of a growing brain glucose and cholesterol dysmetabolism mediated ATP deficit [36]. However, any aggravating impact of downstream effects on both metabolisms were considered when deemed important. The hypothesis that tau toxicity precedes Aβ toxicity was adopted, resulting in an AOP structuring KEs and KERs leading to memory loss (not dementia) driven by tau, hyperphosphorylated tau (p-tau), and toxic tau variants only. Because of insufficient understanding of the complex interplay among the molecular and cellular mechanisms, and the genes and genetic polymorphisms associated with the risk of acquiring sAD, the available gene clustering results were used only to identify and confirm eligibility of affected processes for inclusion in the AOP blueprint [35]. Ca2 + dyshomeostasis is common in neurodegenerative disorders. Since the role of endoplasmic reticulum stress induced Ca2 + release into the cytoplasm is still debated it was not included [48, 49].
BUILDING A TAU-DRIVEN AOP BLUEPRINT (FIG. 1)
The bidirectional relationship between glucose and cholesterol dysmetabolism as hypothetical starting point
The selection of this bidirectional relationship rather than the individual processes is motivated by the tight interplay between glucose and cholesterol (dys)metabolism. This interplay is exemplified by 27-hydroxycholesterol (27-HC), a biomarker for reduced glucose metabolism but also linked to hypercholesterolemia [36], and apolipoprotein E allele 4 (ApoE4) causing deregulation of cholesterol metabolism as well as insulin resistance when overexpressed [50–53]. The next two sections review how glucose and cholesterol (dys)metabolism each may contribute to sAD progression. In the context of the proposed AOP, the evidence supports the assumption that tau-driven memory loss requires both processes to be dysregulated. Furthermore, there is no conclusive evidence showing that alterations are triggered by, e.g., glucose dysmetabolism alone without involvement of cholesterol dysmetabolism.
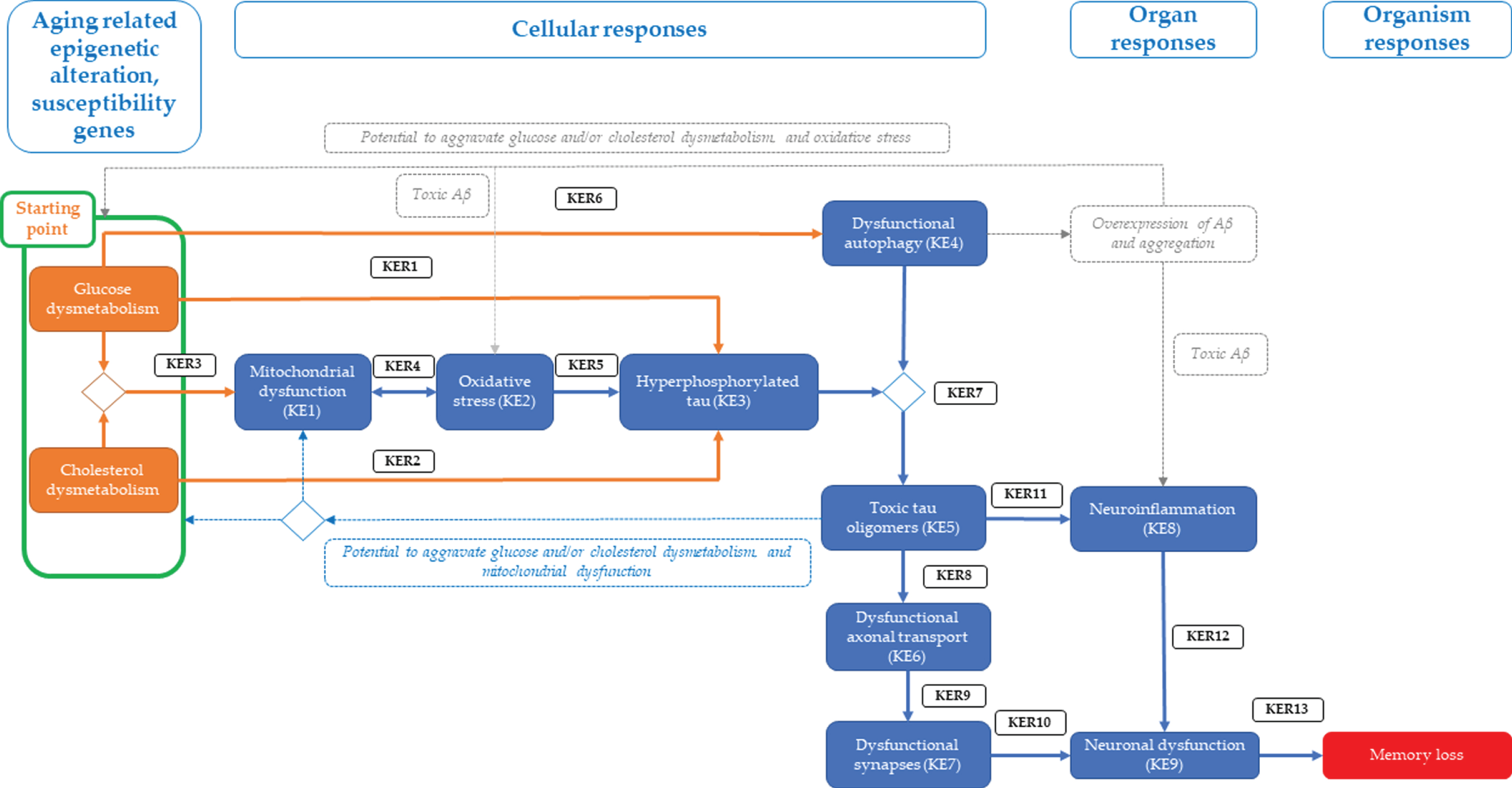
Proposed tau-driven AOP blueprint towards memory loss (AO). Glucose and/or cholesterol dysmetabolism (as hypothetical starting point) linked to hyperphosphorylation of Tau (KE3) via key event relationship 1 (KER1) and/or KER2, respectively. The starting point is also associated with mitochondrial dysfunction (KE1) via KER2, leading to oxidative stress (KE2) via KER4 and subsequently to KE3 via KER5. Glucose dysmetabolism can be also directly linked to dysfunctional autophagy (KE4) via KER6. KE4 can cause accumulation of cytosolic toxic tau oligomers (KE5) via KER7, which in turn impedes the axonal transport (KE6) via KER8, resulting in synaptic dysfunction (KE7) via KER9. KE5 is associated with neuroinflammation (KE8) via KER11. Both KE5 and KE7 can cause neuronal dysfunction (KE9) via KER12 and KER10, respectively. Lastly, KE9 leads to memory loss, the adverse outcome (AO) of the proposed AOP. Alternatively, KE4 can also cause Aβ overexpression and aggregation, leading to toxic Aβ which can either linked to starting point, KE2 or KE8 (shown with grey dotted lines). Also, KE5 can link to starting point or KE1 (shown with blue dotted lines). Diamond shape indicates convergence of different KEs or a KE leading to more than one effect.
