
Abstract
Background:
RE1-silencing transcription factor (REST) is known to silence target genes involved in synaptic plasticity and neuronal differentiation. Although previous studies implicate REST in neurodegenerative diseases, its function in the progression of Alzheimer’s disease (AD) is uncertain.
Objective:
The aim of the present work was to explore the mechanisms of AD and determine whether and how REST was involved in the pathogenesis of AD.
Methods:
We investigated the differentially expressed genes and key transcription factors in AD using bioinformatics analysis. In addition, we assessed the expression of REST under the influence of AD-related factors. Mice overexpressing REST were generated and analyzed by proteomics analysis. We used transmission electron microscopy, Golgi-cox staining, immunohistochemistry, and western blotting to examine the impact of REST on neurons.
Results:
The results of bioinformatics analysis revealed REST as a hub transcriptional regulator in AD. We demonstrate that the mRNA expression of REST was significantly upregulated compared with that in the control groups, not only in AD patients but also in APP/PS1 transgenic mice, lipopolysaccharide-induced neuroinflammatory mice, and oxidative and glutamate stressed neurons. Using proteomics analysis, we showed that the upregulation of REST increased the expression of genes involved in apoptotic and mitochondrial pathways. Long-term overexpression of REST significantly reduced the number of dendritic spines and increased the mitochondrial defect and apoptosis. Reduction of the cofilin phosphorylation may be one of its mechanisms, and cofilin activity could be affected through the P38 MAPK/CREB signaling pathway.
Conclusion:
These results demonstrated the possible mechanism underlying AD and indicated REST as a potential therapeutic target for AD.
INTRODUCTION
Alzheimer’s disease (AD) affects millions of people worldwide. Furthermore, the number of affected patients has been continuously increasing annually. The majority of AD cases are late-onset AD, which is characterized by the accumulation of amyloid-β (Aβ) plaques, hyperphosphorylation of tau, and loss of cholinergic neurons in the anterior basal brain that the cholinergic nuclei project fibers to the neocortex and the hippocampus [1]. The main clinical manifestations of AD are memory loss and cognitive impairment. Moreover, hippocampus is the major functional area responsible for learning and memory, and it is especially vulnerable to the injury associated with AD in the brain [2]. Because the underlying cause of AD remains unknown and AD treatments have not achieved good results, it is necessary to find out the hub genes and pathways in AD. In recent years, bioinformatics analysis and prediction has been well applied in various fields of life science research. Therefore, we tried to find out the cause of AD through bioinformatics analysis in this study.
RE1-silencing transcription factor (REST), also known as NRSF, is a neuron-specific gene silencing transcription factor that plays a strategic role in neurogenesis and neuronal differentiation. REST represses several neural genes involved in synaptogenesis and synaptic plasticity [3]. During neuron differentiation, the expression of REST gradually decreases, consequently increasing the expression of neuron-related genes [4, 5]. Downregulation of REST expression forms an important basis for ensuring correct differentiation of neurons, whereas its overexpression inhibits the expression of neuron-specific genes and causes axonal misorientation [6]. Thus, emerging evidence indicates that REST plays a significant function in neurodegenerative diseases, including AD, stroke, epilepsy, Parkinson’s disease, neuroinflammation, and spontaneous locomotion deficits [7–16]. Lu et al. created a REST overexpression mouse model using Drd2-Cre in adult mice and found that overexpression of REST results in increased REST and decreased DRD2 expression in the striatum and causes spontaneous locomotion deficits [7]. Upregulated expression of REST has been reported in cerebral ischemia, and RNAi-mediated depletion of REST promotes functional recovery after transient focal ischemia in a middle cerebral artery occlusion rat model [8]. Rocha and colleagues reported that the overexpression of REST occurs in an age-dependent manner in the hippocampus of individuals having seizures [9]. González-Castañeda et al. investigated the expression of choline acetyltransferase (ChAT) and REST in frontal, temporal, entorhinal, and parietal cortices of AD patient brains and revealed considerable increases in REST with concomitant decreases of ChAT in AD patient brains [10]. Orta-Salazar et al. observed significant decreases in the number of ChAT-immunoreactive cells in 11-month-old female 3xTg-AD transgenic mice compared with control mice. They also found an increased level of REST protein and a reduction of ChAT protein expression in the 3xTg-AD mice compared with their controls in the Meynert nucleus and of fibers in the frontal motor cortex and hippocampal CA1 area [11]. Buffolo et al. found that neuroinflammation induces synaptic scaling through IL-1β-mediated activation of REST. Their results demonstrated that REST upregulation represents a new pathogenic mechanism for the synaptic dysfunctions observed under neuroinflammatory conditions [12]. However, certain opposing groups argue that REST plays a protective role in AD and Parkinson’s disease. Lu and colleagues showed that REST potently protects the neurons from oxidative stress and Aβ-protein toxicity by increasing the expression of mRNA levels for catalase, superoxide dismutase 1, and FOXO1a and repressing genes that promote cell death and the pathology of AD [13]. Some studies had pointed to a protective function for increased REST expression in dopaminergic neurons or astrocytes through upregulating glutamate transporter EAAT2, tyrosine hydroxylase, or antiapoptotic genes [14–16].
In this study, we investigated the differentially expressed genes (DEGs) and key transcription factors in the hippocampus of AD using bioinformatics analysis and found REST as a key transcription factor. REST levels are increased during aging and can decrease neuronal excitability, however, whether neuronal increases of REST were protective or harmful remained a subject of debate. Specially, we still did not know the effects of REST if it increased to a lager degree or REST-overexpression lasted for a long time. Thus, we subsequently generated REST-overexpressing mice to evaluate the effects of REST by proteomics analysis, transmission electron microscopy (TEM), Golgi-cox staining, immunohistochemistry, and western blotting.
