
Abstract
Alzheimer’s disease (AD), the most common neurodegenerative disorder, is accompanied by cognitive impairment and shows representative pathological features, including senile plaques and neurofibrillary tangles in the brain. Recent evidence suggests that several systemic changes outside the brain are associated with AD and may contribute to its pathogenesis. Among the factors that induce systemic changes in AD, the gut microbiota is increasingly drawing attention. Modulation of gut microbiome, along with continuous attempts to remove pathogenic proteins directly from the brain, is a viable strategy to cure AD. Seeking a holistic understanding of the pathways throughout the body that can affect the pathogenesis, rather than regarding AD solely as a brain disease, may be key to successful therapy. In this review, we focus on the role of the gut microbiota in causing systemic manifestations of AD. The review integrates recently emerging concepts and provides potential mechanisms about the involvement of the gut-brain axis in AD, ranging from gut permeability and inflammation to bacterial translocation and cross-seeding.
INTRODUCTION
Dementia, which refers to various symptoms caused by disorders of brain function, accompanies conditions that affect memory, thinking, behavior, and emotions. Alzheimer’s disease (AD) accounts for 60–80% of all dementia cases and is regarded as the most common neurodegenerative disorder. AD shows key representative pathological features, such as senile plaques composed of dystrophic neurites, reactive glial cells surrounding accumulations of amyloid-β (Aβ), and neurofibrillary tangles (NFTs) containing the hyperphosphorylated tau protein [1]. Centering on these features, the amyloid and tau hypotheses currently represent the main etiological explanation for AD. Mechanisms that constitute these hypotheses are explained below.
In neurons, Aβ generated from amyloid-β protein precursor (AβPP) can accumulate in endosomes, but is mainly secreted from the cells to form oligomers or plaques that are toxic to surrounding cells. Neurotoxic Aβ directly destroys synapses and/or lowers neuronal activity, leading to a decline in cognitive ability. Another causative agent of AD is the hyperphosphorylated and aggregated form of tau protein. In the central nervous system (CNS), tau protein is involved in microtubule assembly, structural stabilization, and molecule transport within neurons [2]. The tau protein also undergoes post-translational modifications. Some of the forms are majorly found in NFTs of the brain tissue and in the blood of AD patients, and thus can be utilized as blood biomarkers [3, 4]. Before the formation of NFTs, hyperphosphorylated tau can be sent from the axonal compartment of the neuron to the somatodendritic compartment, where it impairs synaptic function. Thereafter, the tau aggregates propagate in a prion-like manner from the entorhinal cortex to the neocortex of AD patients, causing serial neurodegeneration [5].
In AD, the accumulation of Aβ plaques and tau aggregates and the resulting neuronal death activate neuroglial cells (astrocytes and microglia) in the brain [6]. Astrocytes provide nutrients from the blood to support nerve cells and/or protect nerve cells by removing surrounding waste through phagocytosis. In the context of neurodegeneration, astrocytes are mainly activated around the brain lesions of patients. Sustained reactivity of astrocytes to remove damaged neurons or lesions generates increased hydrogen peroxide, which is detrimental to neurons [7]. Microglia, which are innate immune cells residing in the brain, maintain brain homeostasis through various cell-surface sensing proteins. Microglia constantly surveil the brain’s microenvironment; when they detect a damage signal, they quickly move to the area and trigger immune responses, such as phagocytosis and/or secretion of pro-inflammatory cytokines. In AD, microglia usually undergo rapid activation to remove misfolded Aβ, tau, or deteriorating nerve cells at the initial stage of disease onset. However, chronic stimulation of microglia degrades their control mechanisms, resulting in an overactive inflammatory response that leads to neuronal destruction [8]. In addition, microglia that fail to remove Aβ aggregates eventually die, contributing to the formation and growth of plaques in the brain [9].
In order to enable patients to recover from the cognitive deficits and functional impairments arising from these major causes, researchers are competing to develop AD drugs. The FDA has approved four drugs to date: memantine, galantamine, rivastigmine, and donepezil [10]. Unfortunately, the approved drugs are only able to slow disease progression by facilitating intra-neuronal signal transduction through N-methyl D-aspartate receptor regulation or cholinesterase inhibition. The FDA also recently approved aducanumab, which is an Aβ-targeting antibody [11]. However, the efficacy of aducanumab is widely debated, and additional longer-term evidence is needed to determine whether it can actually prevent cognitive decline in AD patients and thereby serve as the first disease-modifying drug.
Given the centrality of the Aβ and tau hypotheses, continuous attempts have been made in the pharmaceutical industry to remove the pathogenic proteins from the brain or coax excessive immune activity. However, due to limitations such as the short-lived effectiveness of the drugs and the risk of side effects (e.g., cerebral microbleeds), no successful disease-modifying treatment has been developed thus far. Moreover, while these efforts to directly target pathogenic molecules remain valid, accruing evidence suggests that the pathogenesis of AD needs to be viewed in a broader perspective—one that does not focus solely on the brain, but also views the disease in relation to other organs. Recent experimental and clinical studies suggest that systemic changes outside the brain can be causative factors in AD [12]. In particular, metabolic dysregulation leading to obesity, diabetes, and systemic inflammation has been shown to increase the risk of AD, while physical activity and dietary modulation help lower the risk [13]. Thus, the communication between the brain and the whole body has attracted attention as a new target for AD therapeutics. From this perspective, developing a holistic understanding of the pathways throughout the body that can affect the pathogenesis, rather than viewing AD solely as a brain disease, may be key to developing a successful therapeutic agent. The gut microbiota has been recognized as a “forgotten organ” that regulates host physiology and metabolism [14], and even influences brain development and function [15]. Therefore, in this review, we focus on the role of gut microbes and discuss the potential mechanistic involvement of the gut-brain axis (GBA) in AD. Also, we describe recently emerging concepts regarding GBA involvement in AD, ranging from gut permeability and inflammation to bacterial translocation and cross-seeding.
THE GUT-BRAIN AXIS IN NEUROLOGICAL DISEASE
The gut microbiota is closely connected to the CNS through the endocrine, nervous, and immune systems, together forming the so-called GBA. The bidirectional signaling of the GBA is regulated by the gut microbiota, and many researchers have reported that the gut microbiota plays a significant role in brain development, diseases, and the associated physiological, psychological, and behavioral changes [16]. Many studies examining the effects of the gut microbiota on the CNS have been mainly conducted in germ-free (GF) mouse models. By observing the behavioral and neurological differences in GF and specific-pathogen-free (SPF) mice, researchers have elucidated mechanisms by which the gut microbiota affects brain development and function. For example, the turnover rates of noradrenaline, dopamine, and serotonin were found to be elevated in the striatum of GF mice compared to SPF mice, and GF mice showed altered expression of anxiety- and synaptic plasticity-related genes [17]. In addition, although adult neurogenesis of the hippocampus decreases with age, this age-dependent decrease in neurogenesis was found to be delayed in GF mice, affecting their cognitive functions related to behavior, motor control, learning, and memory [18]. Another study suggested that manipulating the gut microbiota with probiotic bacteria can restore cognitive function, memory impairment, and anxiety disorder, and recover intestinal inflammation in inflammatory bowel disease (IBD) model mice [19]. These findings supported the idea that the gut microbiota contributes to CNS functions, prompting researchers to investigate the underlying mechanisms responsible for regulating the GBA.