The role of glucose dysmetabolism in the onset of tauopathy
A functional brain requires proper regulation of glucose metabolism and insulin signaling to support neurotransmission and ATP production [54], synaptic formation, plasticity, and cognition [55, 56].
The biological plausibility
The molecular mechanisms involved in healthy and pathologic insulin signaling and their contribution to cognition have been extensively covered elsewhere [57–59] and were summarized in Fig. 2. Pathology results from deregulation of the PI3K-AKT and MAPK/ERK pathways, activation of GSK3β and deregulation of cholinergic stimulation, axonal transport, microtubule dynamics, apoptosis, and inflammation [60, 61]. Concomitant deregulation of FoxO and mammalian target of rapamycin (mTORC1) further pushes pathology by impairing neuronal polarity, gene transcription, autophagy, synaptic plasticity, and membrane density of glucose transporters (GLUT) [62].

Overview of the impaired brain insulin signaling pathway known to contribute to tauopathy and cognitive impairment. The changes affected by insulin resistance are indicated by the red arrows. (INSR, insulin receptor; IRS, insulin receptor substrate proteins; Grb2, growth factor receptor-bound protein 2; MAPK/ERK, mitogen-activated protein kinase/extracellular signal-regulated kinase; PI3K, Phosphoinositide 3-kinase; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; AKT, protein kinase B; GSK3β, glycogen synthase kinase 3β; GLUT, glucose transporters; O-GlcNAcylation, O-linked β-N-acetyl glycosylation; p-Tau, phosphorylated tau protein; PP2A, phosphoprotein 2A; Acetyl-CoA, acetyl coenzyme A; ATP, adenosine triphosphatase; NFTs, neurofibrillary tangles).
The empirical support
Increased glucose levels and decreased cortical insulin levels are broadly accepted risk factors for neurodegenerative diseases [47, 63] and relate to defective brain insulin signaling [57, 64–66]. These risk factors may be detectable long before the first signs of AD [27, 67] and deteriorate with age [45–47]. Poor insulin signaling reduces hippocampal GLUT-4 and GLUT-8 membrane density [55, 56] and decreases insulin and insulin growth factor (IGF) levels in frontal cortex, hippocampus, and hypothalamus [58]. The insulin resistance marker, insulin receptor substrate protein 1 (IRS1), and pro-insulin processing enzymes such as pro-protein convertase subtilisin/kexin 1 and 2 (PCSK1 and PCSK2) are expressed at lower levels in AD [68, 69]. Downregulation of the receptor of hepatocyte growth factor in AD brain may reflect downregulation of vascular endothelial growth factor (VEGF), which is inversely correlated to insulin, and responsiveness to insulin [70, 71].
The role of cholesterol dysmetabolism in the onset of tauopathy
Cholesterol, another cornerstone for a healthy brain, is synthesized de novo by brain cells [72]. Impaired homeostasis unambiguously links to initiation and progression of AD [73, 74], irrespective of whether there is too little or too much [36, 75].
The biological plausibility
The molecular mechanisms involved in healthy and pathologic cholesterol metabolism have been outlined elsewhere [76, 77] and were summarized in Fig. 3. The balance between 24S-hydroxycholestrol (24S-HC) and 27-hydroxycholestrol (27-HC) governs the health status of the brain [78] by modulating cholesterol synthesis and transport from glial to neuronal cells through nuclear transcription factor mediated processes resulting in ATP-binding cassette transporters expression (i.e., ABCA1, ABCG1, and ABCG4) and ApoE [79, 80]. 24S-HC provides neuroprotection by activating sirtuin 1, a histone deacetylase protein regulating antioxidant expression, and by inhibiting the generation of Aβ through stimulation of α-secretase and inhibition of β-secretase [81]. In contrast, 27-HC antagonizes these beneficial effects by triggering the production of reactive oxygen species (ROS) and decreasing antioxidants levels [82]. ApoE plays a fundamental role in transport of cholesterol and other lipids to the neurons [83]. ApoE4, a strong genetic risk factor for sAD [84, 85], is less efficient in delivering cholesterol to neurons resulting in reduced neurite outgrowth and neuronal signaling, disrupted cytoskeleton and impaired axonal transport [86–88]. Eventually, AβPP processing is affected resulting in Aβ generation and aggregation [89].
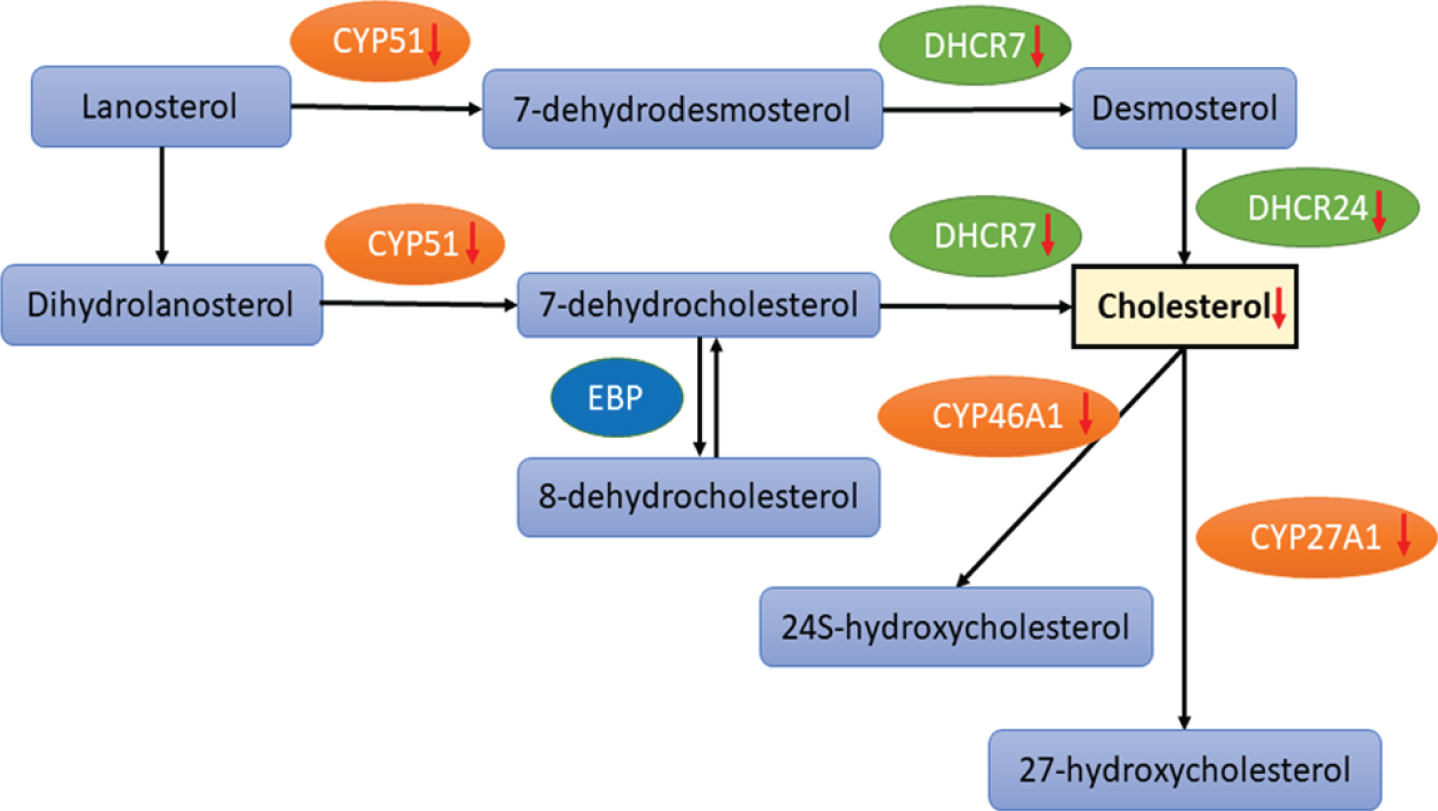
Impaired cholesterol synthesis by inhibition of the key enzymes involved in cholesterol biosynthesis pathway. Red arrows indicate inhibition of enzymatic functions. Modified from [76]. (CYP51, sterol 14α-demethylase cytochrome P450; DHCR7, 7-dehydrocholesterol reductase; CHDR24, 24-dehydrocholesterol reductase; CYP46A1, cholesterol 24S-hydroxylase; cholesterol 24-monooxygenase; CYP27A1, sterol 27-hydroxylase).
