
Abstract
Alzheimer’s disease (AD) is the most common form of dementia with global burden projected to triple by 2050. It incurs significant biopsychosocial burden worldwide with limited treatment options. Aducanumab is the first monoclonal antibody recently approved by the US-FDA for mild AD through the accelerated approval pathway. It is the first molecule to be approved for AD since 2003 and carries with it a therapeutic promise for the future. As the definition of AD has evolved from a pathological entity to a Clinico-biological construct over the years, the amyloid-β (Aβ) pathway has been increasingly implicated in its pathogenesis. The approval of Aducanumab is based on reduction of the Aβ load in the brain, which forms a surrogate marker for this pathway. The research populace has, however, been globally divided by skepticism and hope regarding this approval. Failure to meet clinical endpoints in the trials, alleged transparency issues, cost-effectiveness, potential adverse effects, need for regular monitoring, and critique of ‘amyloid cascade hypothesis’ itself are the main caveats concerning the antibody. With this controversy in background, this paper critically looks at antibody research in AD therapeutics, evidence, and evolution of Aducanumab as a drug and the potential clinical implications of its use in future. While the efficacy of this monoclonal antibody in AD stands as a test of time, based on the growing evidence it is vital to rethink and explore alternate pathways of pathogenesis (oxidative stress, neuroinflammation, cholesterol metabolism, vascular factors, etc.) as possible therapeutic targets that may help elucidate the enigma of this complex yet progressive and debilitating neurodegenerative disorder.
ALZHEIMER’S DISEASE: GLOBAL BURDEN
Alzheimer’s disease (AD) is the most common cause of dementia worldwide [1]. Almost 60–80%of all dementia cases are attributable to AD [2]. As per the World Health Organization (WHO) estimate in 2020, around 50 million people in the world are living with dementia and the number increases by 10 million every year [1]. The prevalence of dementia is expected to triple worldwide by 2050 with higher estimates if a ‘biological definition’ is considered over a clinical criteria [3]. The medical, social, psychological, and economic burden due to AD is well established in the literature [1, 4]. Despite the considerable disease burden, the treatment options of AD are still limited. The drugs hitherto approved for AD, namely the acetylcholinesterase inhibitors (donepezil, galantamine, rivastigmine) and an NMDA-receptor antagonist (memantine), are modestly effective at best. These drugs are targeted at the neurotransmitter derangements in AD, that are, acetylcholine and glutamate respectively. However, these are not curative or disease-modifying treatments, and rather focus on symptomatic management and also help in the behavioral and psychological symptoms of dementia. However, benefits may not be observed in a significant number of patients [5]. Non-pharmacological modalities of treatment which include caregiver education, environmental modification, and behavioral techniques form an important component in AD management. With the unsatisfactory pharmacological management options, research has been dedicated in the last decade to find newer molecules targeting different pathologic pathways responsible for AD. Aducanumab, a disease-modifying monoclonal antibody, recently approved by the United States Food and Drug Administration (USFDA), is one such molecule. This is the first monoclonal antibody approved for AD since 2003 [6]. This paper looks at the evolution of this novel molecule as a therapeutic modality for AD, the underlying evidence and possible implications for future. We provide an open disclaimer that this discussion has received no funding, the authors have no conflict of interest, and we do not approve or disapprove this drug in AD therapeutics. The authors intend to put forward a balanced yet critical discussion of Aducanumab’s trajectory to the final FDA approval, the evidence backing it up, and potential scientific implications which may anchor any future academic and research-based discourse on this monoclonal antibody as well as AD drug development. Throughout the discussion, based on the NIA-AA 2018 criteria we will consider AD continuum and not just AD dementia.
TARGETING Aβ AMYLOID AS A THERAPEUTIC MODALITY: GENESIS OF ADUCANUMAB
Over the years, from the NINDS-AIRDA criteria to the NIA-AA (2011) criteria and finally the NIA-AA (2018) ATN classification, AD has evolved as a postmortem pathological diagnosis to a clinico-biological entity that can be diagnosed during the lifetime with the help of specific markers. Various structural, functional, and neuropathological pathways have been elucidated in the pathogenesis of AD. Among the various theories regarding the pathophysiology of AD, amyloid cascade is one of the time-lasting ones, attracting the attention of the researchers and drug developers for years. First proposed by Hardy and Higgins in 1992 [7], this hypothesis posits that the neurodegeneration in AD is caused by an abnormal accumulation of amyloid-β (Aβ) plaques in the brain cortex. A detailed review of the amyloid cascade hypothesis is beyond the scope of this review, and the readers are encouraged to go through Ricciarelli and Fedele [8] for a comprehensive discussion on the same. Here a brief overview has been provided related to the mechanism of action of Aducanumab. Figure 1 provides a simplified depiction of the amyloid cascade.