The gut microbiota has attracted attention as an active regulator of the GBA in various neurological diseases. For instance, the neurotransmitter, dopamine, is synthesized in the enteric nervous system at a level similar to that in the brain. A metagenomics analysis of 1,054 people found that dopamine-producing bacteria were lacking in the guts of patients with depression [20]. About 90% of serotonin (a hormone that affects appetite, sleep, mood, memory, and learning) accumulates in the intestine, and certain intestinal microorganisms can increase serotonin synthesis [21]. Serotonin can directly stimulate intestinal nerve cells, and the endocrine cells that synthesize serotonin can form a synapse to the brain via the vagus nerve to transmit signals [22]. These findings support the importance of the gut microbiota in maintaining mental health through endocrine regulation, and clearly indicate that intestinal bacteria play active roles in the neural communication between the gut and brain.
The concept that the gut microbiota is a potential regulator of resident immune cell functions in the gut, periphery, and brain suggests that the immune system should be viewed as another mechanism underlying the GBA [23]. Fecal microbiota transplantation from patients with multiple sclerosis (an autoimmune disease) into GF mice increased the number of inflammatory T cells in peripheral lymphoid tissues and accelerated the multiple sclerosis pathology [24]. In a stroke model, researchers elucidated a mechanism by which gut-derived immune cells interact with the CNS via the commensal gut microbiota [25]. Short-chain fatty acids (SCFAs, representative metabolites of gut microbes) are well known to be involved in gene expression and hormone secretion, mainly in the gut. Interestingly, however, the defects in the maturation and function of microglia in the CNS of GF mice were restored by SCFAs [26]. In another work, neuroinflammation was alleviated by binding the tryptophan metabolism by-products of gut bacteria to the aryl hydrocarbon receptors of astrocytes [27]. These studies provide convincing evidence that the metabolites of gut microbes can directly affect the activity of innate immune and glial cells residing in the brain. Alongside this work showing that the gut microbiota can control brain function via the endocrine, neuronal, and immune systems, other studies have shown that, conversely, the brain can alter the gut environment. For instance, stress hormones (e.g., norepinephrine) released into the blood under psychological and physical stress were found to negatively affect the bacterial composition and function of the intestine by regulating bacterial gene expression and inducing community-specific growth [28]. As researchers continued to unravel the detailed mechanisms by which gut microbes regulate signal transduction between the gut and brain, the field increasingly recognized that gut microbes could be associated with various pathologies, such as the neurodegenerative diseases, amyotrophic lateral sclerosis (ALS), Parkinson’s disease (PD), and AD.
ALS is a neurodegenerative disease characterized by the gradual loss of the motor neurons responsible for motor, speech, and cognitive abilities [29]. Transgenic model mice overexpressing mutant superoxide dismutase 1 (SOD1) or chromosome 9 open reading frame 72 (C9orf72), the most common genetic causes of ALS, exhibit pathological changes in reaction to nicotinamide-producing and immune-stimulating bacteria, respectively [30, 31]. PD is another chronic brain disease in which motor and cognitive impairments occur due to alpha-synuclein (α-syn) accumulation and cell death among dopaminergic neurons of the substantia nigra [32]. Enteric α-syn was found to propagate to the brain through the vagus nerve, leading to PD-related neuropathology [33]. The finding led to the hypothesis that the onset of PD starts in the gut, not the brain, and brought about the breakthrough concept that targeting the accumulation of α-syn in the enteric nervous system could be an important strategy for treating PD.
As with the above-described neurodegenerative diseases, alterations in the composition and diversity of gut microbiota in AD patients and animal models of AD have been reported, extending our knowledge on AD pathogenesis and GBA-related AD phenotypes.
THE GUT MICROBIOTA AND AD
AD patients: Evidence from human samples
The advancement of sequencing technology has improved researchers’ ability to analyze bacterial taxa. This has enabled scientists to isolate and distinguish microorganisms, opening up previously unknown realms of diverse microbes. Early studies utilizing this technology identified perturbations in the gut microbiome in various diseases. As the link between the gut and the brain is increasingly being solidified through numerous studies examining the GBA, the gut microbiome’s influence on the overall physiological condition, including that of the brain, is becoming evident. Evidence from human samples of various origins has revealed the implications of the gut microbiome in AD.
Stool (fecal) samples of AD patients
Analysis of human fecal samples is widely used as a proxy for examining the gut microbiota for pragmatic reasons, such as the ease and non-invasiveness of sample acquisition. Moreover, human fecal contents have been solid linked to the composition of the gut microbiome and its metabolic function. For example, in a study involving 713 fecal metabolites, 95% and 82% of the fecal metabolites were significantly associated with 99.7% of the microbial metabolic pathways and 90% of the gut microbial species, respectively [34]. Accordingly, the growing body of human evidence regarding the link between the gut microbiota and AD is mainly centered around evidence obtained from fecal samples.
Vogt et al. discovered that Firmicutes and Bifidobacterium were decreased, whereas Bacteroidetes was increased, in the microbiome of AD patients compared to that of control participants [35]. Zhuang and colleagues also reported that the gut microbiota is significantly altered in AD patients, but these authors observed different alterations: In their study, AD patients showed a mild decrease in Bacteroidetes, whereas Firmicutes were almost identical between the control and AD groups [36]. The caveat of these findings is that discoveries made at this phylum level may be highly variable and controversial, given that Bacteroidetes and Firmicutes make up approximately 80% of the human gut microbiota [37] and the ratio between them changes with the host’s age [38, 39]. Moreover, AD is a multifactorial disease affected by environmental factors; thus, the discrepancy in the findings of the abovementioned studies may reflect regional differences in lifestyle or dietary habits [40, 41]. The discrepancy might also arise from using different sampling or analytic methodologies [42, 43] and possibly, different stages of AD progression that the patients are in. Going forward, further data collection and standardization between institutions could enable researchers to perform efficient meta-analyses and obtain more consistent and reliable results. Cattaneo et al. identified a difference in the abundance of specific bacterial taxa in amyloid negative/positive patients compared to healthy controls. Compared to both healthy and amyloid-negative patients, amyloid-positive patients exhibited a greater abundance of Escherichia/Shigella and a reduced abundance of Eubacterium rectale. Interestingly, the pro-inflammatory cytokines, interleukin 1 beta (IL-1β), NOD-like receptor pyrin domain-containing protein 3 (NLRP3), and C-X-C motif chemokine ligand 2 (CXCL2), were positively correlated with the abundance of Escherichia/Shigella and negatively with that of Eubacterium rectale [44]. These findings suggested that certain microbial species can induce systemic inflammation and impact the brain amyloid burden of the host.
Human fecal samples also provide valuable resources for in vitro applications in mechanistic studies. For instance, in vitro T84 intestinal epithelial cell functional assays performed using patients’ stool samples revealed that P-glycoprotein expression was significantly lower in AD patients [45]. Intestinal P-glycoprotein secretes endocannabinoid into the intestinal lumen to control neutrophil infiltration and is thus considered to be a crucial regulator of intestinal inflammation and homeostasis [46]. The decreased expression of P-glycoprotein in AD patients implies the presence of disrupted homeostasis and increased inflammation. In addition, the authors used metagenomic sequencing to distinguish microbial composition between healthy controls, AD patients, and patients with other types of dementia, and found that AD patients were characterized by lower proportions of species of the critical butyrate-producing genus, Butyrivibrio, and members of the Eubacterium genera. Butyrate derived from microbes directly induces epithelial anti-inflammatory IL-10 receptor α-subunit, resulting in repression of the tight junction (TJ) protein, claudin-2, which is known to promote barrier permeability [47]. As butyrate is essential for the ability of colonic epithelial cells to maintain the gut barrier function, a decreased butyrate level could explain the increased gut permeability observed in AD patients.