Empirical support
Brain cholesterol synthesis decreases with age and exhibits changes in hippocampus, cortex, temporal gyrus, and cerebrospinal fluid (CSF) of patients with AD [90–93]. Elevated serum cholesterol levels are detectable long before clinical manifestations of the disease [94]. In patients with advanced AD, 24S-HC levels are decreased while 27-HC levels are increased, with desmosterol, cholesterol, and 24S-HC levels in CSF inversely correlating p-tau levels [95, 96]. Increased 24S-HC plasma levels occur in early stages of AD, suggesting implication of ongoing demyelination [97], while loss of neuronal cholesterol 24-monooxygenase (CYP46A1) decreases 24S-HC levels in advanced AD [98]. ApoE4 affects memory in an age-dependent manner, with young carriers of ApoE4 performing better than non-carriers, but with poor cognition in elderly carriers [83, 99].
KEs AND KERs TOWARD INCREASED LEVELS OF HYPERPHOSPHORYLATED TAU (P-TAU) (KE3)
Whether glucose and cholesterol dysmetabolism is the consequence of aging or of a series of upstream events caused by systemic factors, it appears to be the ultimate event affecting downstream pathways implicated in tau metabolism either by direct impact on tau phosphorylation, or indirectly via mitochondrial dysfunction (KE1) and ROS production (KE2).
KER1: A direct path from glucose dysmetabolism to p-tau (KE3)
Glucose dysmetabolism may impact tau phosphorylation directly, or through insulin mediated activation of transcriptional factors (e.g., sterol regulatory element-binding proteins (SREBPs)) involved in cholesterol biosynthesis [53].
The biological plausibility
Glucose dysmetabolism and impaired insulin signaling negatively affect PI3K and AKT activity, and trigger GSK3β activation and tau hyperphosphorylation (Fig. 2).
The empirical support
In AD lower GLUT-1 and GLUT-3 levels are associated with downregulation of O-linked β-N-acetyl glycosylation (O-GlcNAcylation) and hyperphosphorylation of tau [100], while impaired GLUT-4 function is related to distorted brain glucose metabolism [55]. Brain insulin resistance and the degree of pathology are inversely related, with an 80%reduction in IRS levels in advanced AD stages, along with decreased IGF1 levels and elevated levels of tau protein [101]. In mice, brain-specific knockout of insulin receptor (INSR) or IRS triggers GSK3β activity, impairs brain growth, and promotes tau phosphorylation [102, 103]. In vitro studies indicate that short insulin treatment of rat primary cortical neurons and SH-SY5Y human neuroblastoma cells increases tau hyperphosphorylation, while a longer exposure results in decreased tau phosphorylation [104, 105].
Overall assessment
Based on the evidence it is plausible that glucose dysmetabolism affects tau hyperphosphorylation. While the current quantitative understanding is a weakening glucose metabolism (e.g., due to increasing insulin resistance) drives p-tau levels, the threshold, magnitude, or duration of the change needed to elicit a detectable effect on KE3 remains to be established. The available studies do not allow a confident decision about the adjacency of both events. Whether glucose dysmetabolism directly affects KE3, or whether the impact is to some extent the consequence of an energy deficit mediated via mitochondrial dysfunction (KE1) and/or oxidative stress (KE2) remains to be clarified.
KER2: A direct path from cholesterol dysmetabolism to p-tau (KE3)
While there is evidence that ApoE4 acts via its cholesterol transporter function, a possible direct or indirect effect via brain glucose dysmetabolism may exist [50–52].
The biological plausibility
During aging the balance between 24S-HC and 27-HC is dysregulated by autoxidation of cholesterol and accumulation of a variety of oxysterols [106–108]. The resulting stress activates glial sterol 27-hydroxylase (CYP27A1) leading to increased 27-HC production in the brain, an increased influx of peripheral 27-HC, aggravation of the oxidative stress [81, 107] and tau pathology [25, 109]. Dysregulation of ApoE4 transport may be implicated by suppressing tau protein phosphatases (e.g., phosphoprotein 2A (PP2A) [110].
The empirical support
Higher levels of ApoE4, tau, and p-tau are observed in CSF from mild cognitive impairment (MCI) patients as compared to controls [111]. Additional studies report decreased levels of PP2A and increased tau phosphorylation in AD brain [112, 113]. Overexpression of human ApoE4 in neurons of a transgenic mouse model induces tau hyperphosphorylation and causes more brain atrophy and neurodegeneration than other alleles of ApoE [87, 114–116]. Overexpression of C-terminal ApoE4 causes hyperphosphorylation of tau and neuronal loss in vitro [87, 117].
Overall assessment
The data provide strong evidence supporting ApoE4 overexpression during cholesterol (and potentially glucose) dysmetabolism as an important cause of elevated p-tau levels. The quantitative understanding is that overexpression of ApoE4 is driving this KE, without any specification on threshold, magnitude, or duration. Both events may be adjacent, but the observed oxidative stress (KE2) implicates a pathway involving KE1 and KE2. These uncertainties remain to be clarified.
KER3 - KER5: linking mitochondrial dysfunction (KE1), oxidative stress (KE2) and p-tau (KE3)
While replicating mammalian cells rely essentially on a glycolytic metabolism, post-mitotic neurons exhibit an absolute energetic dependence on mitochondrial oxidative phosphorylation, while their polarized shape entails compartmentalized and distinct energetic needs. The fundamental pathological pathways driven by mitochondrial dysfunction and oxidative stress, and their implication in neurodegenerative diseases, have been extensively reviewed [118, 119].
KER3: Glucose and/or cholesterol dysmetabolism leads to mitochondrial dysfunction (KE1)
Several studies have shown a strong dependency of these organelles on proper glucose and cholesterol metabolism [56, 121].