The amyloid cascade hypothesis. Early onset AD (dependent on genetic risk factors) and late onset AD (dependent on presence of at least one APOE4 allele and other unknown factors) arise from dysfunctional APP metabolism which leads to Aβ oligomerization and senile plaque formation. Aβ oligomerization forms the triggering factor for the cascade leading to tau hyperphosphorylation, formation and accumulation of neurofibrillary tangles that eventually result in inflammation, microglial activation, synapse loss and neuronal death. With increasing amyloid burden, this cascade progresses further with a vicious cycle of downstream events and progressive worsening of dementia. This process happens maximally in the medial temporal structures (including hippocampus), which are involved in learning and memory. (Adapted from Morris et al., 2014 with changes; https://creativecommons.org/licenses/by/4.0/)
Aβ plaques are made up of Aβ peptides, that are 39–42 amino acid residue peptides generated by the proteolysis of transmembrane amyloid-β protein precursor (AβPP) by cleaving enzymes, namely β-secretase and γ-secretase. Aβ142 isoform aggregates faster, gets deposited, and forms the β-pleated sheet in the plaques. This deposition and plaque formation give rise to a molecular cascade of microglial activation and inflammatory response [9]. Microglial activation and consequent neuroinflammation is a well-known mechanism underlying the amyloid cascade that has gained recent importance [3]. The enzyme α-secretase, on the other hand, cleaves AβPP within the Aβ domain and does not release insoluble Aβ leading to accumulation [10]. The plaques are also known to influence the abnormal phosphorylation of microfibrillary tau-proteins, thereby increasing the amount of hyperphosphorylated tau (p-tau), also implicated in AD pathogenesis [11]. The final pathways in development in AD are endocytosis, cholesterol metabolism, immune/inflammatory mechanisms, Aβ metabolism, and angiogenesis [12]. Biomarkers of AD include cortical amyloid PET-binding and reduced cerebrospinal fluid (CSF) Aβ42/Aβ40 ratio among others [13]. It has been postulated that the Aβ changes in the brain may precede the clinical cognitive decline for many years. Thus, AD has been considered by the NIA-AA research framework to be on a clinico-pathological continuum with the pathological changes being the mainstay of AD categorization, and presentation ranging from preclinical to clinically severe dementia [13, 14]. The central anchor to the amyloid cascade hypothesis is that a “crucial threshold” of Aβ deposition at selective sites of the brain (preferably the medial temporal cortex, entorhinal cortex, hippocampus) is necessary for the subsequent downstream pathogenic processes of AD. Though debated and considered oversimplistic, this hypothesis has been further supported by the autosomal mutations (PSEN 1,2) causing AD, which involve proteins forming the catalytic active site of γ-secretase [15]. Similar evidence is also provided by the APOE mutations [16]. However, though the amyloid cascade hypothesis incorporates several key pathogenic points in the process of AD and provides a practical explanation for the phenotypes, it falls short of accounting for the Aβ interaction with tau, which is a central factor for the downstream neuronal damage in AD [17]. Besides, while the pattern of tau pathology in AD is highly stable, the Aβ pathology is much more heterogenous [18]. In their landmark discussion on Aβ therapeutics in AD, Karran and colleagues (2011) highlight that though Aβ deposition may be crucial in initiating the disease process, there is no conclusive evidence to support that the continuation of this pathological process is driven by further Aβ deposition [17]. This is of vital scientific concern, as in that case an anti-amyloid drug may not be theoretically effective in AD once the disease process has started off. Several studies have also shown that rate and severity of Aβ deposition do not correlate with the extent of neurofibrillary tangles (NFT), neuronal loss, and dementia. This leads to two key questions: at what stage in the AD pathogenesis would an Aβ-targeted therapeutic intervention show maximum efficacy and by how much should Aβ production be lowered or Aβ clearance enhanced to elicit a significant disease-modifying effect in AD?
There have been several drug development attempts targeting Aβ with no considerable success [19]. The failure of Semagacestat, a γ-secretase inhibitor, as a therapeutic modality has led many to question the role of Aβ deposition and amyloid cascade in AD [20, 21]. The main anti-amyloid therapeutic strategies currently used are reducing Aβ production, promoting Aβ clearance, and preventing Aβ aggregation. The amount, rate, site, reversibility, and temporality of Aβ deposition in the brain are the key considerations in all these strategies. Subsequently an immunotherapeutic approach was adopted while developing monoclonal antibodies targeting Aβ epitopes. Passive immunotherapy using anti-Aβ antibodies have been the cornerstone for AD treatment in the recent years. A number of such monoclonal antibodies have been studied, with some partially halted or discontinued due to either toxicity or failure to reach primary endpoint targets [22–24]. There have been several related challenges in anti-amyloid therapeutics, an important one being the “treatment versus prevention” dilemma which is discussed subsequently.
These antibodies also have varied binding properties and Aβ selectivity profiles. Crenezumab and Bapineuzumab usually bind to both monomeric and aggregated Aβ, Gantenerumab binds to soluble and insoluble aggregates, while Ponezumab and Solanezumab are specific to the soluble monomers. It has been postulated that their heterogenous efficacy profile is attributed to this differential binding properties [25]. Aducanumab, or BIIB037, was also developed as a recombinant human IgG1 antibody selectively aimed at the aggregated Aβ, both soluble oligomers and insoluble fibrils [26, 27].
ANTIBODY TREATMENT CHALLENGES IN ALZHEIMER’S DISEASE
Several monoclonal antibodies have been tried in AD. These molecules once referred to as “anti-AD drugs” based on the amyloid cascade have been critiqued heavily [28–30]. Past trials suggested that passive immunotherapy targeting the Aβ oligomers are effective in learning and memory improvement [31]. Drugs like Bapineuzumab, Solanezumab, Ganterenumab, and Crenezumab have been tried, targeting Aβ. However, the Alzheimer’s Disease Neuroimaging Initiative (ADNI) and Dominantly Inherited Alzheimer Network Trial Unit (DIAN-TU) trials with these drugs largely failed over time in producing any clinically significant outcome [32]. Bapineuzumab, despite causing significant reduction in CSF p-tau, was not superior to placebo in terms of cognitive outcomes, and had more serious adverse effects (cerebral vasogenic edema) [33]. Crenezumab failed two phase III trials in meeting the primary endpoint. Amyloid-related imaging abnormalities (ARIA) were observed as a side effect in this, too [34]. A dementia prevention trial with Ganterenumab and Solanezumab in DIAN-TU study has shown Ganterenumab to have a meaningful effect on the Aβ biomarker load, but not on cognitive symptoms. Solanezumab failed to show any improvement on either. Asymptomatic patients were not seen to worsen, but symptomatic patients had further cognitive decline before reaching the target dose [3, 36]. Another molecule, Donamemab, was recently found to produce conflicting results on the primary and secondary cognitive rating scales [37]. Cummings et al. (2020) provides a detailed overview of the AD therapeutics in the drug-development pipeline, which is beyond the scope of this paper [38]. A summary of the monoclonal antibodies targeting amyloid and tau in AD and their present clinical status are presented in Table 1.