MATERIALS AND METHODS
Identification of DEGs and key transcription factors
All AD raw gene expression profiles were retrieved from the Gene Expression Omnibus [17] by searching for the keyword “Alzheimer’s disease”. Considering different platforms and chips, we used three independent validation datasets (GSE5281 [18], GSE48350 [19], and GSE28146 [20]), all of which were based on the GPL570 platform, as well as the Affymetrix Human Genome U133 Plus 2.0 Array for subsequent studies. A total of 90 human hippocampal samples (44 AD and 46 controls) were used for detailed analysis. Raw data were merged using the ComBat package in R language to remove batch effects [21]. The DEGs between patients with AD and healthy elders were analyzed using the limma package of the R language [22]. The threshold was defined as |log2 fold change (FC)| > 0.7 and p-value < 0.05. The string database (version: 11.0) [23] was used to study potential interactions between the DEGs and their targets. Based on the interactions, a protein-protein interaction (PPI) network was constructed using the Cytoscape software (version: 3.82) [24] and analyzed by the cytoHubba plugin [25]. WebGestalt online [26] was used for the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis (Top 20). Key transcription factors were analyzed using the FunRich software (version: 3.13) [27].
Mice
Male C57BL/6 mice (3-month-old), male APP/PS1 mice (10-month-old), and age-matched, wild-type male C57BL/6 mice (10-month-old) were used in this study. All experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals and approved by the Ethics Committee of the Shanghai Institute of Pharmaceutical Industry. Animal surgeries were performed under 10% chloral hydrate, and every effort was made to minimize animal suffering.
Lipopolysaccharide-induced inflammation in mice
To establish the inflammation model, 12 mice were randomly assigned to the lipopolysaccharide (LPS, 5 mg/kg; intraperitoneal injection) group (n = 6) and the control group (n = 6). The control group received an equal volume of phosphate-buffered saline (PBS) [28]. Twenty-four hours after LPS administration, the mice were sacrificed, and the hippocampus was dissociated.
Primary cultured cortical neurons
Briefly, dissociated cerebral cortex neuronal cultures were prepared from E16–19 mice. The neurons were digested with collagenase IV and DNase, gently dissociated, plated on six-well plates coated with polyethyleneimine, and cultured in the neurobasal medium supplemented with 1% B27 (Invitrogen) and 0.5% penicillin-streptomycin (Gibco) for 8–12 days in a 37°C, 5% CO2 humidified incubator. Before each experiment, the neurobasal medium was changed to serum-free medium without B27 supplement. Then, the cultured neurons were randomly divided into 3 groups (n = 6/group): 1) the control group (CTRL); 2) the H2O2 group, in which the cells were treated with 44μM H2O2 for 24 h; and 3) the glutamate group, in which the cells were subjected to 50 mM glutamate for 48 h.
REST-overexpressing mice
REST-overexpressing mice were generated using intravenous injection of AAV-PHP.eB targeting neurons, as previously described [29]. REST (NM_011263.2) was synthesized and cloned into the hSyn promoter-MCS-SV40 PolyA to generate REST-overexpressing AAV-PHP.eB vectors. AAVs were generated by transfecting HEK293T cells with REST-overexpressing AAV-PHP.eB vectors and purified by ultracentrifugation using iodixanol gradients. C57BL/6 mice were intravenously injected with 200μL of AAV-PHP.eB particles in PBS at a viral concentration of 1012 V.G/mL, whereas mice in the control group were intravenously injected with the empty AAV-PHP.eB vector.
Quantitative polymerase chain reaction
Total RNA was extracted from the brain tissues and primary cultured cortical neurons using the TRIzol reagent and reverse transcribed into cDNA using the Takara cDNA Synthesis Kit. Mouse REST (NM_011263.2) was evaluated using FastStart Universal SYBR Green Master (Roche) using GAPDH (NM_008084.2) mRNA as an internal control. Each sample was tested for 3 times. The relative changes in REST gene expression were calculated using the 2(-ΔΔCt) method. The primers designed for REST and GAPDH were synthesized by Sangon Biotech (Shanghai). The following primers were used: REST: 5’-TCATTCAGGTGAGAAGCCATTT-3’ (forward) and 5’-GGACAGGTGGGATGCTTAGATT-3’ (reverse); GAPDH: 5’-CCTCGTCCCGTAGACAAAATG-3’ (forward) and 5’-TGAGGTCAATGAAGGGGTCGT-3’ (reverse).
Western blotting
Mice were anesthetized with 10% chloral hydrate and the brain was dissected quickly. The prefrontal cortex (PFC) tissues were weighed and immediately frozen at –80°C until use for western blotting. To obtain the total protein, the tissue were lysed in RIPA buffer (Sangon Biotech) with protease and phosphatases inhibitor cocktail (Sangon Biotech). The samples were sonicated and centrifuged at 13,500 rpm for 15 min at 4°C, then the supernatants were quantified by BCA assay (Beyotime), mixed with 4×loading buffer (Sangon Biotech) and heated at 95°C for 10 min. The supernatant samples were separated on 10% SDS-PAGE gels and proteins electrophoretically transferred to PVDF membranes. Membranes were washed in TBS containing 0.1% Tween (TBST), blocked with 5% BSA in TBST buffer for 1 h, and incubated with antibodies against REST (1:1000; Proteintech), Cofilin (1:1000; ImmunoWay), p-Cofilin (1:1000; Affinity), p-P38 (1:1000; Cell Signaling Technology), P38 (1:1000; Cell Signaling Technology), p-CREB (1:1000; ImmunoWay), CREB (1:1000; ImmunoWay), GAPHD (1:1000; Servicebio), and β-actin (1:1000; ImmunoWay), followed by HRP-conjugated IgG secondary antibody (1:2000; Jackson). The bands were revealed with ECL kit (Servicebio) and quantified using ImageJ software [30].