Blood samples of AD patients
Blood circulates throughout the body and carries various bacterial metabolites secreted from the gut [48, 49], and is thus a valuable resource for explaining the intricate interplay between the gut microbiome and systemic metabolism [34]. Association studies between amyloid pathology and blood levels of SCFAs and lipopolysaccharide (LPS) suggest that these bacterial metabolites can mediate gut dysbiosis and AD. For example, in a study using florbetapir amyloid positron emission tomography imaging, brain amyloid was positively correlated with blood LPS, acetate, and valerate, but negatively correlated with butyrate [50]. Another study suggested that butyrate-producing microbial species are decreased in AD patients [45], further supporting the notion that gut epithelial integrity is impaired in AD. Furthermore, acetate and valerate showed significant associations with pro-inflammatory cytokines, such as CXCL2. Interestingly, several studies showed that acetate and valerate have anti-inflammatory effects on glial activation [51–53].
As LPS and SCFAs are regarded as metabolites produced almost exclusively by gut microbes in the human body [54, 55], the changes in their level strongly indicate the changes in gut microbes. While the level changes of LPS and SCFAs in blood may not be solely related to AD, their significant correlation with brain amyloid implies that AD pathology is one of the factors attributable for their increase in blood level and AD plays a role in inducing systemic inflammation.
CSF samples of AD patients
The ability of gut metabolites to reach the CNS was confirmed by the detection of these metabolites in cerebrospinal fluid (CSF) [56]. Vogt et al. reported that CSF carries trimethylamine N-oxide (TMAO), which is a microbial metabolite that has been implicated in cerebrovascular and metabolic diseases [57, 58]. Furthermore, they discovered that the TMAO level in CSF is higher in individuals with mild cognitive impairment and AD dementia than in healthy controls, and that it is associated with AD biomarkers, such as phosphorylated tau (p-tau) and neurofilament light chain protein (NfL) [59].
As TMAO is produced from trimethylamine, which is exclusively produced by anaerobic microorganisms residing in the mammalian gut [60], detection of TMAO in CSF provides obvious evidence that bacterial metabolites can directly enter CSF and cause changes to the brain. TMAO level in CSF may not be specifically related to AD. However, based on its significant correlation with canonical AD biomarkers such as p-tau and NfL, it is conceivable that AD can act as one of the contributing factors in allowing the increase of TMAO level in CSF by impairing the gut.
Therapeutic strategies in patients
As the gut microbiota has been increasingly acknowledged as contributing to regulating overall physiologic homeostasis, increasing efforts have been made to treat patients by altering their gut microbiome. In diseases where the link between gut bacterial imbalance and disease onset has been thoroughly investigated, gut microbiome modulation has become a viable treatment strategy. For example, in a gastrointestinal (GI) disorder patient with irritable bowel syndrome (IBS), the administration of gut microbiota from a healthy donor can significantly change the intestinal bacterial profile and improve fatigue and quality of life [61]. For IBD patients, fecal microbiota transplantation (FMT) has been used in clinical trials for over two decades to treat the chronic inflammatory conditions of the GI system [62].
In one brief clinical trial spanning 12 weeks, AD patients administered with probiotic milk containing mainly genus Lactobacillus exhibited improvements in their Mini-Mental State Exam (MMSE) scores, insulin metabolism, and triglyceride levels; however, the changes in the levels of oxidative stress and inflammation were negligible, indicating the need for further studies [63]. The same group also reported that co-supplementation of probiotics and selenium to AD patients for 12 weeks improved their cognitive function, as assessed by MMSE, and reduced the levels of serum lipid metabolites, such as triglycerides, very-low-density lipoprotein, and low-density lipoprotein [64]. One preliminary study found that supplementing AD patients with probiotics influences the gut bacteria composition and tryptophan metabolism in serum [65]. The authors speculated that the increased serum kynurenine levels seen upon probiotic supplementation were caused by macrophage activation, but it remains unclear how this might offer a therapeutic benefit in AD.
AD model mice: Experimental models
There are fundamental differences in the microbiomes of humans and mice. For example, the relative compositions within a phylum can differ, as evidenced by the finding that genera Mucispirillum is exclusively present in rodents [66]. However, there is high similarity between human and murine gut microbiota: Both have Bacteroidetes and Firmicutes as major phyla, and 90% and 89% of microbes are shared at the phylum and genus levels, respectively [67]. Thus, analysis of the mouse microbiome can provide valuable insights into human physiology.
The prominent feature that distinguishes an AD model from others is the accumulation of amyloid in the brain. Thus, most studies using AD models take advantage of amyloid pathology driven by human transgenes, such as APP and presenilin (PS), harboring familial AD mutations known to accelerate the pathology. Early findings by Shen and colleagues showed that the histological and behavioral AD manifestations in APP/PS1 mice depended on the presence of a specific gut microbiome state, and that the gut microbiome exhibited an age-related decrease in diversity. They found that, compared to wild-type (WT) mice, AD mice had significantly increased Odoribacter and Helicobacter at the genus level [68]. Another study in APP/PS1 mice investigated the shift in gut microbiota composition across the lifespan [69]. The researchers found that bacterial profiles were similar between APP/PS1 and WT mice at a young age, before the pathology set in, but started to diverge at 6 months with increases in Proteobacteria, especially genus Sutterella. Interestingly, Sutterella had been shown to correlate with the tumor necrosis factor-alpha (TNF-α) level and aggravated social behavior in a female BTBR mouse model of autism spectrum disorder [70]. This opened the question of whether certain microbes are responsible for inducing inflammatory responses in neurological disorders.
Comparison of the fecal microbiome and SCFAs of APPswe/PS1ΔE9 transgenic model mice versus WT mice revealed differences [71]. The diversity of the fecal microbiome differed, especially at 8-12 months of age, with Proteobacteria found to be dramatically increased in the AD model mice. Interestingly, B. pullicaecorum was reduced in AD mice. This butyrate-producing microbe is known for its potential to strengthen epithelial barrier functions and has been proposed as a promising probiotic candidate for IBD patients [72, 73]. Moreover, butyrate is essential for the maintenance of a healthy gut with an intact barrier, and its level might be disrupted in an AD gut [45]. In Tg2576 mice harboring a Swedish APP mutation known to lead to early-onset AD, the abundance of Bacteroides was correlated with aging and plaque pathology [74]. Interestingly, in humans, neurotoxins from Bacteroides have been hypothesized to drive the AD process [75]. In the same model, another group found that intestinal epithelial barrier dysfunction occurs before cerebral Aβ aggregation is detectible. These changes were accompanied by elevated levels of plasma cytokines, such as IL-9, vascular endothelial growth factor, and interferon-inducible protein 10 [76].