AD, Alzheimer’s disease; ADAD, autosomal dominant Alzheimer’s disease; MCI, mild cognitive impairment; CSF, cerebrospinal fluid.
Financial obstacles, regulatory challenges, selection of appropriate targets and biomarkers to evaluate response, exploring alternative therapeutic pathways, and tackling the adverse effects are some of the important challenges in the development of monoclonal antibodies in AD therapeutics [21, 38]. Research has gradually accumulated to suggest that instead of the sequential cascade of events hypothesized earlier, simultaneous and cumulative changes in multiple pathways (amyloid, tau, neuroinflammation, etc.) are involved in AD pathogenesis [3, 9]. Hence, therapeutic strategies targeting multiple pathways are possibly of greater clinical benefit than a particular molecule or cascade [3, 38]. Of special mention is the “treatment versus prevention” dilemma during the monoclonal antibody trials in AD. Based on the amyloid cascade hypothesis, anti-Aβ therapy is most likely to be efficacious in primary prevention of AD. However, most clinical trials recruit patients with mild cognitive impairment (MCI) or AD over 18–48 months interval, where the observable benefits of these antibodies are minimal [39]. So, the question arises what we are using these anti-amyloid drugs for: to prevent the development or progression to AD versus as disease-modifying agents in AD [9, 21]. The evidence for the latter is dismal. In order to overcome this dilemma, certain strategies have been suggested [21]. The preclinical studies need to be better aligned with the subsequent clinical trials. Either anti-Aβ antibodies can be tried in primary and secondary prevention, or longitudinal designs need to be established to understand their roles in prodromal AD and mild AD. The approval of Aducanumab in spite of these challenges is definitely a positive step, but these ongoing questions and dilemmas may make this accelerated approval look premature.
ADUCANUMAB: THE MOLECULE
Aducanumab was derived from the plasma of healthy and cognitively normal older donors via a process called reverse translational medicine [40]. It was tried in a randomized controlled trial (RCT) due to an analogous molecule showing significant Aβ-plaque clearance from transgenic mice brains [14, 41]. It was found to have a linear pharmacokinetics within the given dose range. The median plasma half-life was 21 days via intravenous route. After repeated dose administration, Aducanumab was seen to have a sustained effect on the plasma Aβ level. On brain imaging, it was observed that it bound preferentially to the parenchymal Aβ over vascular Aβ. The hypothesis on Aβ clearance mechanism considered the role of microglia-mediated phagocytosis [27].
Aducanumab has been among the few anti-amyloid agents with meaningful efficacy in clinical trials. Compared to another monoclonal antibody Gantenerumab which only showed marked biomarker effects, Aducanumab revealed better outcomes both in terms of clinical measures and biomarkers [42]. The ability to bind and target neurotoxic soluble Aβ oligomers is a crucial factor in the efficacy of these antibodies. For Aducanumab, though the phase III trials were stopped after an interim analysis (mentioned in later sections), final results of the EMERGE trial supported efficacy on “surrogate markers” such as p-tau in CSF as well as tau-PET imaging [43]. The claimed target for Aducanumab is thus selective binding of pathogenic parenchymal Aβ oligomers, enabling their clearance which is expected to effect cognitive decline as well as behavioral and psychological symptoms of dementia associated with AD [42].
The PRIME study found out statistically significant Aβ reduction on amyloid-PET in both APOE4 carriers and non-carriers, and dose-dependent slowing of clinical progression on dementia rating scales (Clinical Dementia Rating-Sum of Boxes (CDR-SB) and Mini-Mental State Examination (MMSE)) [26, 27]. The brain Aβ plaque burden was found to be reduced in a dose and time-dependent fashion after Aducanumab treatment. Further, post-hoc analysis revealed that individuals with decreased amyloid plaque burden in the brain after treatment were more likely to experience stabilization in cognitive clinical decline after a year [27]. The most common adverse effects found were ARIA, headache, and infection. ARIA was more common in APOE4 carriers [26]. At present, Aducanumab is available in 100 mg/mL injectable solution, with guidelines for administration as IV infusion every four weeks at least 21 days apart [6, 41]. A vital favorable pharmacokinetic factor for Aducanumab is its “exquisite selectivity” for both insoluble fibrillar aggregates and soluble oligomeric depositions of Aβ found in the amyloid plaques. The structural and kinetic basis for this selective binding of Aducanumab has been demonstrated by Arndt et al. [25] using biochemical and structural analyses. The molecule differentiates between various types of Aβ based on strong selectivity for epitope-rich aggregates, weak monovalent affinity, and fast-binding kinetics. Evaluation of the crystal structure of Aducanumab Fab fragment has shown that in its extended conformation, it binds to the N-terminus of Aβ, which marks its ‘signature’ compared to the other monoclonal antibodies [25]. The epitope-binding among different immunotherapeutic agents decide their overall selectivity and kinetics. It is established that monomeric Aβ possibly have distinct neuroprotective roles in the brain [44] whereas Aβ aggregates lead to the synaptotoxic and neurotoxic cascade seen in AD [45, 46]. Hence, low selectivity for non-pathogenic Aβ monomers while heightened affinity toward pathological aggregations has possibly renewed the promise in Aducanumab research.