The biological plausibility
Cytoplasmic glucose is converted to acetyl-coenzyme A (CoA) and used by mitochondria for ATP production, while intracellular uptake of free fatty acids and their conversion to fatty acyl-CoA and acetyl-CoA is enhanced. Distortion of either of these pathways deregulates mitochondrial function and triggers excessive production of free ROS [120]. Dysfunctional mitochondria are characterized by a weakened mitochondrial membrane permeability transition due to elevated intracellular Ca2 + levels, decreased protein expression, altered redox status of mitochondrial proteins and a dysfunctional electron transport chain (ETC) [122, 123]. ATP deficit and excessive ROS levels trigger cytochrome c and apoptosis inducing factors translocation from the matrix into intermembrane space, activation of the caspase cascade, and apoptosis [124, 125].
The empirical support
It has been suggested that disturbed insulin signaling can cause brain mitochondrial dysfunction in response to glucose metabolism impairment in human AD brain via upregulation of AEBP1 and downregulation of PCSK1 and PCSK2 leading to decrease in insulin production in neurons and downregulation of NEUROD6, resulting in mitochondrial dysfunction [56]. Furthermore, decline in cerebral glucose metabolism is associated with reduced expression of mitochondrial ETC genes and enzyme activity in neurons [118], while mitochondrial ROS production, depolarization, and swelling are reported in brain with insulin resistance (and AD pathology) [126]. There is evident relation between stimulation of brain insulin signaling via PI3K/AKT/GSK-3β and improved brain mitochondrial biogenesis and cognitive function, in rat hippocampus [127]. In type 2 diabetic mice, hypothalamic insulin resistance was associated with decreased expression of mitochondrial proteins, leading to mitochondrial dysfunction [58]. In addition, the impact of cholesterol accumulation in mitochondrial membrane physical properties affecting the transport of glutathione into mitochondria mediated by upregulation StARD1 protein in mice, has been suggested [128].
Overall assessment
The evidence identifies mitochondrial dysfunction (KE1) as a plausible consequence of brain glucose and cholesterol dysmetabolism. Both events are considered adjacent. There is a clear quantitative relationship between increasing glucose and cholesterol dysmetabolism and increasing mitochondrial dysfunction and energy deficit, and the mechanisms involved are well understood. However, threshold, magnitude, and duration that are required to elicit a detectable change in KE1 are also here unknown.
KER4: Mitochondrial dysfunction (KE1) leads to oxidative stress (KE2)
While moderate levels of ROS constitute a defense against external stressors [129], excessive ROS production triggered by mitochondrial dysfunction (KE1) or depletion of glutathione depots causes aggravation of the mitochondrial health status [118].
The biological plausibility
Pathological ROS levels result in oxidation and loss of function of proteins, lipids, and nucleic acids. The resulting oxidative stress directly, or via mitochondrial dysfunction, results in less mitochondrial biomass, intracellular ATP and respiratory complexes, elevated concentration of intracellular Ca2 + and activated Ca2 +-dependent calpains, cellular dysfunction, and eventually cell death [69, 130–132].
The empirical support
Oxidative imbalance is predominant in AD pathogenesis [133, 134]. Accumulated 8-oxoguanine (8-oxoG), oxidized guanine, a marker for nuclear and mitochondrial DNA damage, has been found in postmortem AD hippocampus [135, 136]. Enhanced lipid peroxidation, oxidized protein, and nucleic acids, and a significant decrease in antioxidant enzyme activity are reported in AD [118, 137]. Proteomic analysis shows a 2-fold increase in mitochondrial protein nitration and oxidation in subjects with MCI when compared to healthy subjects, but not in AD subjects, suggesting that mitochondrial dysfunction occurs during early development of disease [138]. Several studies report that neuronal stress increases cytosolic Ca2 +-activated calpain expression leading to neuropathological diseases, such as AD [139–141].
Overall assessment
Data suggest that glutathione depletion and mitochondrial dysfunction are plausible causes for excessive ROS levels, but lack quantitative data about threshold, magnitude, and duration. KER4 already exists in the AOP-Wiki (ID:189), describing the link between the upstream event ‘disruption of mitochondrial ETC’ and the downstream event ‘increase of ROS production’. Both events are considered adjacent, despite the evidence for this relation is classified as moderate due to some inconsistencies.
KER5: Oxidative stress (KE2) leads to p-tau (KE3)
Physiological tau phosphorylation regulates neuronal polarity and axonal growth by supporting transport of mitochondria and lysosomes, proteins, and lipids from soma to axons, dendrites, and synapses [142, 143].
The biological plausibility
The evidence suggest that ROS induced oxidative stress promotes pathological tau modifications and disruption of the mechanisms involved in proper mitochondrial functionality [133, 144–146]. Hyperphosphorylation of tau disrupts its affinity for microtubules, increases its resistance to degradation, and induces conformational changes promoting aggregation [49]. Hence, a proper regulation between tau protein phosphorylation and dephosphorylation of the many phosphorylation sites of tau is detrimental for a healthy neuronal cell physiology [147–149]. Under excessive oxidative stress, Ca2 +-dependent calpains activate cyclin-dependent kinase 5 (Cdk5) and GSK3β, both involved in tau hyperphosphorylation and shown to be important for proper neural development, synaptic signaling, learning, and memory [149, 150]. Cdk5 and GSK3β interact with the truncated regulatory unit of p35 (p25), also a product of calpain activation [151]. Cdk5/p25 and GSK3β/p25 mediated tau phosphorylation decreases the affinity of tau for microtubules, disrupts the cytoskeleton, and causes apoptosis [152, 153]. The GSK3β/p25 complex inhibits PP2A phosphorylation through increased inhibitory Tyr307 phosphorylation and decreases expression of PP2A [154]. The data suggest a GSK3β-Cdk5-PP2A synergy in tauopathy, which is characterized by decreased affinity of tau for microtubules, abnormal hyperphosphorylation, aggregation, and eventually synaptic dysfunction [155–158].
The empirical support
Mitochondrial dysfunction (KE1) and/or oxidative stress (KE2) activate the calpain signaling pathway, a process that precedes p-tau formation during the early stages of AD development [159]. Available evidence suggests that oxidative stress through tau hyperphosphorylation contributes to tau pathology and AD [160]. Calpain mediated activation of the tau kinases Cdk5 and GSK3β correlates with the degree of pathology (Braak stage II-III) and precedes tau phosphorylation and synaptic loss [159]. Proteomic analysis suggests that loss of function of neuronal peptidyl prolyl cis-trans isomerase 1 (Pin1) by oxidative damage, and its downregulation in AD hippocampus, are linked to tau phosphorylation and AD neurofibrillary pathology [161]. The observation that human truncated tau protein expression leads to accumulation of ROS and cortical neuron death in rats, suggests that tau modification may also precede oxidative stress [162].
Overall assessment
Data support that excessive ROS levels are a plausible cause for tau pathology, and that both events are adjacent. Even though it technologically is possible to measure ROS levels in vitro, it was not possible to find threshold and magnitude values, nor duration of exposure, that are required to result in a persistent adverse impact on p-tau levels.