Chen and colleagues extensively analyzed the gut microbiota composition of the well-known 5XFAD mouse model of AD [77]. The microbial communities were the same at 3 months of age and were characterized by a lower abundance of Firmicutes and a higher abundance of Bacteroidetes. At 6 months of age, however, the abundance of the two dominant phyla, Firmicutes and Bacteroidetes, showed a marked difference between the groups: Pro-inflammatory phyla, such as Proteobacteria and Bacteroidetes were drastically increased only in 5XFAD mice, while anti-inflammatory Firmicutes took up most of the portion in WT mice. At the genus level, 5XFAD mice showed increases in Helicobacter, Prevotella, and Sutterella [77, 78].
Although tau is another major pathological hallmark of AD, tau models have been used to a lesser extent than amyloid models in AD microbiome studies. This may be because pathological tau is found widely in other tauopathies and is not as exclusive as Aβ in constituting the AD phenotype. However, evidence on the alterations in the microbiome in tau models of AD has begun to accumulate, and the implications of these studies could also apply to other tauopathies. At 3, 6, and 10 months of age, the P301L tau transgenic mouse model showed increased and decreased abundances of Bacteroidetes and Firmicutes, respectively, compared with WT mice [79]. Interestingly, similar phylum-level trends were seen in humans [35]. Thus, further studies on restoring the balanced ratio between these two dominant phyla in the gut microbiota could potentially lead to the development of a therapeutic intervention for humans.
Meanwhile, in the novel ADLP (AD-like pathology) mouse, which exhibits both amyloid and tau pathology [80], analyses of the gut microbiota revealed chronic intestinal and systemic inflammation resulting from impaired integrity of the epithelial barrier [81]. In triple-transgenic model (3xTg-AD) harboring PS1 (M146V), APP (Swe), and tau (P301L) transgenes [82], relatively decreased abundances of Actinobacteria and TM7 compared to wild-type were observed already at 3 months of age [83]. Also, 3xTg-AD mice had lower concentrations of Anaerostipes, which plays a crucial role in gut health by producing butyric acid than wild-type [84]. Further investigations are required to reveal the synergistic effect of amyloid and tau on altering the gut microbiota.
Evidence from observational studies therefore strongly indicates that there is a significant link between gut dysbiosis and AD. In support of this link, animal models primarily designed to mimic the diseased brain also exhibit alterations in the gut microbiome. Researchers have therefore implemented various strategies to actively manipulate the microbiome in animal models, in an effort to better understand how gut perturbations can induce AD phenotypes.
Germ-free models
GF model animals are bred in isolators that keep them from contacting any microorganism. Using this model, researchers can observe how a pathology changes in the complete absence of microbes or investigate how select microbes affect the disease by generating gnotobiotic animals. When select microbiota is transplanted, the initial colonization effect can be observed without any interference from pre-existing microbes.
Harach and colleagues found that cerebral Aβ pathology is drastically reduced in APP/PS1 transgenic mice that completely lack a gut microbiota. Interestingly, when GF AD model mice were colonized with microbiota from their conventionally raised counterparts, the cerebral amyloid level was increased. The microbiota from the WT counterpart did not induce amyloid pathology as much as that from the disease model, strongly suggesting that the composition of the microbial environment is associated with the development of amyloid pathology [85]. Fujii et al. created a gnotobiotic mouse model by transplanting GF C57BL/6N mice with human gut microbiota from a healthy volunteer and an AD patient. The mice that received the healthy microbiota performed better on several behavioral tests, such as the object recognition and object location tests. Moreover, the fecal levels of neurotransmitters and CNS metabolites, such as gamma-aminobutyrate, taurine, and valine, were significantly decreased mice that received the diseased microbiota [86].
Antibiotics treatment
Antibiotics-treated mice can be a more affordable and easily accessible alternative to GF mice, as the former do not require high-cost facilities and cumbersome isolators. However, antibiotics can selectively deplete specific types of microbes via different mechanisms of action, meaning that the incomplete ablation of microbes should always be kept in mind. Moreover, the potential side effects of antibiotics on pathological phenotypes should be carefully monitored.
In AD research, antibiotics are often used to create dysbiosis within the gut to uncover how the perturbed microbiota impacts disease progression. Minter and colleagues initially reported that antibiotics treatment of APP/PS1 mice perturbed the innate gut microbiome and influenced neuro-inflammation and amyloidosis [87]. The authors found that long-term application of a broad-spectrum antibiotic cocktail significantly shifted the gut microbial composition and diversity. Surprisingly, this modulation of gut microbiota was sufficient to decrease Aβ plaque deposition in the brain, increase the level of soluble Aβ, alter the circulating cytokine signatures, and attenuate plaque-associated glial reactivity. This prompted the same research group to investigate how antibiotics-mediated perturbation of the microbial diversity of APP/PS1 mice during post-natal development impacts amyloid pathology when the animals aged [88]. Contrary to the chronic exposure performed in the former study, acute treatment at this stage was sufficient to reduce brain Aβ deposition in aged model mice. Also, the antibiotics-treated group had fewer plaque-localized microglia and astrocytes. As the early post-natal development period is critical for neuro and immune development, these results showed that the gut microbiota is involved in brain development and the onset of AD.
Although antibiotics treatment studies provide helpful insights, they do not clarify the involved bacterial strain(s) or mechanism(s). For example, while the synergistic alterations of gut microbial consortia achieved by applying an antibiotics cocktail of kanamycin, metronidazole, gentamicin, colistin, and vancomycin was shown to reduce amyloidosis in APPPS1-21 mice, the same effect was not achieved in mice treated with the individual antibiotics, complicating efforts to isolate the causative bacterial strain(s) for AD [89]. Chen and colleagues sought to utilize antibiotics to elucidate the underlying mechanism that links gut dysbiosis and AD. In the 5XFAD model, they initially found that gut dysbiosis was aggravated with age, leading to activation of the CCAAT/enhancer binding protein β/asparagine endopeptidase pathway. The team applied antibiotics to see if gut microbes mediate this pathway, and found that this treatment diminished the signaling and attenuated amyloidogenic processes, ultimately yielding cognitive improvements [77].
Despite the potential beneficial effects of antibiotics treatment in transgenic AD mouse models mentioned above, the debate over the efficacy of antibiotics in treating AD has never reached a consensus. While some studies report that antibiotics effectively removed amyloid plaques from the brain [87, 88], others report that cognitive impairment was worsened following gut dysbiosis [90] or antibiotics treatment had no beneficial effects on cognitive function in AD [91]. The controversy is mainly due to intrinsic limitations and biases regarding the studies. The initial composition of the gut microbiota at the time of antibiotics treatment could be different due to numerous factors such as disease state, environmental factors, and dietary habits. Also, as certain types of antibiotics selectively remove specific bacterial types [92], the effect depends on the type and combination of antibiotics. The treatment time point, period, and dosage could also be essential factors.
Probiotics treatment
Probiotics are live microorganisms that have beneficial effects on our bodies when consumed. Numerous probiotic products are commercially available and consumed by humans as dietary supplements. Since the concept is already familiar to consumers, it would be easy to implement the administration of probiotics as a therapeutic strategy for curing AD.