Quantitative proteomics
A systematic quantitative label-free proteomic analysis was conducted (n = 3/group). Samples were prepared as described previously [31]. Each sample was tested three times and the averages from each set of triplicates were used to calculate p-value. Briefly, proteins extracted from the tissues were reduced and alkylated. Next, the lysate was digested using trypsin, and the digest was subsequently desalted using Sep-Pak C18 cartridges (Waters Corporation, Milford, MA) and dried under vacuum for further analysis. Peptides (200 ng) were reconstituted in buffer A (0.1% formic acid in water) and separated on a 25 cm analytical column with a packed emitter tip (75μm ID, 1.6 C18 beads, Aurora Series with CSI, IonOpticks, Australia). The column was maintained at 50°C in an integrated Toaster column oven. Samples were analyzed on a nanoElute liquid chromatography system (Bruker Daltonics) coupled to trapped ion mobility equipped Q TOF mass spectrometer (timsTOF Pro) with a 100 min gradient at a flow rate of 300 nL/min: Buffer B (0.1% formic acid in ACN) was increased from 2 to 22% in 90 min, 22 to 37% in 10 min, 37 to 80% in 10 min, and was sustained at 80% for 10 min. For mass spectrophotometry (MS) acquisition, the timsTOF Pro was operated in positive ion data-dependent acquisition Parallel Accumulation Serial Fragmentation (PASEF) mode. The capillary voltage was set to 1400 V. The MS and MS/MS spectra were acquired from 100 to 1,700 m/z, and an ion mobility range (1/K0) of 0.75 to 1.4 Vs/cm2 was used. The ramp and accumulation time were set to 100 ms to achieve a duty cycle close to 100%. The PASEF setting was one complete frame with 10 PASEF MS/MS frames, resulting in a cycle time of 1.1 s. Polygonal filtering was applied to exclude low m/z, singly charged ions for PASEF precursor selection. The “target value” of 10,000 was applied to a repeated schedule, and the intensity threshold was set at 2,500. The collision energy was ramped linearly as a function of mobility from 59 eV at 1/K0 = 1.6 Vs/cm2 to 20 eV at 1/K0 = 0.6 Vs/cm2. Data were submitted to Peaks Studio X Pro for protein identification and label-free quantitation. Data were searched against the SwissProt mouse database (17,032 sequences), with trypsin as the protease, allowing two missed cleavage sites; carbamidomethyl cysteine as a fixed modification; oxidized methionine and acetylation of N-terminal as a variable modification; and 15 ppm mass tolerance on precursor ions and 0.05 Da on fragment ions. The false discovery rate (FDR) was set at less than 1% for the peptide sequences. Each sample was tested for 3 times. The threshold of differentially expressed proteins was defined as FC > 1.5 [or < 0.67] and p-value < 0.05. Reactome pathway enrichment analysis was conducted using WebGestalt online tools and illustrated by ggplot2 package in R [32, 33].
Transmission electron microscopy
TEM was conducted to assess the mitochondrial number and size. A total of 12 mice, REST-overexpressing (n = 6) and control (n = 6) mice were used in this study. Briefly, brain tissues from the hippocampus were cut into 1 mm3 thick sections and impregnated in 1% OsO4 (Servicebio) for 2 h at room temperature. The cut sections were dehydrated, resin penetrated, embedded, and polymerized. The resin blocks were cut to 60 to 80 nm on the ultra-microtome (UC7, Leica), and the tissues were fished out onto the 150-mesh cuprum grids with formvar film and stained. The cuprum grids were placed on a grid board and dried overnight at room temperature. The cuprum grids were observed under TEM (HT7800, HITACHI), and images were acquired.
Immunohistochemistry
A total of 12 mice, REST-overexpressing (n = 6) and control (n = 6) mice were anesthetized with 10% chloral hydrate and perfused with PBS and 4% paraformaldehyde. The brains were then dissected and post-fixed in 4% paraformaldehyde for 24 h, dehydrated using a conventional alcohol gradient, embedded in paraffin, and cut into 4μm thick slices. Immunohistochemical staining was performed using the anti-cleaved caspase-3 antibody (1:300; Servicebio), anti-NeuN+antibody (1:500; Servicebio), and DAPI (4’,6-diamidino-2-phenylindole; 1:300; Servicebio).
Golgi-cox staining
Golgi-cox staining was conducted to observe the morphology of neurons and dendritic spines. A total of 12 mice, REST-overexpressing (n = 6) and control (n = 6) mice were used in this study. Briefly, brain tissues from the hippocampus were impregnated in the fixative (Servicebio) for more than 48 h, followed by Golgi-cox staining solution (Servicebio) for 14 days, and 80% glacial acetic acid overnight, and subsequently placed in 30% sucrose. The tissues were cut into 100μm slices using a freezing microtome (Cryostar NX50, Thermo), and panoramic images were obtained by panoramic multi-layer scanning with a digital slide scanner (Pannoramic 250, 3DHISTECH). 10 dendrites which was at least 100μm long and within a single plane of focus in each image of Golgi-cox staining were counted. The average number/10μm of dendritic spines were used to calculate p-value. Spine counts were made by two researchers who were blinded to the group of each image.
Statistics
Data are expressed as mean±standard error of the mean (SEM). Results were analyzed using Student’s unpaired t-test for two groups and one-way analysis of variance (ANOVA) for multiple comparisons. A p-value less than 0.05 (p < 0.05) was considered to be statistically significant.
