
Abstract
Alzheimer’s disease (AD) is the most common age-related neurodegenerative disorder, responsible for nearly two-thirds of all dementia cases. In this review, we report the potential AD treatment strategies focusing on natural polyphenol molecules (green chemistry) and more specifically on the inhibition of polyphenol-induced amyloid aggregation/disaggregation pathways: in bulk and on biosurfaces. We discuss how these pathways can potentially alter the structure at the early stages of AD, hence delaying the aggregation of amyloid-β (Aβ) and tau. We also discuss multidisciplinary approaches, combining experimental and modelling methods, that can better characterize the biochemical and biophysical interactions between proteins and phenolic ligands. In addition to the surface-induced aggregation, which can occur on surfaces where protein can interact with other proteins and polyphenols, we suggest a new concept referred as “confinement stability”. Here, on the contrary, the adsorption of Aβ and tau on biosurfaces other than Aβ- and tau-fibrils, e.g., red blood cells, can lead to confinement stability that minimizes the aggregation of Aβ and tau. Overall, these mechanisms may participate directly or indirectly in mitigating neurodegenerative diseases, by preventing protein self-association, slowing down the aggregation processes, and delaying the progression of AD.
INTRODUCTION
Alzheimer’s disease (AD) is the most common age-related neurodegenerative disorder, responsible for nearly two-thirds of all dementia cases [1, 2] including Lewy body dementia, vascular dementia, and frontotemporal dementia [3, 4]. The prevalence of AD is about 5–10% above the age of 60 years but increases up to 40–50% above the age 85 years [5]. With the aging of the population [6], dementia affects nearly 50 million people worldwide, and is predicted to increase to 152 million by 2050 [7]. The estimated annual healthcare cost is US $1 trillion, which could double by 2030 [7].
The multifactorial disorders of AD, considering genetic and non-genetic components, are clinically characterized by memory dysfunction, loss of lexical access, spatial and temporal disorientation, and impairment of judgement. Although the molecular mechanisms of AD have not been fully elucidated yet, compelling evidence indicates that abnormal proteins accumulation in the brain, such as the intracellular aggregation of the tau [8–10] and extracellular deposition of amyloid-β (Aβ) [11, 12] leads to neuronal loss [13]. However, the heterogeneous nature of neurodegenerative disorders increases the challenges to understand the underlying mechanisms from the initial phases to the progression of AD. Over the years, several pathways have been studied including the Aβ cascade and deposition, abnormal tau aggregation, oxidative stress, mitochondrial dysfunctions, lysosomal alterations, neuroinflammation, and metabolic disorders where all of these converge to neurodegeneration (Fig. 1).

Multiple targets of polyphenols in the integrative amyloid-cascade and tau pathway. Both Aβ and tau pathology are independent and dependent pathways leading to AD. In familial form of AD, different mutations on APP, PS1, and PS2 genes lead Aβ1–42 overproduction while in sporadic form of AD, failure of the Aβ1–42 clearance under physiological conditions induced its gradual rising. The net result is to enhance the production of the putatively neurotoxic Aβ1–42 monomer at the expense of the putatively neuroprotective Aβ40. Aβ1–42 accumulation into soluble oligomers induce oxidative damage, inhibit the activity of Nrf2 and thus the antioxidant genes, activate NF-κB and the production of cytokines and finally cause apoptosis. Aβ1–42, ROS, and oxidative damage can activate glycogen synthase kinase 3β (GSK3β) which phosphorylates tau protein. Both the formation of NFTs due to the hyperphosphorylation of tau and the amyloid cascade lead to synaptic dysfunction, neuronal loss, and finally to learning and cognitive impairment. () Represent different pathways targeted by polyphenolic compounds.
The amyloid-β pathway
Since 1991, the amyloid-cascade hypothesis has provided the main framework to understand the pathogenesis of AD [14, 15]. The basis for this hypothesis was the discovery of autosomal dominant mutations in three genes—APP, PSEN1, and PSEN2 (the latter two encoding presenilin 1 and 2, respectively)—that induce pathogenic Aβ aggregation into neuritic plaques [14, 15]. The amyloid-β protein precursor (AβPP), a type I transmembrane protein, contains a large extracellular domain [16]. In familial AD with APP mutations, the amyloid cascade leads to early onset of cognitive deficits and dementia, likely through complex age-dependent cellular and molecular changes, including the spreading and deposition of neurofibrillary tangles (NFTs). Although mutations in the above three genes do not occur in sporadic AD, similar neuropathological changes in Aβ and tau were observed in both familial and sporadic AD [17–19].
Over the years, the amyloid-cascade hypothesis involved into an integrative model that provides a general framing for other disease mechanisms, e.g., immunoreactivity, microgliosis, mitochondrial dysfunction, oxidative stress, and dysregulation of protein homeostasis [20, 21].
The amyloid-β and tau integrative pathways
It is now thought that Aβ and tau pathologies can follow both independent and dependent pathways leading to AD. Aβ preferentially accumulates in brain regions with high metabolic demand (such as association cortices) and spreads from neocortex to allocortex to brainstem, eventually reaching the cerebellum [22–24]. Tau pathology, by contrast, first becomes evident in the (trans)entorhinal cortex from which it spreads to limbic areas, and from there to the neocortex [25–28]. The finding that Aβ and tau pathologies initially start in different brain regions, referred to as the ‘spatial paradox’, argues against the idea that tau pathology is driven by amyloid pathology occurring in the same local brain area. Aβ and tau pathologies also follow distinct temporal sequences because Aβ in the neocortex is already present 10–20 years before the emergence of clinical AD symptoms and the rate of Aβ accumulation attenuates during the clinical stage of AD [29]. In addition, the extent and locations of Aβ deposition are only modestly correlated with the brain areas affected by neurodegeneration [30, 31]. Both spatially and temporally, tau pathology correlates much more strongly with neurodegeneration and cognitive impairment than Aβ pathology. Increased tau PET signal is associated with worse cognitive performance in both cognitively normal individuals and patients with clinical AD [26, 33]. The spread of tau pathology was associated with a specific gene-expression profile of ‘axon-related’ genes, whereas the spread of Aβ was linked to a different profile of ‘dendrite-related’ genes. A third subset of ‘lipid metabolism-related’ genes was linked to increased spread of both Aβ and tau pathology [34].
Learning, memory, and cognitive deficits characterize AD patients, whereas memory deficits are a hallmark of amnestic mild cognitive impairment. These altered functions largely originate from synaptic dysfunction involving altered synaptic proteomes [35, 36] with the particular contribution of Aβ42 oligomers [35, 37]. These oligomers cause oxidative damage to synaptic membranes [38], suggesting the relation between oligomer-induced oxidative damage and synaptic dysfunction.