In a study conducted with 3xTg-AD model mice, a probiotics formulation of nine live bacterial strains (called SLAB51) significantly reduced oxidative stress in the brain by activating sirtuin 1 (SIRT1)-dependent mechanisms. There was no significant change in brain amyloid, but there were changes in the brain levels of antioxidant enzymes, such as superoxide dismutase and glutathione peroxidase, and consequent alterations in protein and lipid oxidation states. These results indicated that probiotics supplementation could be a promising approach to treat AD and other neurological disorders that involve oxidative stress [93]. In an animal model of AD made by intracerebroventricular injection of Aβ into mice, oral administration of Bifidobacterium breve strain A1 (B. breve A1) had beneficial effects on behavior. Interestingly, while B. breve A1 administration had only minor effects on the overall composition of the gut microbiome, it increased the plasma acetate level and suppressed Aβ-induced gene expression changes in the brain [94]. In Aβ-injected rats, behavioral benefits were observed, and long-term potentiation was suppressed upon administration of probiotics, indicating that probiotics could even exert positive effects on synaptic plasticity [95]. Similarly, treatment with Lactobacilli and bifidobacteria was found to ameliorate memory and learning deficits and reduce oxidative stress in Aβ-injected rats [96].
In a mammalian gut microbiota composed of enormously diverse microbes, it is unlikely that a single pathogenic microbial type could be responsible for the dysbiosis of the entire microbiome. Likewise, it is not expected that a single microbial type will play a vastly protective role in AD. Rather, restoring the overall homeostatic condition of the gut should be the primary focus in efforts to treat AD. Therefore, obtaining well-balanced gut microbiota from healthy controls and settling them in the guts of patients by transplantation could be an effective therapeutic tactic. This tactic is usually implemented in the form of the FMT, as mentioned above. Indeed, FMT has already shown efficacy in mouse models of AD.
In a study conducted by Sun et al., FMT to APP/PS1 mice improved cognitive deficits, reduced the brain deposition of Aβ, and decreased the levels of phosphorylated tau, Aβ40, Aβ42, cyclooxygenase-2 (COX-2), and cluster of differentiation molecule 11b [97]. In an ADLPAPT mouse model exhibiting both amyloid and tau pathology, FMT from healthy WT mice into ADLPAPT mice significantly alleviated AD pathology. The formations of Aβ plaques and NFTs were reduced, glial reactivity was attenuated, cognition was improved, the aberrant gene expression related to intestinal macrophage activity in the colon was reversed, and the proper population of circulating monocytes in blood was restored [81]. These results from FMT studies indicate that colonizing the gut with a balanced set of microbes aimed at restoring the homeostatic condition can be more beneficial to AD symptoms than transplanting a specific subset of microbes.
Modulation of diets
Administration of dietary nutrients, which range from micronutrients (polyphenols, vitamins, minerals, etc.) to macronutrients (carbohydrates, fat, protein, etc.), can significantly alter the microbial composition of the gut [98]. Therapy based on dietary modulation is already being used to treat metabolic diseases, such as obesity, diabetes, and IBS [99]. Supplements comprising metabolites that help restore the homeostatic microbial community in the gut (as an alternative to those containing the organisms themselves) are being widely investigated for their potential to treat AD. For example, sodium oligomannate (GV-971) was found to suppress gut dysbiosis and the associated phenylalanine/isoleucine accumulation in 5XFAD mice, leading to cognitive improvements [100]. Moreover, oligosaccharides derived from Morinda officinalis were found to alter the gut microbiota and metabolome in APP/PS1 mice [101].
Calorie restriction (CR) can be another alternative. In 15-month-old female Tg2576 mice, long-term CR feeding was found to reduce the Aβ plaque load. Sustained CR also reduced the expression of γ-secretase complex components, such as PS enhancer 2 and PS 1 [102]. Similarly, Cox et al. found that CR slowed age-related microbiota changes in Tg2576 mice [74]. The authors postulated that this was related to the type strain of Bacteroides fragilis, which is known to increase Aβ plaques in the brain. The highly varied carbohydrate and polysaccharide utilization genes of these Bacteroides species are essential for their ability to maintain colonization, but the CR diet limits calories specifically from carbohydrates. Thus, the CR diet might have directly impacted Bacteroides.
Together, these findings indicate that there is a solid link between gut dysbiosis and AD. In order to cure AD patients, various research groups have focused on restoring the homeostasis of the gut microbiome. The mechanism that governs the connection between AD and the GBA is unclear, however, and must be elucidated in detail if we hope to refine and optimize related treatment strategies. In the next section, we discuss the possible mechanisms by which gut dysbiosis could lead to AD pathogenesis.
POSSIBLE MECHANISMS THROUGH WHICH THE GUT MICROBIOTA CONTRIBUTES TO AD
Cross-seeding effect
Since the gut microbiota can excrete a considerable amount of amyloid peptide and LPS [103], gut dysbiosis might perturb the overall composition of microbial-derived molecules. The majority of bacterial amyloid peptides contribute to adhesion, host cell invasion, and host-pathogen interaction by forming a biofilm [104]. Biofilm-forming bacteria are much more resistant to antibiotics, viruses, and other harmful substances, which greatly contributes to the protection and growth of bacterial communities [105]. Chapman et al. proposed that curli, the first identified functional amyloid, is mainly produced by E. coli and Salmonella species and shares structural and physical properties with the pathogenic amyloids [106]. Therefore, the biofilm-forming gut bacteria might directly affect the AD pathogenesis through the cross-seeding of neuronal amyloid aggregates by bacterial amyloids. CsgA, which is the major curli subunit, has a typical cross-β architecture reminiscent of human pathological amyloids, and CsgA seeds were shown to accelerate AD-associated Aβ fibrillation [107, 108]. Structural similarity between amyloids is required for the epitaxial heteronucleation that forms the underlying mechanism of the cross-seeding reaction. Importantly, full-length CsgA has a fibril structure sufficiently similar to that of Aβ1–40 and Aβ1–42, enabling it to act as a template for fibril elongation [109]. Gram-negative bacteria produce amyloid peptides for biofilm formation, such as FapC from Pseudomonas fluorescens [110]. Structurally, FapC possesses several Aβ-binding sequences and a region that enables it to be incorporated into Aβ nanofibrils via seeding. FapC was absorbed onto the Aβ fibril surface and altered the morphology and toxicity of Aβ in a zebrafish AD model [111]. Many amyloid peptides are produced by the gut microbiome, and the amyloid-producing enteric bacteria might cause nucleation and fibrillation of brain amyloids in AD [112]. Similar to bacterial amyloid, LPS, which is a major component of the outer membrane of Gram-negative bacteria, was also found to promote Aβ fibrillization in vitro [113]. In vivo, bacterial LPS injection increased Aβ deposition and tau aggregation in the hippocampus of model mice [114, 115]. Given this, it is likely that LPS detected in the brain of AD patients can promote the formation of Aβ plaques [116]. Cross-seeding of bacterial amyloid misfolds and LPS are known to either access the brain directly via the autonomic nervous system in a manner similar to prion disease [117], or cross the defective blood-brain barrier (BBB) in AD.
High gut permeability and gut inflammation
Under the healthy condition, the gut epithelium plays a pivotal role in biological functions, such as the absorption of nutrients, ions, and water, and performs physical barrier functions to protect the host against invasion of gut bacteria and toxins [118]. The gut barrier is the first line of defense against pathogenic molecules and harmful microbes, and an imbalance of the gut microbiota leads to barrier dysfunction [119, 120]. Gut barrier breakdown and increased permeability have been described in GI disorders, including IBD (Crohn’s disease and ulcerative colitis) [121]. Patients with IBD show a higher prevalence of anxiety and depressive symptoms and exhibit issues with concentration, memory, and executive function [122, 123]. The psychomotor and cognitive impairments seen in GI disease support the hypothesis that high gut permeability may negatively impact brain functions.