RESULTS
REST is a key transcription factor in AD
Three gene expression profiles were used to screen for DEGs and key transcription factors. ComBat, an empirical Bayes method, was used to remove batch effects [21]. DEGs in the hippocampus of patients with AD and healthy elderly adults were screened using the limma package in R language, according to p < 0.05 and |log2 FC| > 0.7. A total of 228 DEGs were identified, including 220 downregulated genes in patients with AD (Fig. 1a). The top six downregulated genes were YWHAH, CHGB, CADPS, NSF, CDC42, and SNAP25, whereas the top six upregulated genes were CP, FBXO32, GOLIM4, STON2, ANKRD282, and NIPBL in the hippocampus of patients with AD. Next, we constructed a PPI network using Cytoscape software and obtained the maximum betweenness network analyzed by the cytoHubba plugin. In Fig. 1B, we showed the PPI network with maximum betweenness (TOP 15), including BDNF, CDC42, SNAP25, SYT1, and EGFR. These genes may be the most critical in the pathogenesis of AD. The KEGG and Reactome pathway enrichment analyses using WebGestalt revealed that the DEGs were related to several significant pathways, such as trafficking of AMPA receptors, glutamate neurotransmitter release cycle, synaptic vesicle cycle, glutamate binding activation of AMPA receptors and synaptic plasticity, neurotransmitter release cycle, gluconeogenesis, endocrine, and other factor-regulated calcium reabsorption, transmission across chemical synapses, epithelial cell signaling in Helicobacter pylori infection, GABAergic synapse, and L1CAM interactions. These signaling pathways are mainly associated with neurotransmitter transmission and synaptic receptors. Furthermore, the key transcription factors of the DEGs were analyzed using FunRich. These results suggest REST, DBX1, ONECUT1, MAFB, and POU2F1 as the key transcription factors. We hypothesized that the most significant transcription factor, REST (p < 0.001), could serve as a potential candidate target for AD therapy. Therefore, further experiments are required to confirm the finding.

Summary of differentially expressed genes in the hippocampus. A) DEGs between patients with AD and healthy elderly adults were visualized using a volcano plot with |log2(FC)| > 0.7 and -log10(P.Value) > 1.3. The volcano plot shows relatively high expression (red) and low expression (green) in the hippocampus of patients with AD. B) The key PPI network with maximum betweenness (TOP 15). The PPI network of DEGs was constructed by Cytoscape and analyzed by cytoHubba. C) KEGG and Reactome pathway enrichment of DEGs was analyzed by WebGestalt (FDR < 0.05; Top 20). D) Key transcription factor DEGs were analyzed by FunRich (Top 6). AD, Alzheimer’s disease; KEGG, Kyoto Encyclopedia of Genes and Genomes; DEGs, differentially expressed genes; PPI, protein-protein interaction; FDR, false discovery rate.
REST is upregulated by Aβ, lipopolysaccharide, hydrogen peroxide, and glutamate
Based on the expression profiling data of GSE5281 (10 AD and 13 controls), the mRNA levels of REST were analyzed using GEO2R normalized to GAPDH mRNA. The mRNA levels of REST in the hippocampus of patients with AD were significantly increased (p < 0.01) compared with the healthy elders (Fig. 2A). To assess the relationship between Aβ and REST, we measured the REST mRNA expression in the PFC of APP/PS1 and age-matched mice. We found that the mRNA expression of REST was significantly elevated in the APP/PS1 PFC (p < 0.05; Fig. 2B). Inflammation has previously been identified as a cause of AD [34, 35]. Among the neuroinflammation models for AD, a mouse model treated intraperitoneally (i.p.) with bacterial endotoxin, LPS, is commonly used to study neuroinflammation and neurodegeneration [36]. To determine whether neuroinflammation contributed to REST induction, we examined the expression of REST in the hippocampus of LPS-induced mice. Compared with the control group, the REST mRNA levels in the hippocampus of LPS-induced neuroinflammatory mice showed significantly enhanced REST expression at 24 h post LPS intraperitoneal injection (p < 0.01; Fig. 2C). To determine whether REST could be induced by oxidative stress and glutamate stress in primary cultured mouse cortical neurons, REST mRNA expression was triggered by treatment with hydrogen peroxide (H2O2) (44μM) for 24 h or glutamate (50 mM) for 48 h (p < 0.01; Fig. 2D). These results suggest that Aβ, LPS, H2O2, and glutamate-induced REST expression in the brain.