TRIAL TRAJECTORY
USFDA has approved Aducanumab with a condition of post-approval trial conduction to verify the anticipated clinical benefit, i.e., a phase IV trial. In case of the inability of this phase IV trial to confirm the anticipated clinical benefit, it can be taken off the market by regulatory procedures [47]. The trials that led to the approval of Aducanumab started with a preclinical study on APP-transgenic mice demonstrating its safety and effectiveness. From 2011–2013, phase I safety and pharmacokinetic trial was conducted in a single dose escalation study showing reasonable safety profile of the drug at doses of 0.3 mg/kg to 30 mg/kg and 60 mg/kg [48]. Then a randomized placebo-controlled clinical trial, named PRIME, was conducted from 2012–2014, on patients with prodromal or mild AD with positive Aβ PET scan [41]. The formulation was an intravenous infusion at doses 1, 3, 6, and 10 mg/kg body weight, given monthly for a year. The outcomes measured were adverse events, changes in amyloid-PET imaging, multiple-dose pharmacokinetic serum concentration, and change in serum anti-Aducanumab antibody level [41]. In view of the sustained effect of repeated doses of Aducanumab observed on the plasma Aβ level, Biogen, the manufacturer, announced the ensuing of a phase III trial in 2014. An interim data analysis result of PRIME was published in 2015, showing high-dose of Aducanumab reducing cortical amyloid burden and dose-dependent decrease in CDR-SB score. In 2016, PRIME data were formally published, followed by two and three-year data in 2017 and 2018. They reported of slowing cognitive decline as per exploratory analysis [40]. In 2015, two phase III trials started, named ENGAGE (study 301) and EMERGE (study 302) with identical design [49, 50]. In March 2019, Biogen announced a termination of the trials according to the planned futility analysis. However, in October 2019, another announcement was made regarding addition of further blinded data from the trials showing evidence of efficacy. Subsequently, further subgroup analysis was done, and results were published reporting significant reduction in cognitive and functional decline with 10 mg/kg Aducanumab, and in high-risk groups enrolled later. The high-dose (10 mg/kg) group in only EMERGE met the primary endpoint. Though ENGAGE also showed reduced cognitive and functional decline in the experimental group, the difference was not statistically significant [43]. A phase IIIb open-label safety and tolerability trial, on participants previously responding to Aducanumab in the past trials, started in January 2020 (EMBARK), to be continued till 2023, with similar clinical outcomes and biomarkers [51]. After Biogen applied for a priority review for approval in July 2020, several conflicting opinions came up. However, FDA finally granted the drug an accelerated approval on 7 June 2021 [6]. Accelerated approval is provided “to allow for earlier approval of drugs that treat serious conditions, and that fill an unmet medical need” [47]. The timeline of the trial trajectory leading to the present-day approval of Aducanumab is depicted in Fig. 2.
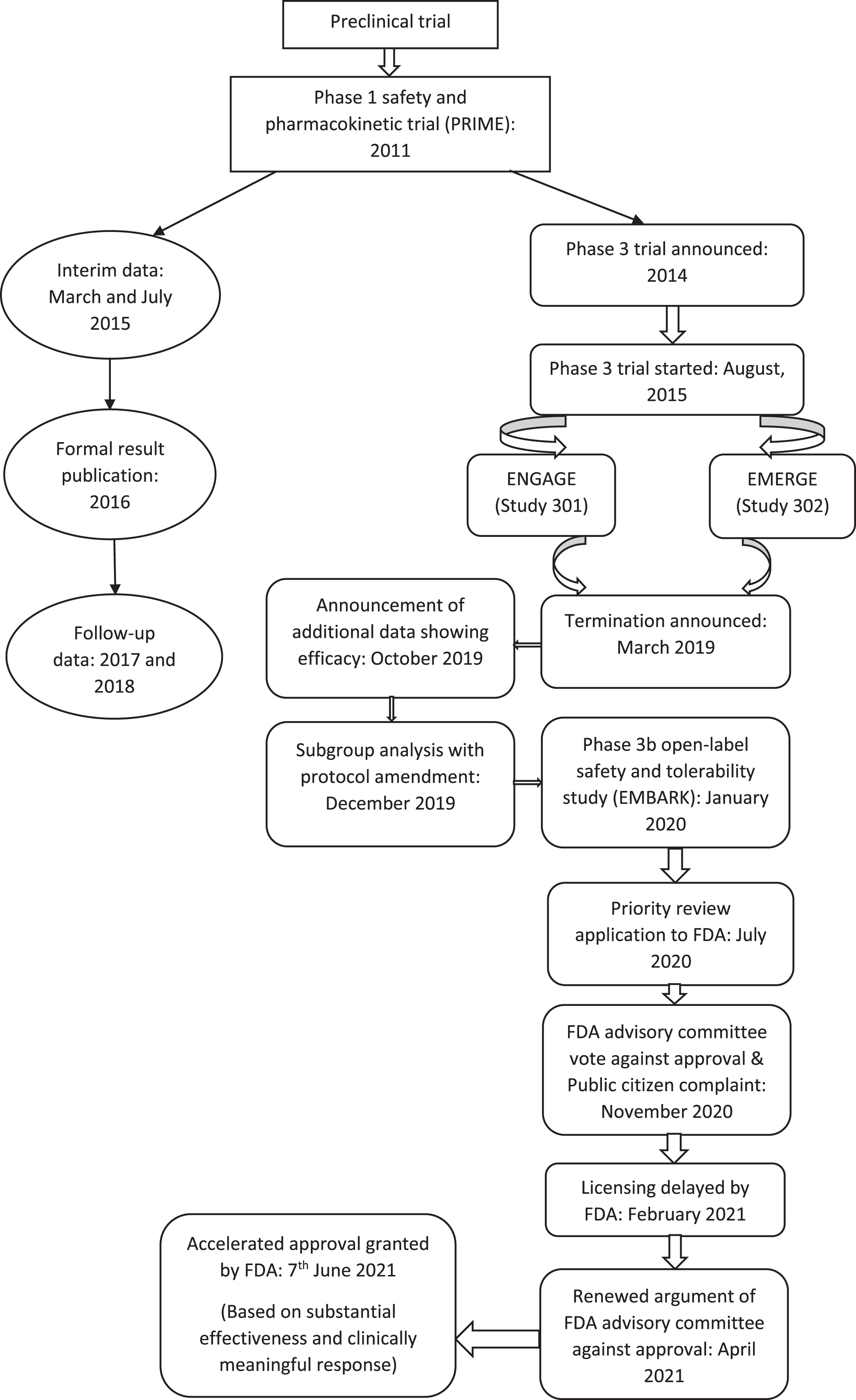
Timeline of trial trajectory leading to the approval of Aducanumab in Alzheimer’s disease.