Role of the amyloid-β oligomers
Many oligomeric Aβ species have been described [39]. Large oligomers of Aβ42 are relatively less toxic whereas small Aβ42 oligomers (e.g., dimers or trimers that easily enter lipid bilayers) appear highly toxic to synapses [40]. Thus, in the absence of amyloid plaques, soluble Aβ oligomers from AD brains have been showed to impair hippocampal synaptic plasticity, decrease synapses, induce tau hyperphosphorylation and neuritic dystrophy, activate microglial inflammation, and impair memory in normal adult rodents [40]. Together, the soluble fraction of high molecular weight oligomeric Aβs are far less bioactive than the smaller oligomers in AD brain. The composition of Aβ plaques is both fibrillar and soluble high molecular weight oligo-meric Aβs. Therefore, it is important to target diffusible Aβ species that are highly bioactive in AD brain. Thus, the neutralization of the toxicity of oligomeric Aβ species was suggested as a chronic therapy for AD [41, 42].
Amyloid-β-targeted therapies
To date, there is no approved drugs that can either revert or cure the AD. The amyloid cascade hypothesis has also guided most drug discovery efforts in both familial and sporadic AD, where the objective was the removal of various forms of cerebral Aβ. Unfortunately, if Aβ-targeted therapies tested in phase III clinical trials (bapineuzumab, gantenerumab, solanezumab, crenezumab, lanabecestat, atabecestat, verubecestat, and elenbecestat) [43] can effectively reduce Aβ load in AD brains, they were unsuccessful in slowing cognitive decline either with mild cognitive impairment or AD patients. In addition, two phase III clinical trials of Aβ-targeted therapy, aducanumab, were halted in March 2019 [44]. Interestingly, in one of the two trials, cognitive decline was attenuated in patients receiving high dose aducanumab, [44]. However, the AD drug candidate aducanumab took a beating from FDA advisors (Science, Nov. 6, 2020) and the FDA’s decision is expected by March 2021 [45].
The lack of beneficial effects on cognitive outcome from these trials could be due to different reasons such as the timing of the interventions, as the studies involved patients in clinically advanced stages of AD, and insufficient dosing or the wrong Aβ species being targeted [46]. Alternatively, the failure of these trials could indicate that removing Aβ from the brain is not sufficient to halt cognitive decline suggesting that tau pathology, tau-mediated neurodegeneration, and other mechanisms in AD are driven partially by Aβ pathways. Most of the unsuccessful therapeutic approaches for AD had focused on Aβ or tau pathways. Interestingly, Hammond et al. [47] suggested that AD treatments may also need to be disease stage-oriented with Aβ and tau as targets in early AD and glucose metabolism as a target in later AD. Recently, the EU-US Clinical Trials on Alzheimer’s Disease (CTAD) Task Force reported that effective treatments should include new biomarkers intervening in early stages of AD, and of combination therapies [44]. This led to the diversification of the development of the drug portfolio.
Protection against amyloid-β by polyphenols
In this regard, several polyphenols have been investigated to promote neuroprotection by targeting Aβ oligomers and attenuated Aβ-induced-reactive oxygen species production, restore the Aβ-induced reduction of antioxidants activities or mitochondrial dysfunctions (Fig. 1) [48–51]. Numerous flavo-noids can interact and destabilize Aβ peptide structures, function of ligand:Aβ molar ratio [52]. Most polyphenols that are Aβ-aggregation inhibitors share a catechol moiety, also related to anti-oxi-dative stress, such as: (+)-taxifolin, myricetin, quer-cetin, (+)-catechin, epigallocatechin gallate (EGCG), anthocyanins [53], OH-tyrosol, tyrosol [54–57], and rosmarinic acid or grape-derived polyphenols [58–61]. As a result, these compounds can efficiently destabilize the β-sheet structures of Aβ and prevent the elongation of Aβ oligomers.
Interestingly, some antioxidant polyphenols, such as resveratrol or quercetin-3-O-glucoside [62], are able to enter the brain to a limited extent and have shown promise for AD treatment [63]. Another mechanism by which polyphenols could exert protective properties is by generation of a hormetic response to their use [64–66], i.e., they generate a mild oxidative stress that the body tries to mitigate by upregulating protective genes. This often leads to increases in the levels of antioxidants such as glutathione and HO-1, mediated by activation of the transcription nuclear factor erythroid 2-related factor 2 (Nrf2) [66, 67] (Fig. 1).
Among the 560 anthocyanins (ACNs) identified in nature, the common anthocyanidin aglycones, e.g., pelargonidin, cyanidin, delphinidin, peonidin, petunidin, and malvidin, can form covalent conjugates with sugars and organic acids to generate a plethora of ACNs. Some ACNs have the ability to cross the blood-brain barrier and reach the brain [68, 69], particularly the hippocampus [70–73]. The type and concentration of ACNs differ widely among different fruits and vegetables, ranging from 1.4 mg/g to 800 mg/g of dry weight [74]. Youdim et al. [75] reported the citrus flavonoids, hesperetin, naringenin, and their relevant in vivo metabolites, as well as the dietary ACNs and in vivo forms, cyanidin-3-rutinoside and pelargonidin-3-glucoside, are taken up by two brain endothelial cell lines from mouse (b.END5) and rat (RBE4).
In this review, we first discussed the biochemical aspects of Aβ and tau which are relevant to β-amyloidopathy and tauopathy. We then reported the potential of AD treatment strategies focusing on natural polyphenol molecules (green chemistry) and specifically on the inhibition of polyphenol-induced amyloid and/or tau aggregation or disaggregation pathways. We discussed how polyphenols can potentially alter the structure and/or delaying Aβ and tau self-association at the very early stage of AD, hence, potentially playing a key role in the progression of neurodegenerative disorders. In addition, we suggest a new concept, referred as “confinement stability”, where the adsorption of Aβ and tau on biosurfaces other than Aβ- and tau-fibrils, e.g., membranes, vessels, red blood cells (RBCs) etc., can either lead to confinement stability or to surface-induced aggregation, depending on the affinity of Aβ and tau to these surfaces.
BIOCHEMICAL ASPECTS OF Aβ40 AND Aβ42
The molecular weights of Aβ40 and Aβ42 mono-mers are 4.33 and 4.51 kDa, respectively. Aβ42 with two additional hydrophobic residues (Ile41 and Ala42) at the C-terminus shows a greater propensity to induce fibrils formation than Aβ40 [76]. The predicted solubility is higher for Aβ40 than Aβ42 being 0.4μM and 0.04μM, respectively [77], consistent with Aβ42 experimental solubility of 0.04μM [78]. Both Aβ40 and Aβ42 monomers have a hydrodynamic radius (Rh) of 0.9±0.1 nm [79], i.e., below the colloidal domain (∼5 nm to 5μm) [80]. The total mass of Aβ is estimated to 6.5 mg in cortical grey matter of AD brain compared to 1.7 mg in control brains [81]. For example, an Aβ rate of mass accumulation of 30 ng/h is enough to place a person on the trajectory to accumulate 5 mg of Aβ in the brain over a 20-year time frame [81]. The in vivo fractional production and clearance rates of Aβ in the human CNS was reported to be 7.6% per hour and 8.3% per hour, respectively [82]. Importantly, Aβ fibrils from human brains are right-hand twisted, quite different from in vitro fibrils [83]; this emphasizes the preferred use of human AD brains in future investigations. Additional biochemical aspects of Aβs can be found in [84].