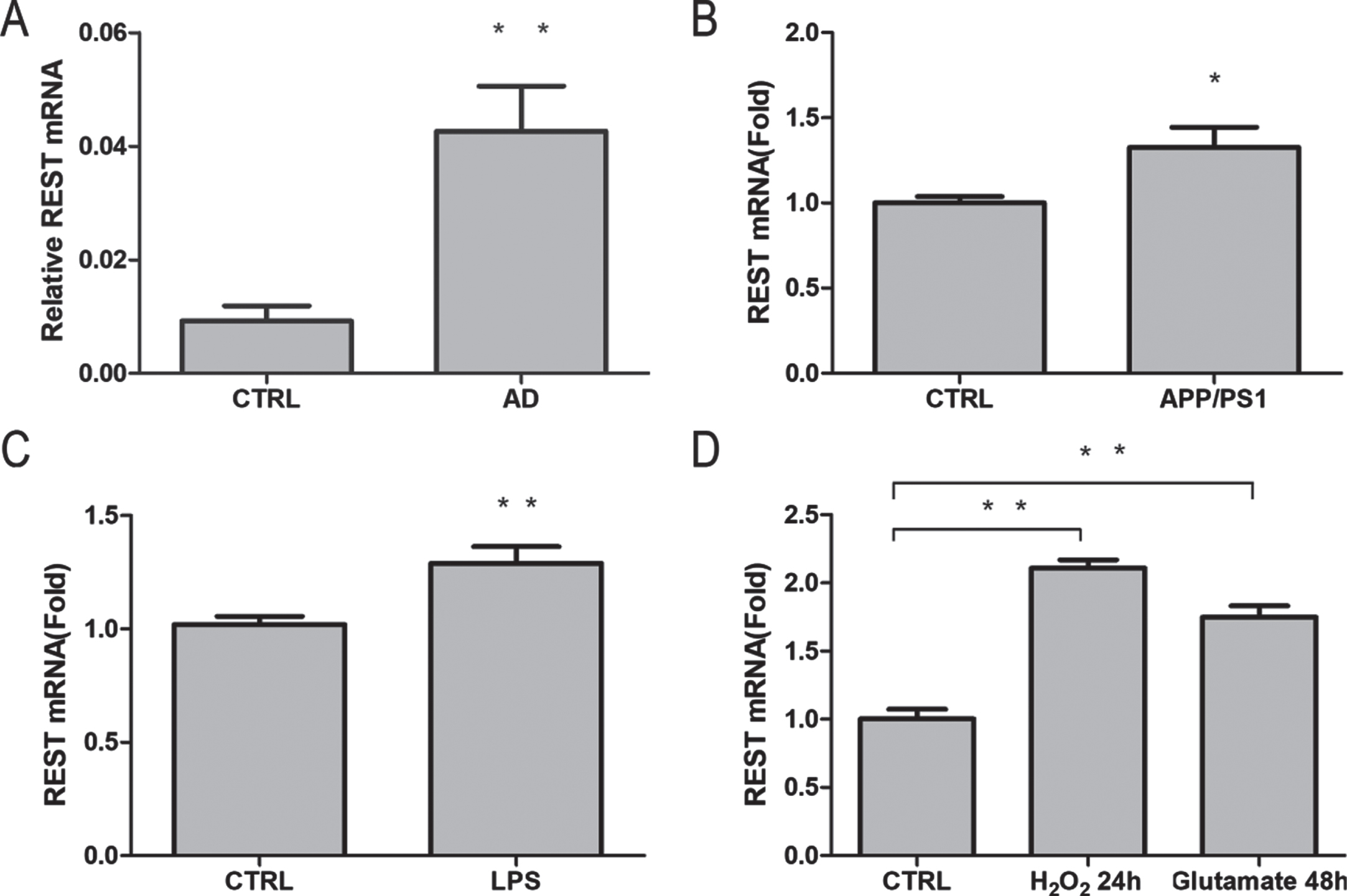
Induction of REST mRNA expression by Aβ, LPS, hydrogen peroxide, and glutamate. A) Relative REST mRNA levels derived from AD and aged hippocampus in GSE5281. B) Quantitative PCR analysis of REST mRNA levels in the PFC of APP/PS1 mice and aged control group (n = 6/group; p = 0.027). C) REST mRNA levels increased in mice treated with LPS (5 mg/kg) compared with that in control mice (n = 6/group; p = 0.008). D) REST mRNA levels increased in primary cultured cortical neurons treated with either H2O2 (44μM) for 24 h (n = 6; p < 0.01) or glutamate (50 mM) for 48 h (n = 6; p < 0.01). Values are expressed as mean±standard error of mean (SEM). *p < 0.05, **p < 0.01 versus control (CTRL).
REST upregulation increases the expression of proteins involved in apoptosis and mitochondria pathways
To assess the function of REST in pathophysiological mechanisms, we produced REST-overexpressing mice using AAV-PHP.eB intravenous injection targeting only the neurons. Compared with the control group, REST-overexpressing mice exhibited significantly increased protein (n = 4/groups; p < 0.05; Fig. 3A, B) and mRNA expression (n = 6/groups; p < 0.01; Fig. 3C) of REST at three months after virus injection. A shotgun proteomics label-free workflow was conducted using liquid chromatography coupled with a timsTOF Pro mass spectrometer. A total of 5,288 proteins were identified using this workflow, and 312 differentially expressed proteins in the hippocampus were shown to be REST-related proteins (p < 0.05 and FC > 1.5 [or < 0.67]), including 178 downregulated proteins and 134 upregulated proteins in REST-overexpressing mice. The top ten downregulated proteins were Aquaporin-1, Chloride intracellular channel protein 6, Claudin-10, Glycine cleavage system H protein mitochondrial, CDGSH iron-sulfur domain-containing protein 3 mitochondrial, TBC1 domain family member 15, Potassium voltage-gated channel subfamily D member 1, Lysosomal acid phosphatase, Potassium/sodium hyperpolarization-activated cyclic nucleotide-gated channel 3, and Integrin alpha-M. The results were visualized using volcano plots (Fig. 3D).

Regulation of REST target genes in the hippocampus of mice. A) Immunoblotting of REST in the PFC of REST-overexpressing and control mice. B) Quantification of REST expression. The expression of REST was significantly increased in REST-overexpressing mice relative to control mice (n = 4/group; p < 0.05). C) The mRNA expression of REST was determined by quantitative PCR in the PFC of REST-overexpressing and control mice (n = 6/group; p < 0.05). Values are expressed as mean±standard error of mean (SEM). *p < 0.05, **p < 0.01 versus the control group (CTRL). D) Proteomics analysis of differentially expressed proteins in the hippocampus of REST-overexpressing and control mice (n = 3/group; p < 0.05 and FC > 1.5 [or < 0.67]). The results were visualized using a volcano plot that showed relatively high expression (red) and low expression (green) in REST-overexpressing mice. E, F) Reactome pathway enrichment analyses of differentially expressed proteins were conducted using WebGestalt online tools (Top 10).
Pathway enrichment provided an overview of the identified differentially expressed proteins. Compared with the age-matched control group, the REST overexpression decreased the expression of proteins involved in vesicle-mediated transport, transcriptional activation of mitochondrial biogenesis, regulation of insulin-like growth factor transport, neutrophil degranulation, presynaptic depolarization and calcium channel opening, GRB2:SOS provides linkage to MAPK signaling for Integrins, role of LAT2/NTAL/LAB on calcium mobilization, and Fc epsilon receptor (FCERI) signaling pathways at three months after virus injection (Fig. 3E). Conversely, REST overexpression increased the expression of proteins enriched in apoptosis-induced DNA damage and programmed cell death pathways (Fig. 3F). These results indicate that REST upregulation suppressed the expression of target genes and induced cell apoptosis and mitochondrial defect.