WEIGHING THE EVIDENCE
The accelerated approval of the drug has stirred significant controversy. Several conflicting viewpoints have come up. In this section, we shall discuss the various reasons for opposing and supporting it.
Arguments against approval
Alternate pathways of pathogenesis
Nearly a decade back, Castellani et al. (2010) highlighted that the “exaggerated importance” given to amyloid oligomers in AD pathogenesis is a “reductionistic distraction” from various other upstream pleiotropic processes like cell cycle dysfunction, apoptosis, vascular remodeling, endothelial dysfunction, inflammation, and oxidative stress [28]. It has been hypothesized by multiple researchers that the long-standing failure of Aβ lowering therapies is mainly due to the fact that the proteinopathy is likely a downstream manifestation of AD, which shows poor correlation with the severity of dementia and can even be present in a cognitively healthy individual [52–54]. Even though amyloid hypothesis has been a time-tested conceptualization of AD pathogenesis, there is a debate between whether it is a triggering factor for further downstream manifestations or it is a downstream final common pathway for injury to the brain [54, 55]. In either case, therapies targeting Aβ alone then will not lead to lasting benefits, which has indeed been the result of many anti-amyloid trials. The same skepticism becomes logical with Aducanumab as well. Furthermore, the correlation between neural functional loss and regions responsible for memory are better with NFT rather than the extent of Aβ deposits. This excessive attention to anti-amyloid molecular targets can impede scientific enquiry toward other potential therapeutic targets as mentioned by an expert group of AD researchers, “It is now long overdue that the neuroscientists avoid the pitfall of perseverating on “proteinopathies” and recognize that the continued targeting of end stage lesions in the face of repeated failure, or worse, is a losing proposition” [28].
One of the main arguments against the amyloid hypothesis, besides failure of drug trials, has been the ‘Aβ deposition paradox’, i.e., Aβ accumulation in cognitively healthy individuals. Nearly 40%of older persons without dementia meet the neuropathological criteria for AD based on Aβ [56]. This has made senile plaques more characteristic of aging rather than AD. Also, evidence has also challenged Aβ being linked with MCI to AD progression. In vivo imaging studies have shown that ‘Aβ plaque burdens’ can be similar in non-demented older persons compared to that with AD [57] as well as the plateauing of amyloid deposition in spite of deteriorating cognition and functioning [58]. However, the other processes of AD pathogenesis such as synaptic loss, microglial activation, vascular inflammation, and NFT deposition correlate well with the disease severity and progression [59]. The various possible explanations of this Aβ deposition paradox are summarized in Table 2. Besides challenging the amyloid hypothesis, these explanations also raise concerns about the efficacy and approval of a sole anti-amyloid antibody for the treatment of AD.
Aβ deposition paradox: Putative explanations in Alzheimer’s disease
Single trial approval
As per the trial trajectory, it is evident that the approval of Aducanumab by the USFDA followed a single placebo-controlled trial demonstrating efficacy. The controversy lay in the “substantial effectiveness” clause required for approval with a single pivotal trial, i.e., a statistically significant result at p < 0.01 [67]. However, the responder analysis of EMERGE study was apparently not submitted to the FDA advisory committee, thereby contributing to the controversy [68].
Close collaboration between the manufacturer and FDA
Manufacturers of Aducanumab worked in close collaboration with FDA to analyze the trial results and the earlier phase Ib study for any statistically significant outcome in the high-dose group. The possibility of a compromised objectivity of FDA, therefore, has been raised [68]. This alleged “collaboration” leading to the fast-tracked approval of Aducanumab has been criticized at multiple fronts. A panel of external experts for the FDA had concluded in November 2020 that pivotal trials of this drug have failed to reveal “strong evidence” that it “actually worked along with potential safety issues” and suggested against its approval. The panel highlighted “questionable efficacy” and “red flags” associated with data analysis in support of their decision [69, 70]. Few members of the panel mentioned in a viewpoint that “there is no persuasive evidence to support approval of Aducanumab at this time”, their argument primarily being that the molecule does not meet the primary endpoint of “significant reduction in clinical cognitive decline” and the risk-benefit ratio is not pragmatic [68]. Considerations for safety were also vital concerns. Even though optimism exists for this drug in a debilitating illness like AD, the fast-tracked approval by FDA based on surrogate biomarkers on the background of an “internal discordance” in opinion has sparked further controversy that taints the official medical use of this monoclonal antibody against Aβ amyloid.