The gut mucus layer
The gut barrier is a multiple-layer system composed of the mucus layer, the epithelial layer, and the underlying lamina propria [124]. The mucus layer consists of an outer layer that is colonized by commensal bacteria and an inner layer that protects the gut epithelial cells from the bacteria and potential pathogens in the lumen. Damage to the inner layer can increase the direct contact between the bacteria and the gut epithelium, resulting in gut barrier dysfunction and allowing bacteria to penetrate through the epithelial tissue into the circulation [125]. The mucus layer comprises large glycoproteins, called mucins, that are predominantly produced and secreted by goblet cells [126]. Therefore, the regulation of the secretory goblet cells is key to maintaining the mucus barrier. Commensal bacteria and their metabolic by-products (e.g., SCFAs such as acetate, propionate, and butyrate) have been implicated in the differentiation of goblet cells to synthesize and secrete mucin [127–129]. Some bacterial residents in the mucus layer can degrade and utilize the mucus component as their carbon and energy sources [130]. Taken together, the available evidence indicates that the microbial composition determines the properties and integrity of the mucus layer, influencing its permeability (Fig. 1).
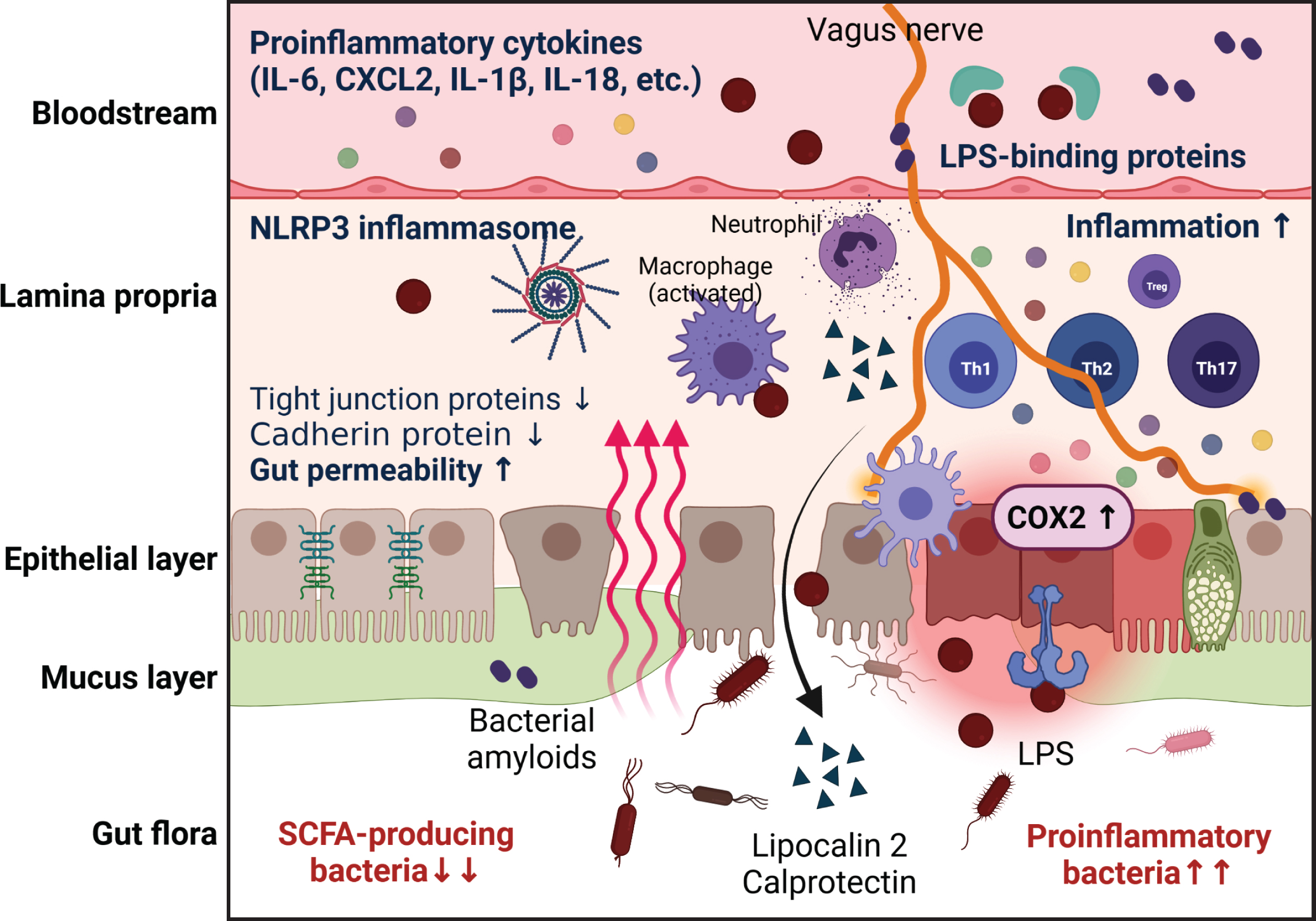
Potential mechanisms through which gut dysbiosis contributes to gut pathology in AD. The imbalanced gut microbial composition induces impairment of the gut barrier; this is accompanied by mucus layer degradation, epithelium damage, and gut inflammation. The gut dysbiosis-mediated increase in gut permeability facilitates the translocation of bacteria into the circulation to cause systemic inflammation. The increased pro-inflammatory bacteria also induce aberrant immune activation, which reinforces and perpetuates the inflammation. High gut permeability and chronic gut inflammation induce translocation of bacteria-derived molecules and inflammatory mediators into the bloodstream and brain.
AD patients are characterized by decreased populations of butyrate-producing species, such as members of genera Butyrivibrio (B. hungatei and B. proteoclasticus) and Eubacterium (E. eligens, E. hallii, and E. rectale), as well as Clostridium sp. strain SY8519, Roseburia hominis, and Faecalibacterium prausnitzii [45]. Another study found that butyrate producers in the gut, such as members of the Lachnospiraceae and Ruminococcaceae families, were decreased in AD patients compared to healthy controls [131]. Furthermore, the potential butyrate producer, Lachnospiraceae NK4A136 group, was found to be reduced in patients with dementia [132]. Consistent with the results from AD patients, Zhang et al. showed that the microbiota composition and diversity were perturbed and the SCFA level was reduced in APP/PS1 mice compared to WT mice [71]. Jiminez et al. provided evidence that supplementing mice with a high concentration of butyrate could reduce the severity of a pathogenic bacterial insult through epithelial barrier repair and mucus secretion [133]. Sodium butyrate treatment was also shown to have profound effects on brain Aβ levels, associative learning, and cognitive function in 5XFAD mice [134]. These findings suggest that increasing butyrate-producing bacteria may be a promising therapeutic strategy for AD.
Mucolytic bacteria, such as Akkermansia muciniphila and Bacteroides thetaiotaomicron, have protective effects on cognitive deficits and amyloid pathology in mouse models of AD [135, 136]. Although it was previously thought that mucin degradation was detrimental to gut health, it is now accepted as a normal physiological process that promotes mucus layer turnover [137]. Research findings that abundant mucin-degrading bacteria can improve gut barrier function [138–140] suggest that such bacteria may be beneficial microbes in AD.