BIOCHEMICAL ASPECTS OF TAU
In the perspective of understanding tau aggregation mechanisms, the following describe some biochemical and biophysical properties of tau protein implicated in AD: structures, domains, phosphorylated and binding residues, solubility, ionic charge, prone to or suppression of aggregation, etc.
The accumulation of misfolded and aggregated forms of tau protein in the brain is a neuropathological hallmark of tauopathies observed in neurodegenerative diseases (NDs), including AD and Pick’s disease [85–87]. The microtubule-associated protein tau (MAPT), identified in mid-1970s [88, 89], encodes the protein tau [90]. Tau is notably characterized by the presence of three or four (according to the isoforms) imperfect repetitions of a motif of about 30 residues, known as the microtubule-binding repeats (MTBRs), and where the N-terminal to the MTBR is a proline-rich region (PRR) [91]. Moreover, tau can be characterized into four sections: N-terminal projection, proline-rich domain, microtubule-binding domain (MBD), and a C-terminal [92]. Tau full length monomer (hTau40wt(441)) has a molecular weight of 45.8 kDa, a radius of gyration (Rg) of 6.5±0.3 nm and a hydrodynamic radius (Rh) of 5.3 nm [93], within the colloidal domain.
Tau is a natively unfolded protein largely found in axons, where it serves to stabilize microtubules that have a diameter of ∼25 nm [94], and shows little tendency for aggregation [95]. Phosphorylation triggers tau-tau self-assembly [96]. Tau pseudophosphorylation on some sites found preceding residue 208 mainly suppresses tau aggregation whereas tau pseudophosphorylation at sites in the C-terminal region preferentially promotes tau self-association, particularly S422 [97, 98]. Tau phosphorylation by GSK3β induced tau aggregation [99, 100]. Two hexapeptides, 275VQJINK280 and 306VQJVYK311, are effective in generating β-sheet structures while processing tau aggregation [101, 102].
Abnormal folding of the MAPT results into paired helical filaments (PHFs) and NFTs [103]. The cores of PHFs and straight filament are composed of eight β-sheets (β1-8) that run along the length of the protofilament, adopting a C-shaped architecture [104]. Hydrophobic clustering, aliphatic stacking (V339, L344, V350, I354), and aromatic stacking (F346) stabilize the interior of β-helix [104]. The existence of in vitro twisted ribbon-like assemblies of tau fibrils was observed, showing corrugations with periodicities of 17.4±2.7 nm (n = 16) in fibrils of human tau40 [105].
In human AD cortex, soluble Aβ dimers induced tau hyperphosphorylation and neuritic degeneration [106]. Therefore, Aβ is upstream of tau in AD pathogenesis and triggers the conversion of tau from a normal to a toxic state, but there is also evidence that toxic tau enhances Aβ toxicity via a feedback loop.
The above biochemical and biophysical properties of Aβ and tau nucleation and growth provide key fundamental molecular insights which are the basis for effective mechanisms in delaying neurodegenerative bioprocesses.
GENERAL STRUCTURE OF POLYPHENOLS
Among 8,000 known polyphenolic compounds, more than 5,000 flavonoids are widely distributed in plants [107–109], e.g., tannins, in particular pro-anthocyanidins with more than 1000 derivatives identified to date. Tannins can be classified into two groups: hydrolysable tannins and condensed tannins [110, 111]. The condensed tannins, also referred as proanthocyanidins, are the most abundant. Hydro-lysable gallotannins contain gallic acid (GA) substituents esterified with a polyol residue (mainly D-glucose). The biosynthetic pathway, starting from D-glucose and after the galloylation reaction, yields di-, tri-, tetra, penta-, hexa-, hepta-, and octagalloyl-glucoses. Gallotannin with 10 (up to 12) units of GA esterified to a single glucose moiety are having many phenolic OHs, e.g., tannic acid (TA) with 25 phenolic OHs. The majority of polyphenols have more than two aromatic rings, essential for π-π stacking with aromatic amino acid residues of Aβ and at least three phenolic hydroxyl groups that can form hydrogen bonds with hydrophilic residues of Aβ [112].
MECHANISMS AND DELAYING AGGREGATION OF Aβ AND TAU
There are various mechanisms for delaying Aβ and tau aggregation in the context of AD. Natural polyphenols are known to strongly associate with these proteins, thus have the potential to prevent protein self-association and the formation of toxic oligomers, fibrils, and plaques. Different aggregation mechanisms occur in human brain, such as: salt-induced aggregation, bulk aggregation, and surface-induced aggregation (either on Aβ and tau fibrils from secondary nucleation, or on biosurfaces other than Aβ- and tau-fibrils). Interestingly, polyphenols are stable in high conductivity environments, such as physiological conditions.
Nucleated polymerization processes are involved in many growth phenomena in nature [113]. For example, the biology of human brain involves molecular and macromolecular growth bioprocesses. This includes different Aβ and tau structures, conformations and shapes, such as aggregate, cluster, dimer, fibril, fiber, monomer, neurofilament light, NFTs, oligomers, PHF, plaque, protofibril, and straight filament. Some of the above are structures within the colloidal domain (∼5 nm to 5μm) [80]. Smaller molecules with less than ten or fifteen amino acids (aas) are below this range, but protein oligomers of Aβ40/42 and larger macromolecules such as tau protein with more than about 100 aas (e.g., tau441), are likely within this range. Nevertheless, the interactions in both ranges are diffusion controlled (perikinetic).
Aggregate denotes dimers, trimers, and higher order assemblies. The term oligomer often refers to aggregates of 2–20mers [114]. Amyloid fibrils are linear aggregates with a repetitive cross-beta structure. Primary nuclei can form during the lag phase from monomers in bulk solution. Then, the proliferation of new aggregates takes place on fibrils catalytic surfaces, referred as secondary nucleation [114, 115]. The lag phase can be minimized by addition of pre-formed nuclei or seeds [116]. Interestingly, a significant delay on the onset of Aβ40 fibrils formation was reported at a low apolipoprotein E3 (apoE3) concentration (40 nM), equivalent to an apoE3:Aβ molar ratio of 1:1000 [117].
Salt-induced aggregation and critical association concentration
The formation of Aβ fibrils and other polypeptide aggregates strongly depends on the physiological and chemical environment, e.g., the type and salt concentration [118]. Jain et al. [119] reported the impact of NaCl on the kinetics of Aβ fibril formation and β-rich oligomer formation, where the aggregation rate significantly increased up to ∼100 mM NaCl and reaches a plateau at about ∼200 mM NaCl. Interestingly, the sodium concentration in human cerebrospinal fluid (CSF) and in serum was reported to be 145.3 to 147.7 mM [120]. Moreover, the aggregation kinetics and thermodynamics measurements yield the following order of chloride cations for Aβ40 peptide aggregation: Mg2+ > Li+ > Na+ > K+, while sodium anions are ranked as: SO2- > I- > Cl- > NO- ≈ ClO- [118]. These series are known as lyotropic series and are predicted from an extension of classical colloidal coagulation theory, by including the effects of electroviscous drag [121, 122].