Surrogate end-point
It has also been argued that the end-point considered by Biogen, the manufacturer, was Aβ clearance and not a clinically meaningful sustainable cognitive benefit. It has been pointed out that no evidence was shown to correlate Aβ reduction to cognitive benefits [71]. Another drug for Duchenne muscular dystrophy was approved in 2016 using a surrogate end-point. A follow-up trial was advised by FDA to confirm the efficacy, results of which are not yet submitted. In the same lines, the final outcome of Aducanumab is also doubted [72]. As mentioned before, Aβ deposition triggers the initiation of the AD process but does not essentially influence the ongoing downstream cascade. Hence, an obvious question arises: will lowering the Aβ load in the brain of an individual who has already been affected with AD lead to a clinical benefit? To put it in simple words, let us take the analogy of treating a person with established atherosclerosis and myocardial infarction with cholesterol-lowering drugs and expecting that the current cardiac condition and longitudinal clinical course would improve. Expectedly, in a protracted disease course such as this, targeting the trigger factor is unlikely to demonstrate efficacy. Indeed, if a hypothetical trial is designed for statins such that it is used in patients in heart failure and morbidity/mortality used as clinical end-points (as opposed to surrogate end-points such as plasma cholesterol or arterial plaque deposition), statins may not turn out to be efficacious. Though statins undoubtedly decrease cholesterol and have protective effects against vascular risk factors, it is challenging to provide evidence that such treatment can reduce cardiovascular morbidity in ‘non-selected’ patient groups. Similar doubts get raised for anti-Aβ Aducanumab, if it can really influence ‘clinical end-points’ or is better suited for preventive purposes, i.e., when the pathogenic process of amyloid cascade has not yet been triggered.
Efficacy dilemma
The EMERGE trial is reported to have met secondary end-points only in the high-dose group. The low-dose group in this trial as well as all the groups of ENGAGE study had failed to show any significance. Moreover, the post-hoc analysis of the statistically significant RCT can introduce significant statistical error in terms of biases in population or sample selection and endpoint. The statistical correction required as suggested should be more robust than simple Bonferroni adjustment. Thus, the post-hoc selection of only the effective RCT (EMERGE) has been questioned [73]. The rapid progressors were excluded from the trial, leading to modest improvement in the drug efficacy. No criteria were specified by the sponsors in identifying such samples for future trial requirements [68]. Knopman et al. suggested that the apparent benefit in the post-hoc subgroup was not due to the drug dose [71]. All these issues have put the efficacy of Aducanumab under scrutiny. The other vital issue like any other clinical trials is that whether the ‘surrogate end-point’ benefit translates to an understandable clinical benefit which helps the affected individuals and their caregivers. Since the trials are restricted in time, the long-term efficacy is also doubted.
Adverse effects
Almost 40%of the samples on Aducanumab developed ARIA, as compared to 10%on placebo. ARIA can be associated with edema, microhemorrhage, or superficial siderosis. ARIA-E was found in 35%and ARIA-H in 21%. The most common associated symptom is headache followed by confusion, disorientation, delirium, dizziness, diarrhea, visual disturbance, and nausea. Headache was reported in 21%, falls in 15%, diarrhea in 9%, and confusion/altered mental status/disorientation/delirium in 8%[74]. The high-dose group was found to have more potentially serious side effects. Another adverse effect noted was hypersensitivity reaction including urticaria and angioedema [74]. Though serious or life-threatening consequence has been reported in only 0.3%of those treated with Aducanumab [74], there is a need to monitor the change with follow-up brain imaging [68, 72]. This can be a potential problem for older adults with AD and their caregivers alike. Besides, in low-and-middle-income countries with a rising AD burden, it may be clinically and practically not feasible. The 10/66 study has shown significant caregiver burden, burnout and costs of care which is only projected to increase [75]. In that background, regular monitoring of serious adverse effects of a costly drug through neuroimaging is definitely not an accessible and affordable option for majority of these populations.
Conflicting evidence for amyloid cascade
Though amyloid cascade has been postulated in the pathogenesis of AD for a long time, subsequent research has shown that it cannot solely explain the disease. Unresolved issues continue to exist in the amyloid hypothesis, as discussed before. As per Morris et al. (2014), alterations in Aβ expression can only be a “partial mediator” or by-product of dysfunctional AβPP metabolism, which affects synaptic functioning in complex multi-factorial ways [30]. Aβ deposition and accumulation are not found to correlate with neuronal loss and cognitive decline [8]. A significant percentage of cognitively normal healthy older individuals have shown amyloid deposition and changes in the brain. Conversely, almost 25%of patients with AD do not show Aβ changes [76]. Other pathways have also been found and molecules are being designed to target at many of them. The approval of Aducanumab, arguably, might hamper the discovery and development of other potential mechanisms and drugs [72]. Genetically-modified mouse models that show increased senile amyloid plaque formation, have not showed a consequent increase in tau deposition as well as nerve death [64]. Further, in Aβ-overexpressing BRI2- Aβ mice, Aβ oligomers and plaques were observed but in the absence of neuronal loss or cognitive decline [77, 78]. Mice models in which amyloid-targeted immunotherapies have been tested did not reveal improvement of clinical symptoms or NFT accumulation [79, 80]. These results further indicate that Aβ42 (oligomers and amyloid fibrils) are potentially not cytotoxic and have poor correlation with tau, which is an important marker of AD pathogenesis [66]. The conceptualization of AD as a disorder triggered by dysfunctional AβPP metabolism and propagated by tau pathology and neuroinflammation [66, 81], further makes an exclusive Aβ-targeted therapeutic modality questionable in the long run. Kepp (2017) highlights ten different challenges of the amyloid hypothesis based on current-day research, necessitating newer approaches addressing data heterogeneity and using aging phenotype as a background model for AD [54]. This will unify the understanding of genetic and non-genetic risk factors in the complex genesis of a major neurodegenerative disease like AD, which also has a potential management value. “The amyloid hypothesis on trial” by Makin (2018) in Nature mentions that though Aβ is an obvious therapeutic target, there is an “awful more lot” that needs to be added for a breakthrough in management [82]. Evidence also suggests that Aβ can have a protective effect against oxidative stress and free-radical damage [83]. In that sense, the age-related accumulation of amyloid and tau are actually protective homeostatic responses to cumulative metabolic demands in the neurons. In the words of George Perry, the Editor-in-Chief, Journal of Alzheimer’s Disease, “the current approach is based on the idea that Aβ is bad. My idea is quite the opposite. Are we going to tie up the whole field for another 15 to 20 years?” [82]. Perry argues for exploring alternate pathways in AD therapeutics rather than persistent “obsession” with the amyloid cascade.