The gut epithelial layer
The gut epithelium is a single-cell layer that separates the luminal gut microbiota from the host. Its integrity and permeability are maintained by adhesive complexes, such as desmosomes, adherens junctions (AJs), and tight junctions (TJs) [141]. Epithelial TJs, which represent the uppermost apical component of intracellular junctions, consist of transmembrane proteins (claudin, occludin, etc.) and cytosolic scaffold proteins (zonula occludens (ZO-1)). TJs regulate the paracellular movement of ions, solutes, and water across the gut epithelium, and also constitute biological barrier against pathogens and their toxins. AJs and desmosomes, which are located beneath TJs, provide essential adhesive and mechanical properties that also contribute to barrier function [142]. The gut epithelium is not a static system relative to the external environment; rather, its structure is greatly affected by enteric pathogens and their secreted toxins. The interaction of gut bacteria with epithelial cells has been proposed as a major contributor to TJ remodeling and gut permeability [143, 144], and specific bacteria-induced mechanisms of TJ disruption have been elucidated. For example, Clostridium difficile and Escherichia coli-induced TJ disruption was suggested to reflect changes in the cytoskeleton and the redistribution of proteins, including occludin and ZO-1, from TJs. Other enteric pathogens, such as Bacteroides fragilis and Vibrio cholera, were found to secrete metalloprotease and disrupt TJs by proteolytic degradation of TJ proteins [143]. As researchers recognized that the intestinal epithelial response to enteric pathogens can lead to perturbation in barrier functions, studies began to focus on the relationship of gut dysbiosis to the leaky gut. Increased gut permeability has frequently been reported in AD patients and model mice with imbalanced gut microbial composition and gut inflammation, and the involvement of increased gut permeability in disease progression is a research topic of growing interest (Fig. 1).
In APP/PS1 mice, findings of dilated TJs and downregulation of TJ proteins, including occludin, claudin 1, claudin 5, and ZO-1, indicated that intestinal barrier integrity is impaired. Observed increases in serum diamine oxidase and d-lactate, which are indicators of gut permeability, further supported the idea that there is severe gut barrier dysfunction in these mice [145]. To determine the effect of AD conditions on the integrity of colonic epithelial TJs and AJs, distal colon cryosections from young 5xFAD and their age-matched non-Tg littermates were stained for TJ (occludin and ZO-1) and AJ (E-cadherin and β-catenin) proteins. The results revealed that the junctional distributions of occludin and ZO-1 were reduced and the distal colon expression levels of E-cadherin and β-catenin were decreased in 5xFAD mice [146]. Fluorescein isothiocyanate-dextran assay of plasma, which can easily measure the pericellular permeability of the gut epithelium, revealed that AD model mice, including 5XFAD, 3xTg-AD, ADLPAPT, and APP knock-in mice, showed higher gut permeability than control WT mice [77, 81]. Due to the high gut permeability seen in AD patients, gut bacteria-derived pathogens, such as LPS and endotoxins, may be translocated into the systemic circulation and/or brain [147]. In AD brain lysates, the presence of Escherichia coli K99 and bacterial LPS was identified, and LPS was found to localize with Aβ1–40/42 in amyloid plaques and around vessels in the brain [116, 148]. A 16s rRNA sequencing analysis showed that the bacterial load is higher in AD brain compared to control brain, and the top-hit bacterial species were proposed to be microbial contributors to AD-associated neuroinflammation [149]. Taken together, these data indicate that the gut microbial environment in AD favors gut barrier disruption, which in turn induces the infectious agents to translocate into the blood and brain (Fig. 1).
The gut lamina propria
In healthy conditions, the gut immune system prevents the invasion of pathogenic bacteria. However, when dysbiosis occurs, harmful bacteria-derived enterotoxins perturb the immune system, causing chronic inflammation to increase gut permeability [150]. Located underneath the epithelial layer, the lamina propria is a thin layer of connective tissue that consists of innate and adaptive immune cells, such as neutrophils, macrophages, T cells, and B cells. The lamina propria plays a pivotal role in the communication between the microbiome and immune cells to maintain gut barrier homeostasis [151]. Pattern recognition receptors (PRRs) on the gut epithelial cell surface detect bacterial antigens and transmit signals from bacteria to adjacent immune cells [152]. The PRRs sense microbial antigens through pathogen associated molecular patterns (PAMPs), such as bacterial glycans, nucleic acids, and peptides. Upon the recognition of a PAMP, PRRs trigger intracellular signal transduction that leads to the upregulation of inflammatory genes, the production of pro-inflammatory cytokines, and the recruitment of innate and adaptive immune cells [153]. Macrophages can phagocytose the infectious agents and play a critical role in initiating and resolving inflammation by shifting between the pro-inflammatory M1 and anti-inflammatory M2 phenotypes [154]. Activated macrophages and dendritic cells present the antigens to naive T cells, leading to the differentiation and activation of various effector T cell subsets (Th1, Th2, Th17, and regulatory T cells (Tregs)) or the production of IgA from B cells [155, 156]. The gut immune cells are involved in host defense against pathogenic bacteria and govern the sophisticated modulation of unnecessary immune activity to maintain tissue homeostasis (Fig. 2).
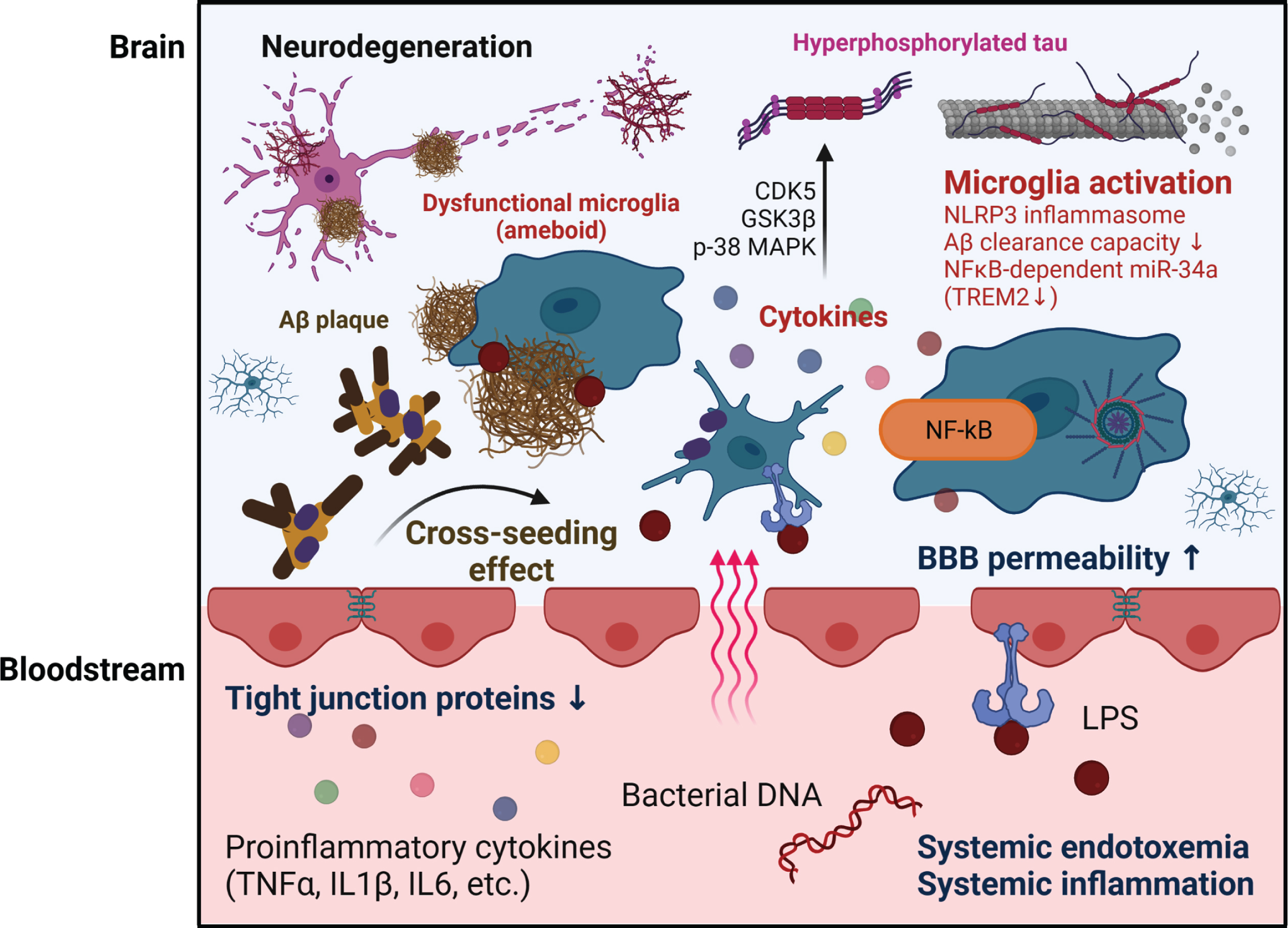