KEs AND KERs TOWARD MEMORY LOSS (ADVERSE OUTCOME (AO))
Abnormal tau phosphorylation prompts conversion of normal tau into several cytosolic tau variants prone to polymerize, to aggregate (neurofibrillary tangles, NFTs) or exert neurotoxicity [163]. Evidence suggests that formation of intermediate pre-fibrillar cytosolic tau oligomers precedes NFT formation, and accounts for the pathogenic effects in AD development [164–170].
KER6: Glucose dysmetabolism leads to dysfunctional autophagy (KE4)
Autophagy is a physiological ‘housekeeping’ process involving lysosome-dependent intracellular degradation of long lived, aggregated and misfolded proteins, and clearing damaged organelles (e.g., mitochondria). Integration of autophagy into these processes is regulated by transcriptional factors and requires proper insulin mediated signaling [171, 172]. The removal of protein aggregates and damaged organelles, through the ubiquitin-proteasome system (UPS) and/or the autophagy/lysosome machinery, is crucial and defects in either is deleterious at cellular and organism level [173, 174].
The biological plausibility
Impaired autophagy correlates with accumulation of lipofuscin in lysosomes and with reduced ability of lysosomes to fuse with autophagosomes [175, 176]. Lower numbers of autophagic vacuoles and disturbed lysosomal degradation are also associated with age-related glucose dysmetabolism [177, 178] and subsequent decline in autophagic markers such as Beclin 1 [179], Atg5, ATG7 [180], LC3, HDAC6 [181], PINK1 [182], and Bcl-2 [183]. Impaired autophagy results in inefficient degradation of intraneuronal protein aggregates, including p-tau and Aβ, and accumulation of toxic tau variants and toxic aggregates of AβPP cleavage products [184]. Tau-mediated axonal transport deficiency affects AβPP delivery into axons and dendrites, causing AβPP to accumulate in the neuronal cell body [185], where the resulting Aβ oligomers trigger neuronal and synaptic dysfunction, and eventually memory loss [186].
The empirical support
Impaired autophagy and reduced mTOR activity are strongly associated with defective insulin signaling and AD pathology [187]. In the inferior parietal lobule and medial temporal cortex neurons in human AD brain reduced mTOR activity correlates with suppressed autophagy and increased tau and p-tau levels [172, 188–191]. In early AD, autophagy is triggered by mitochondrial dysfunction (KE1) and oxidative stress (KE2) to remove damaged organelles and protein aggregates. In contrast, later AD stages exhibit dysfunctional autophagosomes and increased numbers of autophagic vacuoles [22], indicative of dysfunctional autophagy [22, 192].
Overall assessment
The data provide evidence in favor of a direct link between dysfunctional autophagy and age-related glucose dysmetabolism. While the available studies suggest that these events are adjacent, they do not exclude that this dysfunctionality is mediated by mitochondrial dysfunctionality (KE1) and/or oxidative stress (KE2) either as defective signal of activation or as cause of the dysfunction due to reduced oxidative phosphorylation and ATP production. As for the previous KERs, threshold, magnitude, nor duration required for elicitation of a detectable effect in KE4 have not yet been defined.
KER7: Dysfunctional autophagy (KE4) leads to cytosolic toxic tau (KE5)
Consequences of the failing intraneuronal waste clearance are tau oligomers and aggregates triggering axonal dystrophy, neuronal dysfunction, and eventually memory loss [184, 193].
The biological plausibility
Progressive dysfunction of neuronal autophagic capacity contributes to the formation of an initiating substrate complex needed for the initial seeding (or nucleation) of tau (and Aβ) aggregation. As a result, cytosolic tau oligomers are formed, followed by UPS-mediated autocatalytic propagation of tau aggregation [33]. At synaptic sites, accumulated tau oligomers are correlated with accumulated ubiquitinated proteins, proteasomes, and related chaperones [194].
The empirical support
Cytosolic toxic tau oligomers are observed at very early stages of the AD [167], with human AD brain containing more tau oligomers than control samples [195]. Inhibition of autophagy in a neuroblastoma cell model of tauopathy results in elevated levels of soluble and insoluble forms of tau [193]. Observations of internalized tau aggregates colocalizing with lysosomal markers suggest a plausible role of autophagy in tau degradation or lack thereof when dysfunctional. Supporting evidence is provided by a study showing that intracellular tau seed-induced aggregate formation is inhibited by activation of autophagy with rapamycin [196].
Overall assessment
It is plausible that a decreasing capacity to exhibit effective autophagy (KE4), in an environment of elevated p-tau (KE3), plays an important role in the accumulation of cytosolic pathologic tau variants (KE5). While the data indicate that defective autophagy, elevated p-tau and cytosolic tau levels are adjacent, there were no quantitative data found concerning threshold, magnitude or duration required to observe an adverse effect driving the development of memory loss.
KER8 –KER10: Cytosolic toxic tau (KE5) leads to dysfunctional axonal transport (KE6), which leads to dysfunctional synapses (KE7) and neuronal dysfunction (KE9)
Accumulation of cytosolic tau oligomers in somatodendritic compartment in neurons may cause cytoskeleton disruption, leading to progressive synaptic loss by impeding the axonal and dendritic vesicle transport, resulting in synaptic starvation.
The biological plausibility
Tau protein drives cognitive impairment by the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-D-aspartate (NMDA) receptor pathways [197]. In the neuronal dendrites, p-tau targets Fyn kinase, a substrate for the NMDA receptor at postsynaptic compartments [198, 199]. This results in delocalization of tau from axon to synapses and somatodendritic compartments, which causes NFT formation and eventually synaptic dysfunction [147, 165]. Soluble tau oligomers exert more acute toxicity than the insoluble ones [200]. Especially dimers are effectively self-associating into large oligomeric tau nuclei of aggregation. These pathological tau aggregates are cytosolic, but also appear in the extracellular space, and correlate with synaptic dysfunction, neuronal toxicity, and degeneration [200, 201]. In human induced pluripotent stem cell-derived neurons, induction of tau oligomers, but not monomers, drives pathological p-tau aggregation and causes neurite retraction, synaptic loss, neurotransmitters imbalance, and neuronal cell death [196]. In mice, subcortical injection of oligomers reduces the expression of synaptic vesicle-associated proteins leading to synaptic dysfunction. Pathogenic tau oligomers also negatively affect mitochondria, suggesting an amplifying circle of toxic Ca2 +-mediated events linking KE3 and KE5, and leading to mitochondrial dysfunction and synaptic loss [202, 203]. Tau-induced mitochondrial dysfunction (KE1) is characterized by a decrease in mitochondrial complex I levels, activation of caspase-9 and the apoptotic mitochondrial pathway [202]. Toxic Tau35 may be implicated in intraneuronal insulin accumulation and impaired insulin signaling through interactions with phosphatase and tensin homolog protein (PTEN), which inhibits dephosphorylation of PIP3 to PIP2 [204, 205].