Cost
The drug is much criticized for its poor cost-effectiveness ratio. This has been argued to have more severe financial impact on the families of patients with AD, without a substantial improvement in cognition [72].
Along with the many stakeholders raising objections, the Institute for Clinical and Economic Review (ICER) has stated that FDA has “failed in its responsibility to protect patients and families from unproven treatments with known harms.” The ICER has estimated a price of $2,500 –8,300 per year for this treatment [84]. For many nations with developing economies, monitoring ARIA through regular neuroimaging may not be cost-effective compared to its benefits. This may also involve significant logistic difficulties. Also, even though the expected use is in early stages of AD, the current regulations do not limit its use based on the stage of severity. It is estimated that even if 5%of the people living with AD in the United States receive this treatment, the revenue generated will be nearly $17 billion annually, making it the second most-sold drug, with respect to revenues [85]. The question with the current dataset is, however, whether this generated cost-burden is worth it. There is no conclusive answer to that yet.
Arguments for approval
Evidence of benefit
The addition of blinded data after the planned futility analysis and termination of trial in 2019 showed some evidence of efficacy. Hence, the analysis was continued further and the EMERGE trial showed a significant reduction (22%) on CDR-SB in the high-dose Aducanumab group [43]. Watson et al. have argued that CDR-SB is a good measure of cognitive and functional status, having a direct bearing on the patient’s condition [86]. Moreover, small changes on this instrument are considered to be of clinical significance. Similarly, all other measures like MMSE, Alzheimer’s Disease Assessment Scale-cognitive subscale (ADAS-Cog), Alzheimer’s Disease Cooperative Study Activities of Daily Living (ADCS ADL), and Neuropsychiatric Inventory (NPI) showed significant benefit with the drug (40%reduction on ADCS ADL). Caregiver distress, on NPI, was also found significantly lower in the treatment group [43, 73]. Thus, the reduction, though seen in only one group, is argued as considerable, providing an evidence of substantial effectiveness [87].
Amyloid and p-tau hypothesis
Aβ plaque reduction with Aducanumab on amyloid PET scan is found to be significant in both EMERGE and ENGAGE trials. Though amyloid hypothesis cannot be considered a stand-alone in explaining neurodegeneration, it has one of the long-standing and definite implications in AD pathogenesis, acknowledged in the NIAAA framework 2018 [13]. Aβ deposition is known to affect p-tau changes, which, in turn, impact the cognitive decline [13]. EMERGE has also shown a statistically significant correlation between Aβ reduction and clinical outcome, and Aβ reduction and CSF p-tau changes [73, 87].
Adverse effects
As observed in the trials, most people who developed ARIA were without any substantial symptom. Cummings et al. has argued that the adverse effects are manageable and even considerably less than some anticancer drugs [87]. In addition, it has not shown any deleterious consequence and there is no evidence that it will in the long run.
Clinical considerations
In response to the argument of premature approval of Aducanumab by FDA, it has been mentioned that the decision has largely been based on the clinical significance of the results. Considering a large number of patients seeking treatment of dementia and the dearth of existing treatment options, provision of an alternative treatment option has been a welcome step toward better dementia management [87].
Collaboration and not compromise
The close collaboration between FDA and Biogen is claimed to be aimed at hastening the analysis to achieve a goal of public health importance, and ensuring clear communication regarding the trial results and interpretations [87]. The possibility of a compromise in neutrality has been refuted by FDA and other stakeholders. Further refuting this “alleged collaboration”, patient advocacy groups worldwide have heavily supported the approval of Aducanumab, especially because of its “novel status” in a progressive condition with limited therapeutic choices [88]. Various other organizations like the Alzheimer’s Association, Alzheimer’s Foundation of America, and Alzheimer Society of Canada also advocated for this approval [89–91].
Implications for future research
Tacrine, the first ever molecule approved for the treatment of AD, eventually went out of use due to its hepatotoxicity, but gave way to acetylcholinesterase inhibitors, the only group of FDA-approved drugs for AD today. It is argued that a denial of approval despite considerable advancement of knowledge regarding amyloid pathway and Aducanumab would have stalled future research on drug development in the similar line. Thus, this acknowledgment has paved the way forward for better drug development and AD neurobiology research. The scientific community is hopeful about further development of monoclonal antibodies against various targets of amyloid and tau pathways in the near future.
In addition to these points put forward in favor of Aducanumab, the approval of Aducanumab is also contingent on the result of a phase IV post-marketing trial [6]. This potentially increases the window of opportunity for the current claims to be either proved or disproved.