The critical association concentration of Aβ, or (CAC)Aβ, represents the minimum concentration of Aβ leading to self-association, which depends among other parameters on the salt concentrations. For example, the experimental (CAC)Aβ40 was reported to be: ∼0.2μM [123], ∼0.7–1μM [124], 0.88 ± 0.07μM [125], and Hellstrand et al. [126] showed that spontaneous aggregation only occurs when Aβ42 concentration is ∼0.2μM, all in the presence of salt. Novo et al. [127] reported that the in vitro Aβ42 critical aggregation concentration, under physiological conditions, is about 0.091±0.014μM. However, since the experimental solubility of Aβ42 is 0.04μM [78], (CAC)Aβ42 in the absence of salt should be less than 0.04μM.
Bulk-induced nucleation and growth
This type of mechanism suggests that Aβ and tau association and aggregation occur in bulk solution, e.g., physiological fluid such as CSF and blood environment. In the case of Aβ42 aggregation, the key amyloid formation steps have been reported [128, 129] as: 1) “the primary nucleation of new aggregates from monomers in solution” [130–132], 2) “the addition of monomers to fibrils ends resulting in their elongation” [76, 134], and 3) “the secondary nucleation of monomers on the fibrils surface” [128, 136]. All the above three microscopic processes occur during all three macroscopic phases (lag, growth and plateau), albeit at different rates, as governed by the rate constants, and concentrations of reacting species at each point on time [114, 138].
In bulk solution, the secondary nucleation process shows a much lower energy barrier than primary nucleation (∼10 times) [139], where the surfaces of existing amyloid fibrils is catalyzing the formation of new pre-fibrillar aggregates from the soluble peptides [139, 140]. Moreover, Cohen et al. [129] reported the free-energy landscape for secondary nucleation.
Strikingly, even though the oligomers are the key source of fibrils, less than 10% of Aβ42 oligomers successfully converted into fibrillar species, whereas the remaining 90% of the oligomers dissociated back to the monomeric form [141]. Michaels et al. [141] suggested that oligomer dissociation is ‘spontaneous’, whereas oligomer upconversion involves additional interactions with monomers, and may occur in bulk solution in contact with the fibril surface [142]. As for Aβ42 and Aβ40, the vast majority of oligomers do not form fibrils, but rather dissociates back to monomers. This type of mechanism has been ascribed to a non-classical nucleation process for Aβ42 amyloid fibrils [141], but alternatively can be readily described by a dynamic equilibrium between oligomer formation and break-up. Aggregates break up with a certain inherent rate constant and are formed by collisions, which involve interactions between particles or molecules in solution. These processes are pH dependent, as are other aggregation processes of proteins in general.
Frankel et al. [143] investigated the mechanisms underlying Aβ42 aggregation (0.8–10μM) in human CSF through the kinetic experiments, though in healthy humans Aβ42 concentration in CSF is around 250 pM. They also found that the aggregation process involves the same microscopic steps in CSF as in pure buffer, but the secondary nucleation rate constant is decreased [143].
Natural polyphenolic molecules can alter and/or delay Aβ self-association and the growth of oligo-mers/fibrils in bulk solution (Fig. 2).
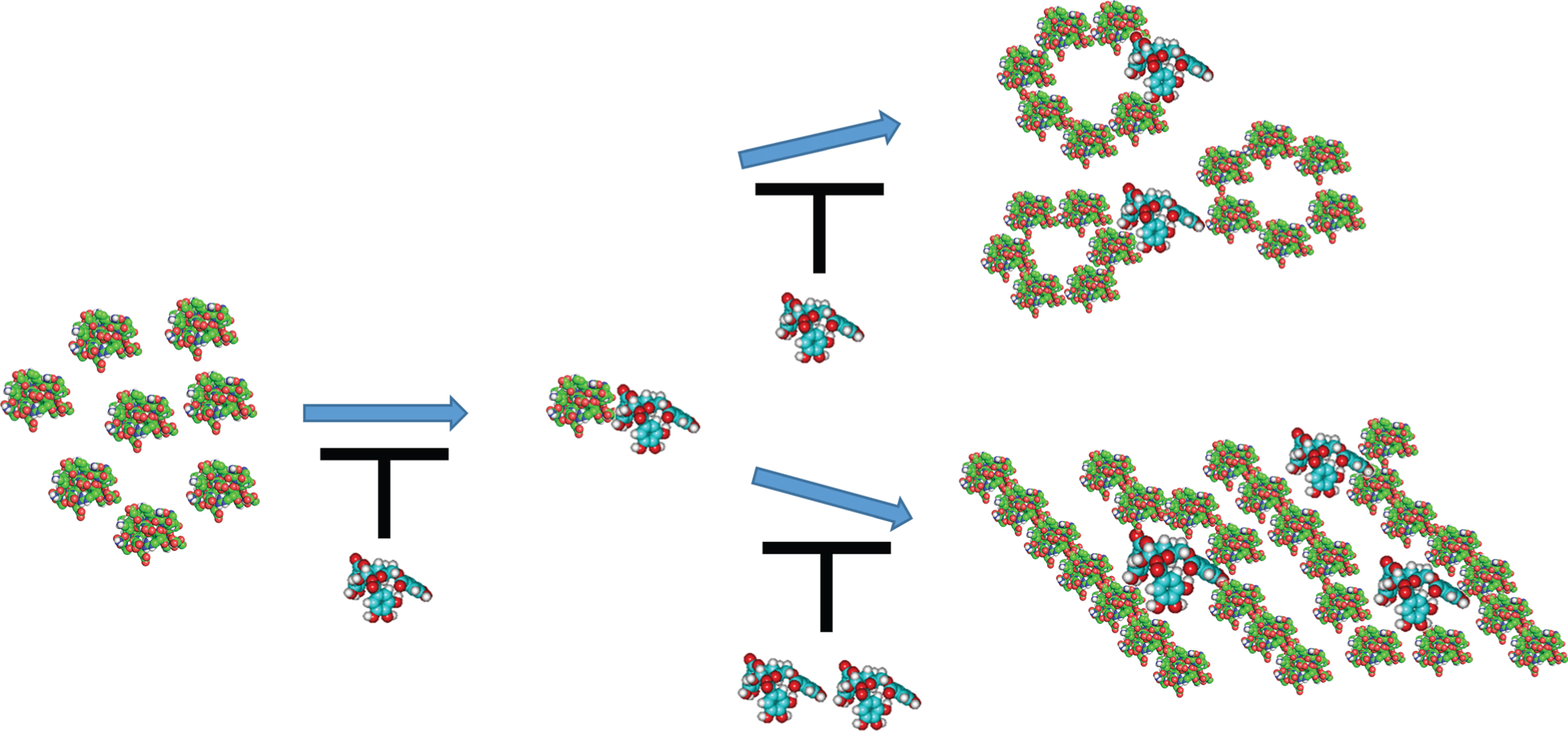
Hypothesis of phenolic-induced altering and/or delaying amyloid-β (Aβ) self-association and the growth of oligomers/fibrils in bulk solution: (left) monomers of Aβ; (middle) Aβ/phenolic ligand complex; (right-top) Aβ-oligomers and complexed with phenolic ligands; and (right-bottom) Aβ-fibrils and complexed with phenolic ligands. The Aβ protein is green/red colored, and the phenolic ligand is blue/red colored. The black T-shape symbol refers to inhibition/delaying the growth of oligomers/fibrils. This cartoon is not to scale, i.e., the phenolic ligand is much smaller than Aβ-protein.