The empirical support
Synaptic loss is associated with early cognitive decline in the neocortex and limbic system, with reductions in synaptic density at preclinical and terminal stage in AD pathology of 25%and 55%, respectively [206, 207]. The levels of markers for presynaptic terminals, synaptic vesicle, and synaptic protein are reduced in early stage of AD [208]. Along the same line, a tau transgenic mouse was found to exhibit a decreased expression of synaptic proteins, such as synaptophysin, synapsin, synaptojanin, and synaptobrevin [197]. An acute exposure to extracellular human tau oligomers caused memory impairment in mice [200] probably through inhibition of IRS1 and PTEN activities and subsequent insulin resistance. Abnormal inhibitory serine phosphorylation of IRS1 by INSR has been linked to brain insulin resistance in tauopathy, including AD pathology [205].
Overall assessment
The evidence supports enhanced cytosolic toxic tau levels (KE5) being adjacent to dysfunctional axonal transport (KE6) which results subsequently in synaptic (KE7) and neural (KE9) dysfunction. Thresholds values, degree, and duration of dysfunction of these Kes required to drive this series KEs toward memory loss are not known.
KER11 –KER12: Cytosolic toxic tau (KE5) leads to neuroinflammation (KE8) which leads to neuronal dysfunction (KE9)
Activation of innate immune response is observed before the deposition of NFTs, suggesting a profound role for tau soluble variants in early tauopathy [209].
The biological plausibility
Small soluble tau oligomers cause inflammatory signaling in the brain by activating microglia. These inflammatory responses are mediated by activated inflammasome and promote proinflammatory interleukin 1β (IL-1β) release, which is controlled by activation of caspase-1 [210]. Activation of microglia and astroglia, and subsequent release of proinflammatory cytokines, occur in the brain of humans and mice exposed to p-tau [211, 212]. Colocalization of activated microglia and astroglia, and proinflammatory cytokines with tau oligomers has been observed in mouse brain, suggesting that tau oligomers play a role in neuroinflammation and in accelerating neuronal dysfunction and neurodegeneration [213]. Tau oligomer levels correlate with High Mobility Group Box 1 (HMGB1) levels, an important pro-inflammatory marker in the brain [213], with recruitment of brain T-cells being linked to tau pathology and neuroinflammatory processes [214, 215].
The empirical support
Neuroinflammatory response in AD brain is driven by potent inflammatory mediators [216] and free radicals [217]. In a cross-sectional study of elderly adults with normal cognition and impaired cognition, six CSF neuroinflammatory markers (interleukin 15 (IL-15), monocyte chemoattractant protein-1 (MCP-1), vascular endothelial growth factor receptor 1 (VEGFR-1), soluble intercellular adhesion molecule-1 (sICAM1), soluble vascular cell adhesion molecule-1 (sVCAM-1), and vascular endothelial growth factor-D (VEGF-D)) correlated with tau levels in CSF, while none correlated with Aβ levels in CSF [218].
Overall assessment
There is strong evidence that processes resulting in increased cytosolic toxic tau levels (KE5) are adjacent to neuroinflammation (KE8), a condition that is widely accepted to play an important role in memory loss and neurodegeneration. The levels of cytotoxic tau, nor the duration of exposure, required to obtain a detectable inflammatory response sufficient to drive neuronal dysfunction are not known.
KER13: Neuronal dysfunction (KE9) to memory loss, the AO for the proposed AOP
Neuronal dysfunction is one of the most well-characterized hallmarks in AD pathogenesis. Particularly, loss of memory at early stage of the disease is associated with neuronal dysfunction in the upper layer of entorhinal cortex, an early affected brain region in preclinical state of the disease [219]. Synaptic dysfunction results in cognitive impairment and neuronal cell death [196, 221]. Brain insulin signaling impairment decreases AKT signaling, which is crucial for cell survival and function [222] and negatively affects synaptic plasticity and memory [223, 224]. A toxic relationship exists between soluble tau oligomers and neuroinflammation, which cause eventually neuronal damage, activating inflammatory mediators and free radicals [213, 217]. In AD mouse models, chronic neuronal tumor necrosis factor α (TNF-α) expression correlates with neuronal death [225]. Significant loss in neuronal density occurs in the hippocampus and cerebral cortex of AD patients [226–228], and is AD-stage dependent [229].
Overall assessment
Despite the lack of data on quantity and temporality data, the evidence supports neuronal dysfunction (KE9) to be adjacent to memory loss.
PLAUSIBLE PLUG-INS FOR ENVIRONMENTAL NEUROTOXICANTS
To portray how neurotoxicants suspected of contributing to sAD progression, the applied search methodology was expanded to include molecular and cellular mechanisms triggered by chemicals that experimentally and/or epidemiologically have been associated with neurotoxicity and possibly with AD development. Thus far, 27 plausible MIEs potentially affecting any of the KEs captured in this AOP have been identified. Table 1 is an effort to summarize to the extent possible the complexity of the acquired information in a simplified way linking target (MIE), cellular effect and relation to the KEs of the proposed AOP. Subsequently, these chemical-triggered MIEs have been mapped on the tau-driven AOP (Fig. 4). This mapping suggests that environmental chemicals and drugs primarily contribute to glucose/cholesterol dysmetabolism (the starting point), mitochondrial dysfunction and oxidative stress (KE1 and KE2), p-tau formation (KE3), dysfunctional autophagy (KE4), dysfunctional synapses (KE7), and neuroinflammation (KE8).