Long-term overexpression of REST increases mitochondrial defect and cell apoptosis
As overexpression of REST increased the levels of proteins involved in apoptosis and mitochondrial pathways, we next examined the effects of REST overexpression on mitochondrial defect and apoptosis in the hippocampus. To determine the effects of REST on mitochondrial number and length, we used TEM to study the hippocampal CA1 region in REST-overexpressing and control mice (n = 6/groups). As shown in Fig. 4A and B, the number of mitochondria in the CA1 region of REST-overexpressing mice significantly reduced relative to that in control mice (p < 0.05). Furthermore, the mitochondrial length was significantly decreased in the hippocampal tissues of REST-overexpressing mice (p < 0.01) relative to that in the control mice.
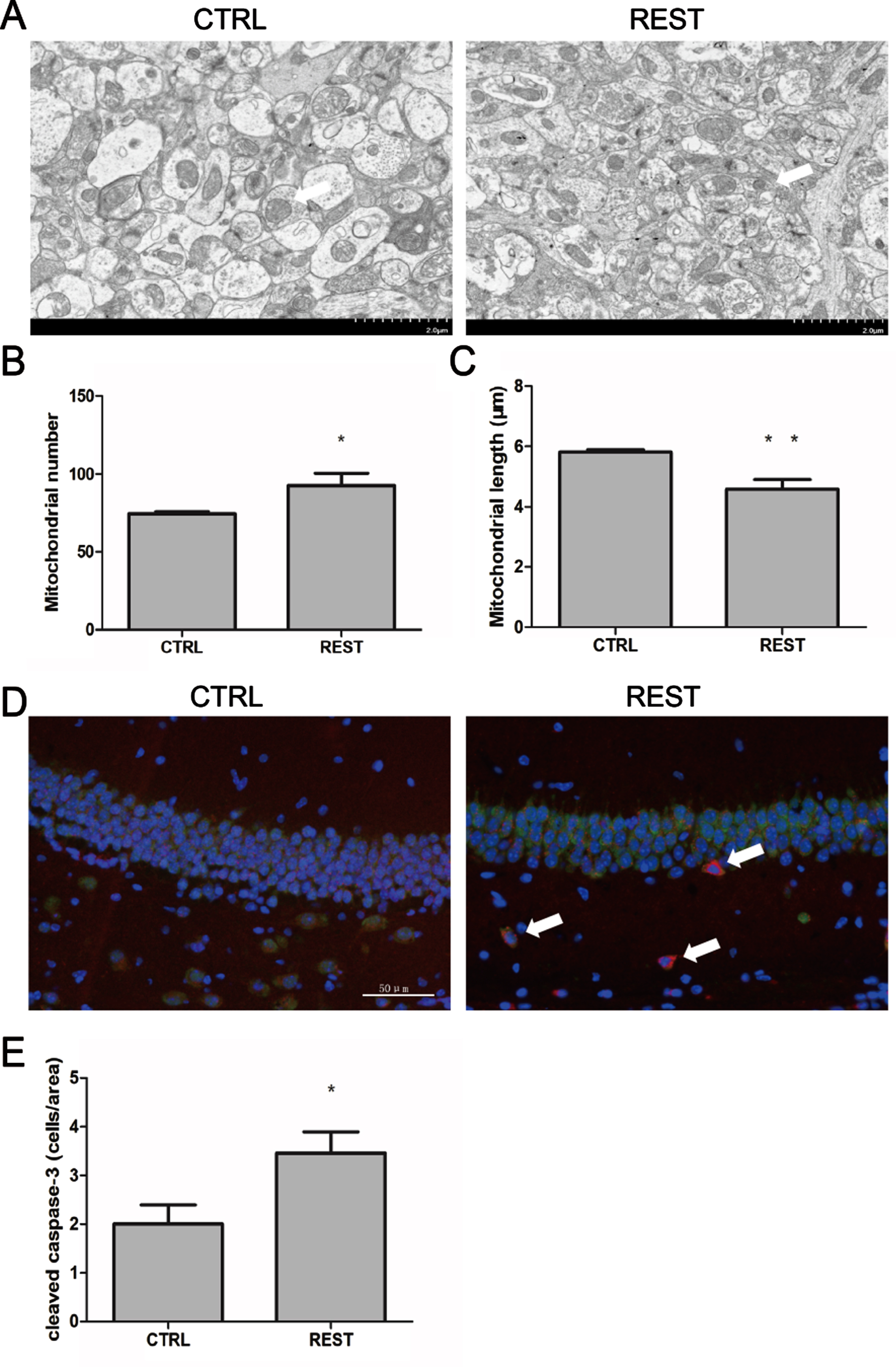
Overexpression of REST increased mitochondrial damage and apoptosis. A) Representative mitochondrial images in the hippocampus of REST-overexpressing and control mice. Scale bar = 2μm. B, C) Mitochondrial number and length in the hippocampus of REST-overexpressing and control mice. A significant increase in the number (p < 0.05) and decrease in the length (p < 0.01) of mitochondria was found in the hippocampus of REST-overexpressing mice relative to age-matched control mice (n = 6/group). D) Immunofluorescence labeling using the apoptosis marker cleaved caspase-3 (red), the neuronal marker NeuN+(green) and DNA (DAPI, blue) in the hippocampal CA1 region. Arrows indicate cleaved caspase-3-positive cells. Scale bar = 50μm. E) Quantitative immunofluorescence analysis of apoptotic proteins in REST-overexpressing and control mice (n = 6/group). The levels of cleaved caspase-3-positive neural cells significantly increased in the hippocampus of REST-overexpressing mice (p < 0.05). Values are expressed as mean±standard error of mean (SEM). *p < 0.05, **p < 0.01 versus sham control (CTRL) by Student’s unpaired t-test.
To determine the effects of REST on apoptosis, we performed immunofluorescence analysis of cleaved caspase-3 in the hippocampus in REST-overexpressing and age-matched control mice. As shown in Fig. 4C and D, immunohistochemical analyses showed increased cleaved caspase-3-positive neural cells (red, marker for apoptosis; p < 0.05) in the hippocampus of REST-overexpressing mice.