It is noteworthy that using AFM and mica sheets functionalized with 1-(3-aminopropyl) silatrane (APS) showed that surface-induced aggregation occurs at a concentration at which no aggregation in solution is observed [144], e.g., likely below the critical association concentration of Aβ. The experiments were performed with full-size Aβ protein (Aβ42, 0.1μM), a decapeptide Aβ14–23 (0.1μM) and α-synuclein (0.01μM); all three systems suggest a significant preference of the on-surface aggregation pathway compared to the aggregation in the bulk solution [144].
Surface-induced aggregation on biosurfaces other than Aβ- and tau-fibrils
So far, we have reported Aβ elongation/aggrega-tion on fibril surfaces which is consistent with the related AD literature. However, other possible mechanisms must be highlighted since many more biosurfaces in human brain are available for Aβ/tau adsorption and aggregation, thus probably relevant to neurodegenerative diseases, such as AD.
In other words, aggregation can also occur on biosurfaces other than Aβ- and tau-fibrils, e.g., membranes, vesicles, endosomes, exosomes, mice-lles, erythrocytes/RBCs, platelets, albumin, and blood vessels. The total length and surface area of human brain capillaries are ∼600 km and ∼20 m2, respectively [145, 146]. For example, Aβ can be cleared via perivascular drainage pathways or depo-sited as neuritic plaques in the brain parenchyma or as cerebral amyloid angiopathy (CAA) along vessel walls [147]. When Aβ deposition occurs in brain capillaries (CAA type I), it tends to be widespread in the neocortex and hippocampus [148, 149], and associated with severe AD pathology [149, 150].
The human tau (hTau40) is also highly surface active and preferentially interacts with negatively charged membranes [151, 152]. Georgieva et al. [153] reported that lipid membranes efficiently facilitate in vitro tau aggregation. For their part, Yu et al. [154] reported that human islet amyloid polypeptide (hIAPP) aggregation was strongly enhanced by negatively charged membranes.
In addition, Aβ peptides interact with plasma proteins and RBC surface [155–158]. In the human body, 84% of the blood cells are RBCs and about 50% of the volume of blood (hematocrit) consists of RBCs [159], having an overall negative charge [160]. In human blood, circulating blood cells are exposed to nanomolar levels of soluble Aβ40/42 [161]. Interestingly, Aβ deposits in the extracellular space of the brain and on the walls of cerebral blood vessels, mainly capillaries. A dynamic equilibrium between brain Aβ and plasma Aβ has been reported by DeMattos et al. [162]. Lan et al. [157] showed that 98% of AD peripheral RBCs were amyloid binding-positive. Kiko et al. [163] provided evidence that Aβ40/42 were detected in RBCs. Moreover, Aβ42 interacted with RBCs more avidly than Aβ40 [164]; in vitro and in vivo experiments suggested that Aβ induces oxidative damage to RBCs [156, 164]. Morphological changes induced in RBCs, triggered by Aβ binding, was also observed [157, 165]. Remarkably, Aβ and tau as well as alpha-syn/Aβ and alpha-syn/tau heterocomplexes were also observed in RBCs [166, 167].
Interestingly, Koren et al. [168] postulated that circulating erythrocytes and likely also other blood cells might be coated by polyphenols from nutrients. The binding of polyphenols [168] and hydrolysable tannins [169] with RBC surface membrane have also been observed. RBCs and lipoproteins in blood showed to be reservoirs and transporters of polyphenols [170]. Harbi et al. [170] also determined the concentration of polyphenols associated with RBCs (intracellular+surface-bound); e.g., EGCG binds to RBC surface (33%) and intracellular (43%).
Moreover, Aβ peptides interact with platelet sur-faces in a highly specific manner [171], with plate-lets being 4.9% of the number of cells in human body [159], and the main source of Aβ peptides in blood (∼90%) [172, 173]. Wolozin et al. [171] also found that low levels of soluble Aβ (0.1–1 nM) augment adenosine-diphosphate(ADP)-dep-endent platelet aggregation. However, the ingestion of the polyphenol quercetin-4′-O-β-D-glucoside inhibited platelet aggregation in humans [174]. Nevertheless, the impact of polyphenols on aggregation might differ whether platelets are activated or not [175].
Biere et al. [176] found that the large majority of Aβ (∼89%) is bound to albumin and specific lipoproteins in human plasma. Albumin is also an Aβ carrier [177]. Interestingly, Yeggoni et al. [178] showed that the natural polyphenol corilagin binds to human serum albumin and found the experimental binding constant of the complex to be 4.2±0.02×105/mol with a free energy of –7.6 kcal/mol, also supported by their computational MD results.
Real-time precise determination of the growth rates of protein aggregates on surfaces can be measured using a quartz crystal oscillator (surface) [179]. Although the quartz crystal microbalance with dissipation (QCMD) method [180] proved to be reliable to study Aβ aggregation [181, 182], there are very few publications on the interactions between polyphenols and Aβ on surfaces. For example, Wang et al. [183] showed a reduction of ∼65% of the growth rates of Aβ monomer on curcumin-induced aggregates. By using QCMD combined with liquid AFM, Yu et al. [154] observations pointed toward a surface-involved pathway of protein adsorption and 2D amyloid aggregation. Surface plasmon resonance also showed to be an effective method to study the kinetic of Aβ42 aggregation on carboxylated dextran-modified surfaces [184].
Surface-induced aggregation
Considering that Aβ [144] and tau [151–153] proteins are highly surface active and preferentially interact with negatively charged membranes, this can trigger adsorption on membranes followed by surface-induced aggregation where protein can interact with other proteins and polyphenols. Importantly, human physiological Aβ [143, 186] and tau [187] concentrations are in the low nanomolar range. In 2017, Barnejee et al. [144] reported that on-surface in vitro aggregation of Aβ and α-synuclein occurs at a concentration at which no aggregation in bulk solution is observed. Interestingly, in 2005 we proposed that surface-induced aggregation/clustering occurs, causing an induction period [188, 189].
Consequently, a strategy where surface-induced aggregation occurring below the critical aggregation concentration of Aβ and tau, treated at the earliest stage, might result in minimizing protein self-association, slowing down the aggregation processes and delaying the progression of AD.
Proposed concept: confinement stability
The adsorption of Aβ and tau on biosurfaces other than Aβ- and tau-fibrils, e.g., membranes, vessels, albumin, RBCs, etc., can lead to confinement stabilization, a new concept recently proposed [190]. For example, Fig. 3 shows that Aβ and/or tau proteins (left) and phenolic ligands (center) are confined and stabilized on RBC biosurfaces (red), which then prevents protein self-association and slows down the aggregation process, hence delaying the progression of AD. Moreover, polyphenolic ligands can associate with Aβ and/or tau proteins, forming complexes (right), and thus prevent or inhibit self-association and aggregation of these proteins.