Plausible plug-ins for environmental neurotoxicants
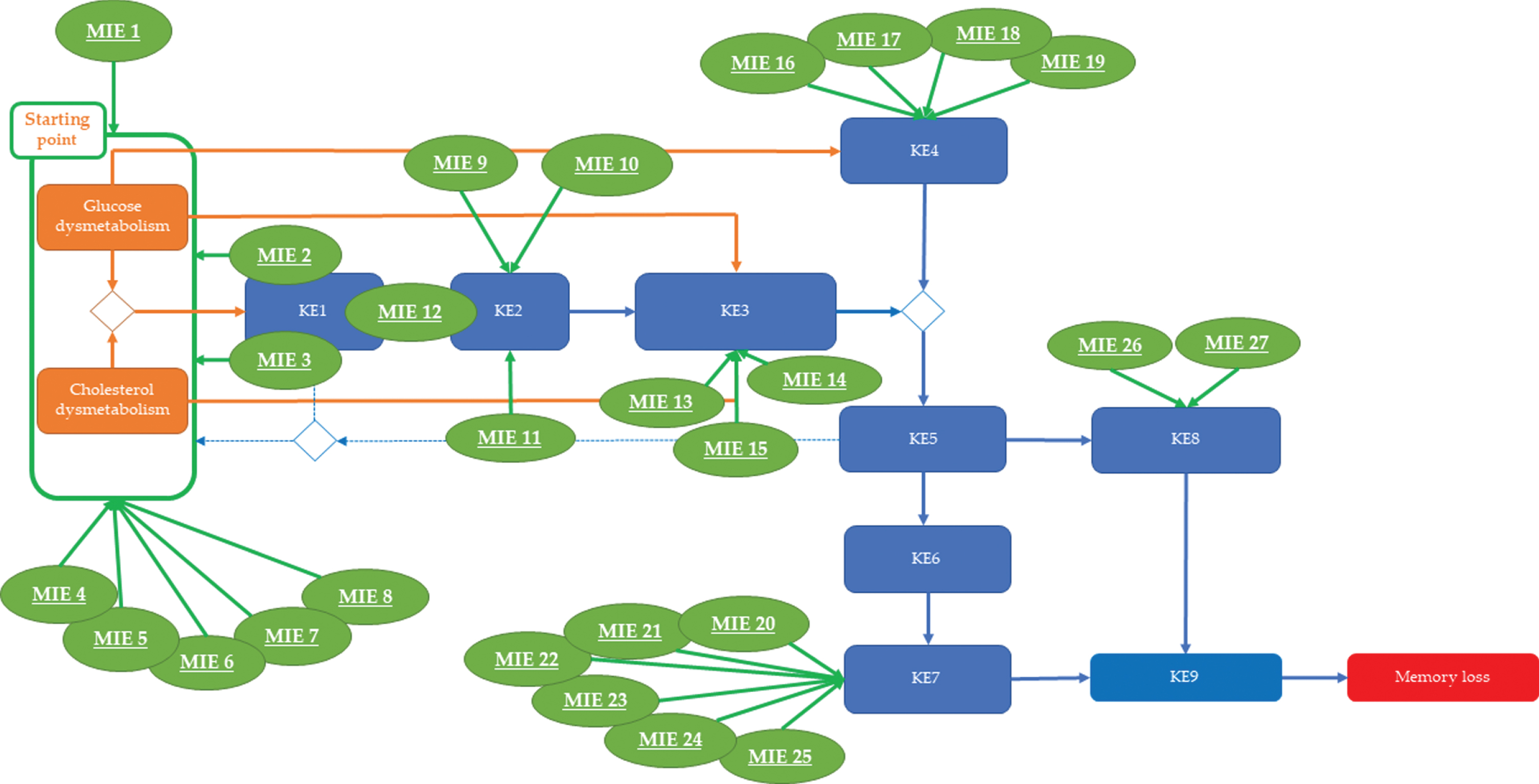
Overview of the Molecular Initiating Events (MIEs) triggered by environmental neurotoxicants that may plug into sAD processes. Based on the mechanistic information on how environmental neurotoxicity link to the presented molecular events of the AOP blueprint (provided in Table 1), 27 plausible chemical-triggered molecular initiating events (e.g., MIEs) are proposed as plugs-in for this AD AOP.
Chemicals triggering MIE1-8 appear to contribute to sAD progression by further destabilizing glucose and cholesterol metabolism in a direct manner (MIE1 and MIE4–8). With respect to MIEs 2 and 3 (i.e., targeting of mitochondrial complexes I and III), this effect may be an indirect consequence of mitochondrial dysmetabolism and/or insulin-mediated phosphorylation of AKT. The available information is not clear about the relation between MIE2-3 mediated mitochondrial dysfunction and suppression of AKT phosphorylation. Therefore, it cannot be excluded that the observed mitochondrial dysfunction is the consequence of a suppressed phosphorylation of AKT with glucose dysmetabolism as a result. Chemicals triggering MIE9-12 seem to affect processes that are critical for proper mitochondrial function and control of ROS production. Taken together, chemicals triggering MIE1-12 potentially have a significant impact on post-mitotic neurons, which are highly dependent on mitochondrial oxidative phosphorylation for their energetic needs.
Another group of chemicals was found to affect tau hyperphosphorylation (KE3) via MIE13-15. For chemicals acting via MIE13 and MIE15, the experimental data revealed, beside hyperphosphorylation of tau, evidence for mitochondrial dysfunction (KE1) and excessive ROS production (KE2). This observation supports a causal relation between energy production, oxidative stress, and p-tau.
Several chemicals were also found to negatively affect genes and processes that are involved in autophagy regulation (MIE16–19). The acquired experimental data reveal that this effect may be associated with oxidative stress (KE2) and p-tau formation (KE3). Since autophagy is energy dependent, its loss of functionality is believed to be the consequence of an age-related increase in glucose/cholesterol dysmetabolism (starting point), mitochondrial dysfunction (KE1), and excessive ROS production (KE2).
While the previous MIEs directly or indirectly involved energy production, which opens for speculation of the specificity of some of them, the next set of chemical-triggered MIEs (MIE20-25) were found to specifically perturb synaptic function by affecting the expression of well-known synaptic markers and the functionality of NMDA-mediated signaling.
Finally, some chemicals triggered neuroinflammation via MIE26-27, a condition that is generally accepted as being an important risk component for sAD progression.
DISCUSSION
A series of BioMed21 workshop produced a set of recommendations with the potential to enable new target discovery and accelerating more effective and efficient translation of novel therapies. One of these recommendations is the adaptation of the AOP concept, used in toxicology, to map perturbations of normal human physiology during disease development [40]. A recent example of an application of the AOP concept to portray complexity and multifactoriality is the ongoing EU activity aiming at ‘Modelling the pathogenesis of Covid-19 using the AOP framework (CIAO)’ (https://ec.europa.eu/jrc/en/event/webinar/intro-webinar-ciao-project). In analogy, we addressed sAD, which, in contrast to Covid-19, is a non-communicable and chronic disease.
Here, for the first time, a proposal for a tau-driven AOP, applicable for sAD, is described. Designing an AOP that captures the full complexity of such a multifactorial disease is a challenging task [41]. Therefore, it was decided to focus on one specific aspect of sAD progression: tau-mediated memory loss. The decision to focus on tau-mediated effects was triggered by the inherent desire to better understand initiation and early progression, and by research efforts showing that tau oligomers and aberrant phosphorylation of tau protein, rather than Aβ, are linked to early tau pathology and sAD development [13–15, 33]. The presented non-linear AOP blueprint is constructed in compliance with the criteria for weighing available evidence, with biological plausibility, essentiality, and empirical support considered as the most critical ones [40]. Thus, possible sequential events, linked to each other through KERs, for which causality and essentiality are supported by published scientific literature based on experimental evidence mainly derived from human data. Non-human studies were also considered mainly to fill possible knowledge gaps.
Typically an AOP conceptual framework starts with an MIE, a well-defined molecular initiating event (e.g., inhibition/activation/de-regulation of a given receptor, an enzyme) that kicks off specific downstream molecular and cellular events leading to a specific adverse outcome [230]. Identifying such a specific molecular event for the initiation and progression of tau-driven memory loss related to AD is not evident. Several risk factors play an important role in the onset of sAD, including aging, lifestyle related factors, genetic and systemic factors, and metabolism-related events, such as cholesterol and glucose dysmetabolism. Notably, aging and genetic background are widely accepted risk factors for sAD development, and aging was reported to affect several downstream processes that are recognized as KEs for sAD progression. The understanding of the molecular and cellular relation of genes and genetic polymorphisms and how they associate with the risk of acquiring sAD initiation and progression is still limited [35]. Therefore, both were ruled out for further consideration. The OECD database for AOPs (https://aopwiki.org) was consulted for specific MIE affecting, e.g., cholesterol and/or glucose metabolism in a sAD relevant way but did not provide any useful candidates. Finally, deregulation of ApoE4 and GLUT membrane density was considered as potential starting point; however, the available evidence suggests that deregulation of ApoE4 and GLUT is a consequence of cholesterol and glucose dysmetabolism, not the trigger. Therefore, these options were not pursued. Considering the multifactorial nature of this human disease, it was decided to adopt a more flexible approach when designing the proposed AOP blueprint, and therefore the bidirectional relationship between glucose and cholesterol dysmetabolism was considered as the event closest to the onset of sAD (here defined as hypothetical starting point). It is recognized that both phenomena are complex and account for many signaling pathways and associated molecules, and that several upstream systemic factors (e.g., hyperhomocysteinemia [231], diabetes [232]) may contribute. The main arguments in favor of this selection are the followings: i) brain glucose and cholesterol dysmetabolism are generally accepted to increase with age, ii) production and clearance of tau and Aβ are affected [45–47], and iii) relevant downstream KEs, contributing to disease progression and preceding the onset of memory deficit, are triggered [36].