Long-term overexpression of REST decreases the number of dendritic spines and modulates cofilin phosphorylation
Because REST is directly associated with the repression of synaptic plasticity [3, 4], we examined the effects of its overexpression on neuronal morphology. We quantified dendritic spines using Golgi-cox staining in the hippocampus of REST-overexpressing and age-matched control mice. As shown in Fig. 5A and B, we found a significantly decreased number of dendritic spines in REST-overexpressing mice (p < 0.05) compared with that in control mice, indicating that the overexpression of REST reduced dendritic spines in the hippocampus.

Overexpression of REST decreased dendritic spines and cofilin phosphorylation. A) Representative Golgi-cox staining in the hippocampus CA1 region of REST-overexpressing and control mice. B) Quantification of spine density in REST-overexpressing and control mice. Significantly reduced dendritic spines were found in REST-overexpressing mice (n = 6/group; p < 0.05) than in control mice. C) Immunoblotting staining of pCofilin and Cofilin. D) Quantification of cofilin phosphorylation. The levels of pCofilin/Cofilin were significantly decreased in REST-overexpressing mice relative to those in control mice (n = 4/group; p < 0.01). Values are expressed as mean±standard error of mean (SEM). *p < 0.05, **p < 0.01 versus sham control (CTRL) by Student’s unpaired t-test.
To identify the underlying mechanisms, we performed an unbiased western blotting screen and subsequently found that cofilin phosphorylation was significantly reduced in REST-overexpressing mice relative to control mice (p < 0.01, Fig. 5C, D). The levels of cofilin phosphorylation in human AD brains were reduced relative to those in non-AD individuals [37, 38]. Moreover, cofilin has been shown to affect actin dynamics, resulting in damage to synaptic integrity, mitochondrial translocation and mitochondria-mediated apoptosis [39], suggesting the implication of REST in AD.
Reduced cofilin phosphorylation is associated with MAPK/CREB pathway in REST-overexpressing mice
Cofilin is activated by REST, but how cofilin phosphorylation was reduced in REST-overexpressing mice remains unknown. Previous studies have shown that cofilin acted as a downstream molecule of P38 mitogen-activated protein kinase (P38 MAPK) and cyclic adenosine monophosphate (cAMP) response element binding (CREB) pathway [40–42]. It was also found that protein activation of CREB was mediated by P38 MAPK signaling pathway [43]. Brain-derived neurotrophic factor (BDNF), mediated by CREB, affects Rho GTPases and downstream target proteins, including cofilin [41, 42]. Therefore, the expression of p-P38 and p-CREB, normalized to their total levels, was further examined in this study using western blotting (n = 6/group). We found that the levels of p-P38 and p-CREB in prefrontal cortexes of REST-overexpressing mice significantly decreased as compared with control (Fig. 6A, B), implying that P38 MAPK/CREB signaling regulated cofilin phosphorylation.
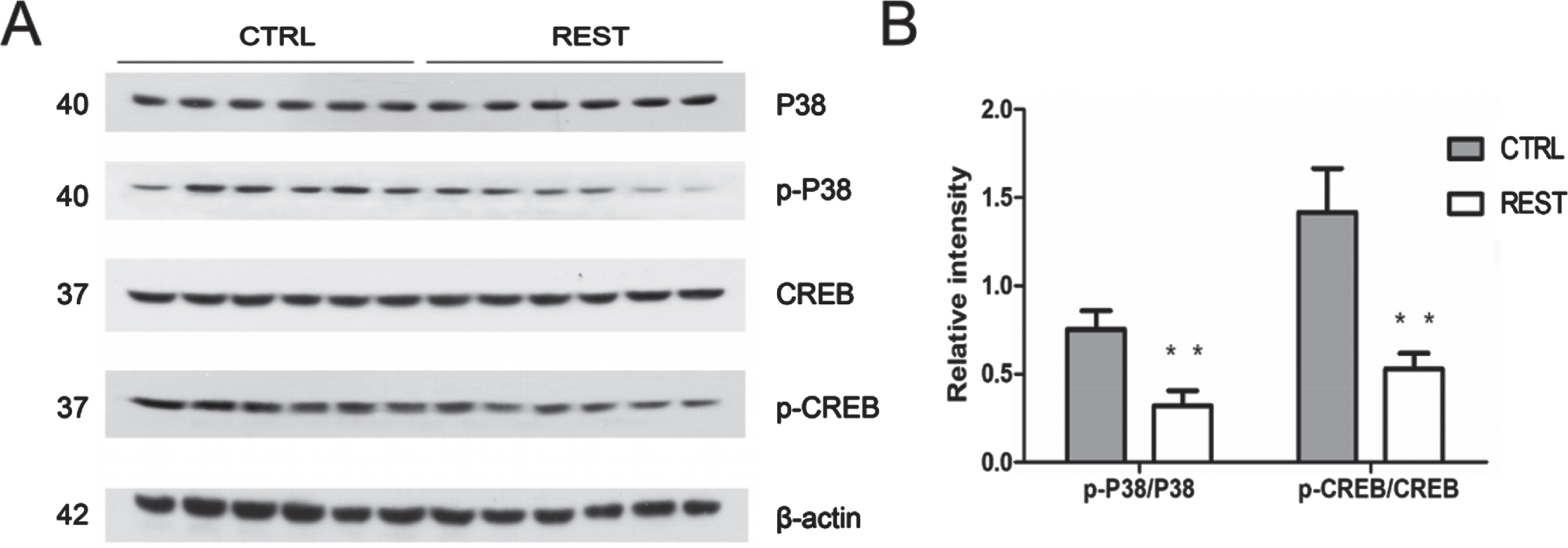
Reduced Cofilin phosphorylation was associated with P38/CREB pathway in REST-overexpressing mice. A) Immunoblotting staining of p-P38 and p-CREB. B) Quantification of p-P38 and p-CREB. The expression of p-P38 and p-CREB, normalized to the total levels, was significantly decreased in REST-overexpressing mice relative to that in control mice (n = 6/group, p < 0.01). Values are expressed as mean±standard error of mean (SEM). *p < 0.05, **p < 0.01 versus sham control (CTRL) by Student’s unpaired t-test.