A schematic of confinement stability: Adsorption of Aβ proteins (left), phenolic ligands (center) and Aβ/phenolic ligand complexes (right) on human RBC biosurface (red), results in stability by confinement, i.e., these proteins and ligands are unable to aggregate due to temporary or permanent confinement. This cartoon is not to scale, i.e., Aβ, ligand and complexes are much smaller than indicated (RBC diameter ∼7.5–8.7μm [268]).
The adsorption and confinement of soluble Aβ and tau on biosurfaces potentially influences a number of phenomena that might occur, such as structural changes, reconformation, increased local protein concentration, decreased protein entropy, alteration of physiological functions, and formation of toxic Aβ-oligomers and/or protofibrils, tau NFTs, etc. Moreover, these confined and stabilized proteins are unable to diffuse and collide with other proteins and/or cells in bulk solutions. Confinement also occurs in hydrogels and tissues where it modifies the folding and aggregation of proteins [191–193].
In AD, the impact of small molecules (proteins, polyphenols) and nanoparticles that are stabilized by RBCs/platelets/vessel walls/etc., through confinement stability, is still to be enlightened. Although RBC perturbation by Aβ protein has been reported [156], our hypothesis is that the overall impact of polyphenols will be beneficial since they will bind to Aβ and/or tau proteins, e.g., resulting in less oxidative damage.
We expect that our proposed concept of “confinement stability” highlights new and additional mechanisms implicating Aβ and tau pathways in the physiopathology of NDs, such as AD. Consequently, research is necessary not only to elucidate direct but also indirect pathways involving surface-induced processes occurring on biosurfaces other than Aβ- and tau-fibrils.
THERAPEUTIC STRATEGIES FOR DECREASING Aβ AND TAU PRODUCTION AND AGGREGATION
The complexity of Aβ/tau production and aggregation pathways, the role of the monomer/oligomers or aggregated forms of Aβ/tau, and the unknowns regarding the progression of AD highlight the challenges for the discovery of effective treatments to delay or prevent this disease. For the amyloidopathy, some strategies targeting the amyloidogenic pathways (e.g., β-secretase (BACE) inhibitor [194–197], the non-amyloidogenic pathways (e.g., α-secretase activator), as well as the γ-secretase inhibitors and modulators (e.g., [198]) were developed with limited success. For the tauopathy, some of the most promising therapies have been discussed by Simic et al. [199]: minimize tau phosphorylation, proteolysis, aggregation, clearance of intra- and extra-cellular tau, and microtubules stabilization. Nevertheless, the need for fundamental understanding of Aβ and tau kinetics as well as aggregation inhibitors is imperative.
One strategy to mitigate AD may be the development of neuroprotective agents to prevent protein self-association, reduce Aβ and tau aggregation and/or induce the formation of non-toxic Aβ oligomers/tau NFTs. For example, polyphenolic ligand might prevent or delay Aβ and/or tau self-association at the earliest stage, e.g., below the critical association concentration of Aβ and tau proteins.
Table 1 shows molecular and pharmacological properties of some natural polyphenols and one commercial synthetic drug for comparison, e.g., donepezil (Aricept), an acetylcholinesterase inhibitor [200–203]. As expected, polyphenols are far less toxic than donepezil. For example, corilagin (β-1-O-Galloyl–3,6-(R)-hexahydroxydiphenoyl-D-Gl-ucose) [204, 205] has an LD50 between 3500–5000 mg/kg b.wt. [206], suggesting a very low toxicity even at high dosages [206, 207] (Table 1). Moreover, corilagin, a hydrolysable tannin, shows numerous pharmacological properties [208–213]. Corilagin (more rigid) is a close analogue of 1, 3, 6-tri-O-galloy-β-D-glucose (TGG) (more flexible), with similar molecular properties (Table 1) [84, 204]. This type of molecules also has remarkable properties for stabilizing and/or destabilizing colloidal systems [188, 189]. For example, we proposed a surface-induced clustering mechanism where corilagin/poly(ethylene oxide) clusters slowly built on micro-cellulose surfaces (corresponding to the induction period) [189]. Very little or no research has been performed on the inhibiting effects of corilagin and TGG on the kinetics of Aβ and tau aggregation/disaggregation bioprocesses. Nonetheless, their complementary structures (rigid versus flexible) might be relevant in the context of AD [84]. For comparison, 1,2,3,4,6-penta-O-galloyl-β-D-glucopyra-nose (PGG) alone inhibited: i) Aβ40/Aβ42 fibril formation; ii) Aβ aggregation at low concentrations (IC50 = 3μM); and iii) neurotoxic Aβ oligomer formation [214]. Interestingly, both TGG and PGG molecules share similar properties, e.g., hydrophobicity and flexibility [84, 204]. Consequently, both natural polyphenols corilagin and TGG have these physicochemical attributes, thus potential candidates for AD treatment.
Molecular and pharmacological properties of some natural polyphenols related to Aβ and tau aggregation/disaggregation and comparison with donepezil
Corilagin, β-1-O-Galloyl-3,6-(R)-hexahydroxydiphenoyl-D-Glucose; TGG, 1,3,6-Tri-O-Galloyl-β-D-Glucose; EGCG, epigallocatechin-3-gallate; aBinding with Aβ based on theoretical calculations (MD and HREX); MD, molecular dynamics; HREX, Hamiltonian Replica Exchange; b[269]; cPEP, Propyl endopeptidase enzyme [270]; d[218]; eDecreases the level of Aβ by inducing non-amyloidogenic cleavage of AβPP [271, 272]; fCorilagin hydrolyzed metabolites (EA, GA, and M3 (C20H18O14) [273, 274]); gPrimary metabolite in human liver (Trans-RES-3-O-glucuronide) [274, 275]; NA, not available; EA, ellagic acid; EGC, epigallo-catechin; PY, pyrogallol; EGC-M5, 5-(3′, 4′, 5′-trihydroxyphenyl)-γ-valerolactone and 5-(3′, 4′-dihydroxyphenyl)-γ-valerolactone (human major urinary metabolites of tea polyphenols); PBS, phosphate-buffered saline; DHC, dehydrocurcumin; THC, tetrahydrocurcumin; 4-OMGA, 4-O-methylgallic acid.
In addition, the effective concentrations (EC50) of TA on the formation, extension, and destabilization of preformed Aβ fibrils (fAβ40 and fAβ42), are in the order of less than 0.1μM [215], and the in vitro IC50 are 0.012 and 0.022μM for Aβ40 and Aβ42, respectively [216]. TA also inhibited the in vitro aggregation of tau peptide R3, with an IC50 of 3.5μM [217]; however, the inhibitor GA was less effective with an IC50 of 92μM [217]. Moreover, TA shows low toxicity, e.g., LD50 of 2260 mg/kg (oral rat) (Table 1).
EGCG, with an LD50 of 2170 mg/kg (mice), remodels large oligomers/fibrils into less toxic off-pathway assemblies [218]. Among fifteen secondary metabolites from plants, EGCG, myricetin, silibinin, and luteolin lowered the Aβ aggregation below 40% [219]. Moreover, EGCG inhibited the in vitro tau aggregation [220], in addition both EGCG and curcumin facilitated clearance of hyperphosphorylated tau [218].