Therapeutic implications for Alzheimer’s disease
The therapeutic implications of this drug include a reduction in the amyloid burden in brain of patients with AD with an improvement in the cognitive and functional outcome. The trials are all done in patients with MCI or mild AD, and thus, the expectation is a delay in further cognitive worsening. Though research is at a premature stage to comment about the long-term outcome, the data released so far by Biogen show a slower progression of the cognitive and functional deficits as measured by standardized assessment tools. Improvement is also found in the neuropsychiatric and behavioral symptoms of AD and caregiver burden from the three RCTs mentioned before (PRIME, ENGAGE, and EMERGE). Cost-effectiveness can be a particularly relevant issue in lower- and middle-income countries where the burden of dementia is significant while economic disadvantages are considerable in the most. Thus, this FDA decision can be considered “dual-edged”. As much as it has been important to obtain a disease-modifying molecule for AD, the concerns are critical. These have prompted significant debate in the scientific community. Subsequently, three FDA advisory board members have resigned [92]. Public statements have been made by certain stakeholders like ICER deploring the approval [84]. On the other hand, it has been appreciated by organizations like Alzheimer’s Disease International (ADI) [93]. The International Psychogeriatric Association (IPA) has released a position statement which is hopeful but guarded considering the need for further evidence of the drug [94]. We have elaborated the possible reasons above as to why we need to be skeptical about the approval of Aducanumab. When a progressive neurodegenerative disease affects millions leading to significant morbidity and impairment in quality of life, a ‘novel’ drug definitely generates hope, which is indeed reflected in the statements of various global psychogeriatric organizations. However, the question lies whether Aducanumab is “an old wine in a new bottle”, since researchers have time and again criticized the therapeutic potential of anti-amyloid drugs. To add fuel to the fire, data supporting efficacy of Aducanumab is equivocal and the approval tainted with controversy. Notwithstanding any of these, whether this monoclonal antibody emerges as a time-tested treatment of AD is left to the future. But clearly it is time to rethink and explore future approaches targeting alternate pathways of AD pathogenesis (discussed above), especially age-related vulnerability of the neuronal environment to oxidative stress, vascular remodeling and neuroinflammation. In the words of Joseph et al. [95], “solve the problems of aging and by extension those of AD will also be reduced”. This involves non-pharmacological measures as well that sustain and promote healthy aging. Eventually, aging is the most pervasive core of this disease and Aβ is just one of the many intriguing facets.
CONCLUSION
Amyloid inhibiting drug trials have always been fraught with ambiguity. Echoing the words of Bart De Strooper, director of the UK Dementia Research Institute, London, “The problem with most of the amyloid trials is that they didn’t disprove anything. They just proved that a drug, in the way it was applied, didn’t work.” [85]. A decade back, Castellani et al. (2010) mentioned that the AβPP molecule research has “encapsulated” AD so much so that the disease itself has become secondary. To quote the authors, “such a mindset tends to ignore not only competing reductionist theories, but the complexity of chronic diseases in general. Moreover, referring to any one approach as ‘once controversial’ highlights the magnitude of the problem, and the necessity to return to objective review of data that are generally soft and manipulable. In effect, how many patients need suffer an untimely demise at the hands of clinical trials driven by schools of thought that are no longer controversial, before a paradigm shift occurs?” [28] The recent approval of Aducanumab does not answer this question but raises many more related ones.
In summary of the discussion above, it can be said that Aducanumab is a drug targeting the amyloid deposition and possibly tau abnormality, the core pathology of AD. It is a promising avenue for a progressive illness with limited therapeutic options and high treatment burden; however, the premise for such ‘promise’ is based on equivocal evidence. The clinical improvement is not apparent consistently across all the clinical trials and the different dose-groups. High dose of Aducanumab has also been found associated with a greater occurrence of adverse effects like ARIA. Moreover, there are several controversies raised due to the conflicting findings obtained from the clinical trials of Aducanumab. Definite concerns exist in terms of need for regular brain imaging monitoring, poor cost-effectiveness, and doubtful long-term efficacy. Though it is an important step to acknowledge the current evidence, equally important is the pursuing of more robust scientific knowledge to establish the safety and efficacy beyond questions. The NIA-AA research framework has laid down diagnostic criteria of AD based on the pathology only. However, it has strictly been designed for the research and not for clinical purpose. We believe that a similar strategy can be applied in furthering the evidence for Aducanumab before any large-scale clinical use. Aducanumab may not be the ‘absolute’ answer to AD pathology, and any new drug is bound to be fraught with debate. Beyond the academic and economic discourse, approval of a new molecule for the mild stage of a devastating neurodegenerative disease like AD generates caution and hope parallelly. The authors do not endorse or refute the approval of this novel molecule but rather attempt to provide a balanced perspective on the trajectory of its approval and the evidence backing it up, which can serve as a potential framework for further research and academic discourse related to its use in AD. Since disease-modifying treatments for AD are immensely important considering the disease burden, scientific research should be simultaneously targeted at exploring all possible pathogenic mechanisms and the relevant drug development, inclusive of but not limited to Aducanumab. To revisit the common themes of Castellani et al. [28] and Makin [82], papers written a decade apart, “there is still an awful lot more to the amyloid hypothesis if we are to understand AD. Lot of ideas are well-supported but few people work on them.” It is for us to decide whether we want to tie up the field of AD therapeutics within certain specified limits or cast a wider net to form a bigger set of ideas, being within scientific, ethical, and pragmatic considerations.
DISCLOSURE STATEMENT
Authors disclosures available online (https://www.j-alz.com/manuscript-disclosures/21-5065r1).