The proposed AOP addresses the KEs and KERs considered to drive toxic tau production as well as KEs and KERs affected by these toxic variants and resulting in neuronal dysfunction and memory loss (AO). The upstream biological events of the tauopathy, characterized by the formation of cytosolic pathogenic tau proteins in neurons, are consisted of possible intermediate KEs that are interrelated via causal links. Additionally, cytosolic toxic tau variants are causally linked to downstream events that eventually cause the targeted AO. The tau-driven AOP also collects molecular and cellular information supporting the tau cascade hypothesis. Indeed, the available information suggests strong tau involvement in deregulation of Aβ and tau metabolism [16] directly or by affecting downstream processes (e.g., Ca2 + homeostasis [17], oxidative stress [18], neuroinflammation [19], cell cycle regulation [20], apoptosis [21], autophagy [22], synaptic function [23], glucose [24], and lipid metabolism [25]). While Aβ is not specifically addressed in this AOP, its impact on tau production and toxic tau formation has been acknowledged in this study, wherever deemed appropriate. The quantitative relationships between the identified KEs are defined in terms of correlations and response-response relationships only. Information on the magnitude and duration (time/scale) of changes in the upstream KEs, required to trigger adversity in the downstream KEs, was not found in the literature reviewed to build this study.
As the proposed AOP captures the defects that can occur before the manifestation of the disease, these depicted sequences of events, which can eventually lead to the targeted AO, may be helpful in defining the early stage(s) of AD development. The application of the AOP conceptual framework may help identify new biomarkers for early diagnosis, new druggable targets, and develop novel therapies; however, it should be considered that this area of study is still in its infancy, and no diagnostic tests or therapeutic approaches suitable for AD have been derived from AOPs thus far.
Subsequently, the AOP blueprint for sAD was used to interrogate currently described molecular and cellular events underlying effects of environmental chemicals, which have been associated with the risk of sAD acquisition, either mechanistically (in in vivo and in vitro studies) or epidemiologically (Table 1). The hypothesis is that plugging the available information on environmental chemicals into the proposed AOP blueprint may provide hints as how neurotoxicity induced by chronic exposure to environmental chemicals may antedate sAD initiation and/or aggravate disease progression. Pesticides, drugs, metals, nanoparticles, air pollution, and various industrial chemicals, among others, exhibit direct or indirect effects on several sAD related KEs through specific molecular interactions. Yet, 27 potential and tentative MIEs have emerged. Several of these identified MIEs exacerbate glucose and/or cholesterol dysmetabolism (hypothetical starting point) in a direct way, while others have a strong impact on various mitochondrial complexes (KE1) and ROS production (KE2) directly or indirectly (e.g., via starting point), and drive p-tau (KE3). The production of cytosolic toxic tau variants seems to be driven by MIEs resulting in autophagy dysfunction. Other MIEs may also have direct effects on synaptic function and plug into KEs that are already mentioned in the OECD AOP database, e.g., KE 386 (Decrease of neuronal network function), KE 618 (Decreased, Neuronal network function in adult brain), and KE 1664 (Induce Neuronal dysfunctions) (https://aopwiki.org/events).
It must be acknowledged that for several of the identified MIEs more research is needed to clarify whether they represent a real independent and direct event. Many studies were designed to study specific cellular or molecular mechanisms and did not explore general cellular or organ functionality. Thus, several in vivo or in vitro studies report the effect observed upon chemical exposure on, e.g., mitochondrial, autophagic, or synaptic function, and show data reflecting changes in functionality only for these cellular endpoints. Regarding MIEs involving deregulation of transcription or expression of specific nuclear factors, direct effects of chemicals have not always been established. For these tentative MIEs, it cannot be excluded that the observed molecular changes are secondary to, e.g., glucose and/or cholesterol dysmetabolism (hypothetical starting point), mitochondrial dysfunction (KE1), or ROS (KE2).
The available studies did not identify the quantitative changes, nor their timescale, required to obtain a detectable adverse effect of the upstream KE on the downstream KE. Only relative changes (defined in terms of low or high, down- or upregulation, decrease or increase), occurring in an upstream KE leading to relative changes in a downstream KE (e.g., in KER1, downregulation of GLUT or IGF1 are associated with increase of tau hyperphosphorylation) were available. Despite these uncertainties, the potential of environmental chemicals to modulate the quantitative and response-response relationships between two KEs is highlighted with relevant supporting information regarding the way the modulating factor is expected to alter the relationship.
The proposed AOP blueprint may help stimulating the discussion on the topic and allow for prioritization of research activities aiming at better understanding the interaction between aging, and genetic and environmental risk factors in sAD initiation and early progression. In this context, the use of human-relevant methods and integrated approaches should be prioritized as these could enable mechanistic understanding of the effects possibly induced by the identified environmental factors possibly associated with the risk of sAD. It may also trigger the development of a similar AOP blueprint capturing the KEs and KERs driving, e.g., Aβ pathogenesis that can be plugged into the proposed tau-driven AOP. Subsequently, the growing AOP network could be linked with the Aggregate Exposure Pathway conceptual frameworks to bridge possible (knowledge) gaps, covering also higher levels of biological complexity (e.g., organism, population), and help define the role and impact of environmental (neuro)toxicants on sAD risk.
CONCLUSIONS
A putative tau-driven AOP blueprint toward the AO of memory loss has been proposed for sAD pathogenesis. The hypothetical starting point (cholesterol and/or glucose dysmetabolism) has been linked to downstream intermediate KEs through KERs, to increase the current mechanistic understanding in early stage of AD pathology. In this context, environmental neurotoxicity is also discussed for its potential impact on sAD development, depicting implicated neurotoxic-induced MIEs and the perturbed KEs in the proposed AOP. More research is needed to support the suggested links between the molecular and cellular events and AO of the proposed AOP, and eventually untangle the complexity of sAD pathogenesis.
Footnotes
ACKNOWLEDGMENTS
This study was financially supported by EU Interreg, Vlaanderen-Nederland (https://grensregio.eu/projecten) (Project Herinneringen), ToxGenSolutions BV (https://toxgensolutions.eu), and 3Rs Management and Consulting ApS (![]() ).
).