There is a wide variety of mechanisms by which polyphenols show neuroprotective effects [221]. For instance, earlier studies have reported IC50 values of myricetin against of Aβ40 and Aβ42 aggregation within 0.2 to 0.9μM [222, 223]. Moreover, fisetin (3, 3′,4′,7-tetrahydroxyflavone) inhibited Aβ42 aggregation [224]. Their results suggest that the 3′,4′-dihydroxyl group, but not the 3- or 7-hydroxyl group, is critical for the inhibitory effect on the formation of Aβ42 fibrils [224]. The polyphenol isomers vescalagin and castalagin protect SH-SY5Y neuroblastoma cells by reducing the toxicity of Aβ42 oligomers [225]. Vescalagin totally inhibited aggregation at an Aβ42:polyphenol ratio of 1 : 1. Both vescalagin and castalagin decreased the amount of parallel β-sheets, and induced rearrangement of peptides into helix, anti-parallel β-sheets and other secondary structures [225]. Morin attenuates tau hyperphosphorylation by inhibiting GSK3β, implicated in AD pathogenesis, and showed the strongest inhibition in the GSK3β activity assay [226].
Potential strategies for effective anti-dementia drugs do not only focus on the inhibition of oligomer/fibril formation, but also the destabilization/disag-gregation of pre-aggregated Aβ- and/or tau-fibrils, or a combination of them. Unsurprisingly, numerous polyphenols have this ability as evidenced in the following section.
DESTABILIZATION AND DISAGGREGATION OF Aβ AND TAU
Fibrils destabilization serves the dual purpose of deformed fibrils becoming non-neurotoxic themselves and further inhibiting the formation of higher-order aggregates [227, 228]. Freyssin et al. [229] reported the significant role of polyphenols on aggregation and disaggregation of amyloid peptides, tau, and α-synuclein, in line with dispersive properties of polyphenols such as tannins [230].
Bieschke et al. [231] showed EGCG to inhibit Aβ42 fibrillogenesis, but also the ability to convert large, mature Aβ fibrils into smaller amorphous protein aggregates. Immuno-infrared sensor data are consistent with the degradation of Aβ fibrils induced by EGCG [232]. Gallic acid was shown to inhibit amyloid fibril formation (molar ratio Aβ:GA of 1:2) and to disaggregate preformed fibrils [233, 234]. Adding GA to the aggregated Aβ42 fibrils for 2 h clearly reduced the Aβ42 fibril particle size from predominantly 100 nm fibrils to ∼60 nm [234]. Fujiwara et al. [214] showed, in vitro and in vivo, that PGG disaggregated preformed Aβ fibrils. After incubation of 25μM of fresh fAβ40 and fAβ42 with 50μM TA, Ono et al. [215] showed that TA destabilized preformed Aβ fibrils. Liu et al. [233] proposed that the gallate group in GA (and related compounds) is the structural motif that prevents fibril formation.
Curcumin showed in vivo, and in vitro [227], the ability to inhibit Aβ aggregation and to disaggregate preformed Aβ fibrils. The activity on insulin-degrading enzyme toward Aβ42 in the presence of resveratrol results in a substantial increase in Aβ42 fragmentation compared to the control [236]. Sun et al. [237] demonstrated that tau alone gave long fibrils, while 50μM resveratrol induced the formation of short tau fibrils, and 200μM resveratrol led to smaller aggregates. Vion et al. [238] showed that both resveratrol and trans ɛ-viniferin, at 1μM, induced disaggregation of pre-aggregated Aβ42 peptide. Caillaud et al. [239] also reported that trans ɛ-viniferin reduces the size and density of amyloid deposits and decreases reactivity of astrocytes and microglia.
Khan et al. [240] provided a synopsis on the relationship between quercetin and cognitive performance in AD. Quercetin and rutin inhibit the formation of Aβ fibrils and disaggregated Aβ-fibrils [195]. Moreover, quercetin displays fibril destabilizing effects on preformed fibrillar Aβ, reversing Aβ-induced neurotoxicity [241]. Dihydroquercetin (Taxifolin), also disassemble Aβ in vitro, reduced levels of Aβ oligomers in vivo, and restored decreased cerebral blood flow as well as cerebrovascular reactivity in Tg-SwDI mice [61].
Dihydromyricetin or anthocyanins/anthocyans also reduces fibrils formation and disaggregates preformed fibrils [242–244]. Interestingly, only amorphous aggregates were formed when the molar ratio of Aβ40 to dihydromyricetin was 1:3 [243].
Although AD pathways and the related polyphenols are constantly investigated, the Gordian knot has not been untied yet. To solve this problem, we still need additional scientific knowledge and tools. For example, many questions related to NDs cannot be handled experimentally and could benefit by using advanced computational molecular methods.
COMPUTER SIMULATION OF PHENOLIC LIGANDS WITH AβAND TAU
After decades of experimental research where ma-jor advancements have been achieved, we still need to identify the real cause of AD. Consequently, computational methods, often named ‘in silico’ approaches, can accelerate the development, where scientists can generate experimental data while molecular theoretical calculations are processing. However, even after almost 20 years of amyloid aggregation MD simulations, there are still limitations, more specifically: force field, protein concentration, and simulation length challenges [245].
Molecular modeling (MM) encompasses all theoretical and computational methods used to model or mimic molecular behavior. MM development began in the early 1960s, although the underlying math originated much earlier. The common feature of all MM methods is the atomistic-level insights it provides. Numerous methods exist: e.g., molecular mechanics [246], semi-empirical [247, 248], density functional theory [249], molecular dynamics (MD) [250], ab-initio [251], as well as sampling methods such as replica exchange molecular dynamics (REMD) [252–254], and molecular docking [255–257]. The selection of methods hinges on whether a quick answer from classical methods (e.g., molecular mechanics) is desired, as opposed to highly accurate results from quantum mechanics-based calculations (e.g., ab-initio molecular orbital calculations) but time-consuming.
The therapeutic area where computational methods impact most is in the small-molecule drug discovery area, e.g., polyphenols. Virtually all small molecule drugs work by binding to proteins, enzy-mes, receptors, and ion channels, and sometimes DNA or RNA. The binding of small molecule, or ligand, to the targeted protein induces a biological response. Small molecule drugs are popular, because they are easy to produce, distribute and administer, and easily chemically modified to fine tune the effects of the drug. Modelling methods can guide chemists to synthesize molecules with improved binding to the protein, its activity. By improving the binding of the target molecule with the desired protein target and reducing binding with undesirable related protein targets, ligands can be made more selective, an attribute which reduces many side effects. Generally, pharmacokinetic properties can be predicted with an in-silico method, allowing researchers to avoid wasting resources on compounds that will either be too toxic, or have the wrong biological transport properties for a successful drug. In silico methods are a way of reducing the chemical search space, by helping to design experiments, and glean as much information as possible, from existing experimental data.