DISCUSSION
We showed that REST plays a critical role in AD. We used bioinformatics analysis and found that neurotransmitter transmission and synaptic receptors signaling pathways were significantly downregulated in the brain of patients with AD, indicating the reduced activity of hippocampal neurons in these patients. PPI network analysis revealed that BDNF, Cdc42, SNAP25, SYT1, and EGFR were key genes in AD brains. Furthermore, REST was identified as the key transcription factor by transcription factor analysis. Lu and colleagues showed that REST potently protects neurons from oxidative stress and amyloid-β protein toxicity, and it is highly depleted from the nucleus in AD [13]. However, REST is a transcription factor that suppresses the expression of neuro-specific genes in neurons, and we found that REST-repression genes significantly decreased in AD, suggesting elevated expression of REST in AD. Prolonged upregulation of REST in neurons might be harmful and contribute to neural circuit injury because of the downregulated expression of REST target genes.
Although little is known about the pathogenetic mechanisms of AD, emerging evidence indicates that Aβ deposition, neuroinflammation, oxidative stress, and glutamate toxicity are closely related to AD [1]. Our results demonstrate that REST is upregulated after Aβ deposition, neuroinflammation, oxidative stress, and glutamate toxicity. However, the reason for the upregulation of REST by these possible sources remains unclear, and further research is warranted. Subsequently, we constructed REST-overexpressing mice, and after three months of feeding, we examined the effects of long-term overexpression of REST. Proteomics analysis revealed that vesicle-mediated transport and presynaptic depolarization proteins were downregulated, whereas cell apoptosis and mitochondrial translation termination proteins were upregulated. Thus, we examined the effects of REST on dendritic spines, cell apoptosis, and mitochondrial defect. Finally, we showed that long-term overexpression of REST not only decreased the number of spines but also increased the mitochondrial defect and apoptosis in the hippocampus. Impaired mitochondrial dynamics (increased fission and decreased fusion) are observed in neurons of AD patients and APP transgenic mice [44]. A significant increase in the number and decrease in the length of mitochondria was found in the hippocampus of REST-overexpressing mice relative to age-matched control mice, indicating defective mitochondrial function.
To identify the underlying mechanisms of REST effects, we performed an unbiased western blotting screen. We propose an abnormal association of REST with pCofilin/Cofilin to induce defective spine biogenesis in AD. The REST-overexpressing mice showed a specific decrease in the phosphorylation (inactivation) of cofilin. In the brains of patients with AD, the level of cofilin phosphorylation, normalized to that of total cofilin, was strongly reduced by about 38% compared with that in healthy age-matched control brains [45]. Moreover, cofilin has been reported to regulate both mitochondrial morphology and apoptosis. Activated cofilin is critical for mitochondrial dysfunction via direct translocation to the mitochondria, inducing cell death via mitochondria-mediated apoptosis [46]. Mitochondrial dysfunction resulting from the translocation of active cofilin eventually produces high reactive oxygen species levels that cause the release of cytochrome C, promoting neuronal cell apoptosis [47].
Cofilin could be phosphorylated by its upstream kinase LIM kinase 1/2, which is activated by p21-activated kinase (PAK) [48]. PAK is phosphorylated by small GTPases of the Rho family, such as Rac1/Cdc42 [49]. In addition, cofilin is activated through dephosphorylation by phosphatases, such as protein phosphatase 2A (PP2A) [50, 51]. Further studies revealed that the inactivation/phosphorylation of PP2A was mediated by activated P38 MAPK. Co-immunoprecipitation showed a direct interaction of PP2A with P38 MAPK, so cofilin acted as a downstream molecule of P38 MAPK pathway [40]. Furthermore, CREB and BDNF have been reported to affect Rho GTPases and downstream target proteins, including cofilin [41, 42]. We found that the expression of p-P38 and p-CREB normalized to their total levels in REST-overexpressing mice significantly decreased as compared with control mice. Our results showed that P38 MAPK and CREB could serve as critical components of the cofilin signaling network. Thus, we conclude cofilin as the key regulator of actin skeleton, and, at least in part, cofilin activity could be affected through the P38 MAPK/CREB signaling pathway, resulting in dendritic atrophy and spine loss, and ultimately mitochondrial defect and apoptosis. However, further studies were needed to clarify whether direct inhibition of cofilin activity could restore REST damage in REST-overexpressing mice.
The expression of REST increases in the presence of some external stimulus, and REST could decrease neuronal excitability and protect neurons from damage [13]. However, it would be harmful if the expression of REST increases to a lager degree or REST-overexpression lasts for a long time. For example, the expression of REST is continuously upregulated in epilepsy and ischemia patients. Encouragingly, several compounds such as valproic acid and X5050 that target REST have been shown to have clinically therapeutic effects on seizures or Niemann-Pick type C disease [3]. Our findings strongly indicate that REST is not only a therapeutic target for epilepsy and cerebral ischemia, but it is also a novel potent therapeutic target against AD and other neurodegeneration. More importantly, our findings suggest for the first time that high REST levels are related to low cofilin phosphorylation levels. These results demonstrated the possible mechanism of AD and indicated that cofilin could also be a potential therapeutic target for AD.
Conclusion
Our study shows that overexpression of REST is a promoter of brain damage and consistently upregulation of REST promotes neurodegeneration by modulating several factors. This study provides a novel explanation of the mechanism underlying AD. Thus, REST and cofilin could be potential therapeutic targets for AD.