Given the experimental difficulties in terms of predictability from mouse to human in vitro/in vivo clinical experiments, computer modelling (in silico) has emerged as a reliable tool to elucidate brain chemistry. Due to their importance and size, both highly intrinsic disordered Aβ40 and Aβ42 proteins have been extensively studied. For example, using a coarse-grained force field coupled to Hamiltonian-temperature replica exchange MD simulations, the equilibrium structures of Aβ40, Aβ42, and Aβ40 (D23N) monomers [258], and dimers [259], were determined. They observed striking morphological differences [258], and they also showed that Aβ42 dimer has a higher propensity than Aβ40 dimer to form β-strands at the central hydrophobic core (residues 17–21) and C-terminal (residues 30–42), i.e., critical segments for Aβ oligomerization. Chiricotto et al. [260] applied the multi-scale Lattice Boltzmann Molecular Dynamics method (LBMD) to study the initial phases of the hydrophobic central core of amyloid peptide (Aβ16–22; KLVFFAE) aggregation.
Using MD simulations, Zhao et al. [261] studied the early adsorption and conformational change of Aβ oligomers from dimer to hexamer on three different self-assembled monolayers (SAMs) terminated with CH3, OH, and COOH groups. Combining with experimental results, all SAM model surfaces exhibited a seeding effect for Aβ polymerization [261].
Results from the virtual oligomerization inhibition were in excellent agreement with the experimental results, of the performance of six known Aβ aggregation inhibitors: brazilin, curcumin, EGCG, ELND005, resveratrol, and tacrine [262]. However, only EGCG is still active at phase III, while some of them were terminated due to the lack of efficacy. The EGCG ligand strongly interacted with most residues of Aβ16–22, notably F19 and F20. Interestingly, with EGCG the oligomerization time was significantly delayed: e.g., i) control (7, 20, and 48 ns), and EGCG-Aβ16–22 (16, 50, and 106 ns), for dimer, trimer, and tetramer, respectively [262]. Using a combination of in vitro experimental measurements and in silico methods, Acharya et al. [232] found that the most favorable GlideScore (–6.9; docking site #1) was obtained when EGCG binds inside the fibril, stabilized by π-stacking interactions with residue F19 [232]. Zhan et al. [263] showed both EGCG and EGC disruptive capacity on the newly cryo-EM resolved LS-shaped Aβ42 protofibrils by breaking the hydrogen bond between H6 and E11 through π–π interactions with residues H14/Y10 and hydrogen-bonding interactions with E11 [263].
The interactions between dihydromyricetin and Aβ40 trimer were mainly nonpolar, and where MD simulation showed the key Aβ40 interacting residues are V18, A21, and D23 [243]. Mechanistic insights from MD suggest that morin can penetrate into the Aβ42 hydrophobic core to disrupt the Asp23-Lys28 salt bridge and interfere with backbone hydrogen bonding [264]. Also, Lemkul et al. [264] reported that morin inhibits the early stages of Aβ peptide aggregation. Gargari and Barzegar [265] showed that flavonoids (myricetin, morin) exert dual and more effective functions against monomeric aggregation-prone state (fibrillogenesis suppression) and remodel the Aβ aggregation pathway (fibril destabilization).
Cyanidin-3-O-glucoside (Cy-3-G) inhibits Aβ40 fibrillogenesis, disrupts the β-sheet structure, disaggregates preformed fibrils, and reduces amyloid cytotoxicity [266], e.g., when the Aβ40:Cy-3G ratio was 1:3, the inhibitory effect on Aβ40 fibril formation was about 95%. Cy-3G mainly interacted with: N-terminal region, central hydrophobic cluster and β-sheet region II via hydrophobic and electrostatic interactions [266].
In vitro and in silico results from Guéroux et al. [267], showed the ability of some polyphenols from the procyanidin family, to specifically bind the proline-rich region of tau. Interestingly, the galloylated procyanidins ECG and EGCG exhibit a higher affinity with respect to the non-galloylated procyanidins [267]. Theoretical and experimental results indicated that tau interacts with TA by forming a hairpin structure, hence, a feature for inhibiting tau polymerization [217].
More than ever, computing capacity and MM methods are transforming our understanding of the brain chemistry and more specifically the underlying mechanisms of amyloid peptides aggregation and disaggregation involved in NDs, such as AD.
CONCLUSIONS
Physicochemical interactions between protein and natural ligands play a major role in numerous bioprocesses. Those have to be addressed to better understand the mechanisms and responses of natural polyphenolic ligands/protein complexation. The Aβ peptide is an endogenous compound involved in several NDs, such as AD which nucleates decades before a conclusive diagnostic. The amyloid-cascade hypothesis has provided the main framework for understanding the AD pathogenesis, where the basis is the pathogenic Aβ peptides aggregation into neuritic plaques. Yet, this hypothesis must integrate the contributions of other proteins such as tau, which is involved in more than 20 neurodegenerative diseases, including AD. Consequently, it is critical to intervene at the earliest stage (e.g., nucleation, induction period) of the disease and to select the effective chemistry with the right mechanisms. This complex task not only needs highly sophisticated medical experimental knowledge, but also theoretical molecular methods which helps to explore the underlying mechanisms. To achieve this, a multidisciplinary approach, combining experimental and theoretical methods, can be more effective to characterize the biochemical and biophysical interactions between proteins and phenolic ligands.
Many pathways have been investigated to mitigate and delay amyloid- and tau-opathies, e.g., targeting amyloidogenic pathways (e.g., BACE inhibitor), and non-amyloidogenic pathways (e.g., α-secretase activator) as well as γ-secretase inhibitors and modulators, inhibit the nucleation process of formation of oligomers/protofibrils/fibrils, alter/reduce oligomer/NFTs cytotoxicity, disaggregate pre-aggregated fibrils, etc. Nonetheless, one critical pathway is to prevent self-association at the early stages of AD (induction period), and below the Aβ and tau critical association concentrations.
The pathways described in this review are mainly related to amyloid- and tau-based surfaces (e.g., pro-tofibrils, fibrils, fibers, NFTs, etc.). However, our review suggests a sub-ensemble which includes surface-induced processes occurring on biosurfaces other than Aβ- and tau-fibrils, e.g., membranes, vesicles, blood vessels, RBCs, platelets, and likely relevant to NDs. For example, the adsorption of Aβ and tau on RBC biosurfaces, can either lead to confinement stabilization or to surface-induced aggregation, depending on the affinity of Aβ and tau, and Aβ/phenolic ligand complexes, to these surfaces. Bioprocesses occurring on surfaces other than Aβ- and tau-fibrils, can be analyzed under a new concept, referred as confinement stability (Fig. 2). For example, adsorption of Aβ/tau proteins, phenolic ligands and Aβ/phenolic ligand complexes on human RBC biosurface, results in stability by confinement, i.e., these proteins, ligands, and complexes are unable to aggregate due to temporary or permanent confinement. Overall, this sub-ensemble may also participate indirectly in mitigating neurodegenerative diseases, by preventing protein self-association, slowing down the aggregation process, and delaying the progression of AD.
In any case, it is imperative to develop strategic pathways that will work at the very early stages and below the CAC of Aβ and tau, i.e., even before they form dimers, trimers, and oligomers.
Footnotes
AcknowledGMENTS
The authors are grateful for the generous financial support by the R. Howard Foundation and La Fondation Famille Lemaire. Professors Mousseau’s, Ramassamy’s and van de Ven and Dr. Gaudreault’s work is also supported, in part, by grants from the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Research Chair Louise & André Charron on Alzheimer’s disease.