Potential mechanisms through which gut dysbiosis contributes to brain pathology in AD. The crossing of bacteria-derived amyloids and LPS through the permeable BBB can promote amyloid aggregation and provoke microglia activation in the brain. The cross-seeding ability of bacterial amyloid involves a synergic effect on Aβ aggregation in the brain. The loss of BBB integrity due to bacterial LPS and/or circulatory inflammatory cytokines initiates the activation of microglia, which are resident immune cells in the brain. The persistent microglial activation driven by chronic systemic inflammation due to gut dysbiosis and gut permeability results in uncontrolled inflammatory responses and loss of microglia function. This neuroinflammation increases Aβ accumulation and facilitates tau phosphorylation, synaptic loss, and neuronal damage in AD.
The gut microbiota composition is crucial for regulating the balance and homeostasis of various helper T (Th) cell populations in the lamina propria. During the colonization of the ubiquitous gut microorganism, Bacteroides fragilis, in animals, a bacterial polysaccharide presented by intestinal dendritic cells mediates the establishment of a proper Th1/T-h2 cell balance, which supports healthy immunologic functions [157]. In adaptive immunity, the balance achieved by reciprocal regulation between Tregs and Th17 cells is vital to preventing a chronic, over-active, or ineffective immune response. In particular, colonic Tregs, which are important for maintaining gut immune homeostasis via protective immunosuppressive molecules, can be activated or expanded by SCFAs, gut microbiota-derived bacterial fermentation products, and specific spore-forming bacteria (Clostridium clusters IV and XIVa) [158, 159]. The differentiation of Th17 cells was found to correlate with the presence of Gram-negative members of the cytophaga-flavobacter-bacteroidetes (CFB) phylum in the intestine [160]. These findings indicate that commensal bacteria are pivotal in developing the immune system and maintaining immune homeostasis, whereas an imbalanced gut microbiota activates an uncontrolled immune response that leads to prolonged production of inflammatory cytokines. This is accompanied by deterioration of the barrier function of the gut epithelium via the disassembly of TJs, and eventual cell death [161]. Consequently, inflammatory mediators are permitted to enter the circulatory system and cause systemic inflammation, which is the major hallmark of chronic inflammatory disease.
In the elderly, an aging-induced shift in the normal gut microbiota composition is associated with the manifestation of immunosenescence and the development of a pro-inflammatory environment (inflamm-aging) [162]. The contributions of pro-inflammatory intestinal bacterial taxa and the peripheral inflammatory state have been reported in the elderly suffering from brain amyloidosis and cognitive impairment. A positive correlation was found between pro-inflammatory cytokines (IL-6, CXCL2, NLRP3, and IL-1β) in the blood and the number of pro-inflammatory intestinal bacteria belonging to the Escherichia/Shigella taxon in stool samples of patients with dementia. As expected, a negative correlation was found between pro-inflammatory cytokines and the number of intestinal bacteria of the E. rectale taxon, which are known for their anti-inflammatory effects [44]. In AD patients, the numbers of pro-inflammatory bacteria, such as Gammaproteobacteria, Enterobacteriales, and Enterobacteriaceae of phylum Proteobacteria, were reported to increase with clinical severity [131].
Calprotectin is a cytosolic protein that is released by neutrophils in response to inflammation. It is noticeably elevated in both plasma and stool samples of patients with inflammatory conditions, including IBD [163]. The gut inflammatory state can be indirectly assessed by evaluating the level of calprotectin. This level was found to be significantly increased in the CSF and brains of AD patients, and calprotectin was found to promote the co-aggregation of Aβ [164]. Another study found that the fecal calprotectin level was elevated in nearly 70% of AD patients, prompting the researchers to hypothesize that the calprotectin found in feces may have been translocated from the leaky gut into the blood, ultimately contributing to neuroinflammation [165]. Another representative biomarker for gut inflammation, lipocalin 2 (LCN2), is secreted primarily from intestinal epithelial cells and myeloid cells in response to tissue injury, bacterial infection, or inflammation [166]. A higher concentration of fecal LCN2, indicative of intestinal inflammation, was found in ADLPAPT mice compared to WT mice [81]. Moreover, the immunoreactivity of the general intestinal regulator and pro-inflammatory protein, COX-2, was increased in the intestinal tissues of AβPP/PS1 mice compared to those of WT mice [167].
NLRP3, a member of the NLR family of innate immune receptors, induces the assembly of inflammasome complexes in the presence of microbial ligands, triggering the activation of caspase-1 and the secretions of IL-1β and IL-18 [168]. Many previous studies have reported that the NLRP3 inflammasome contributes to intestinal inflammation and the pathogenesis of IBD [169]. Shukla et al. observed increased NLRP3 inflammasome expression and IL-1β production in the gut of 5XFAD mice with compromised gut barrier function, as evident from the loss of TJ and AJ proteins [146]. In another study providing direct evidence, fecal transplantation from AD patients to APP/PS1 transgenic mice was reported to induce intestinal NLRP3 inflammasome activation and pro-inflammatory cytokine production to exacerbate neuroinflammation and cognitive deficits [170]. Thus, NLRP3 inflammasome activation caused by gut dysbiosis plays a critical role in neuroinflammation and AD-related neuropathology. In sum, the information gathered above suggests that the imbalanced composition of gut microbiota in AD induces neuropathology through gut inflammation and the concomitant release of inflammatory mediators (Fig. 1).
Bacterial translocation and systemic inflammation
The loss of gut barrier integrity is detrimental to the host because it allows bacteria-derived molecules to be translocated into the circulatory system and consequently induces systemic endotoxemia [171]. For example, in the peripheral blood of patients with IBD, several studies detected live intestinal bacteria by assessing the bacterial DNA concentration in patients’ sera [172–174]. Thus, leaky gut syndrome is considered to be an initial event that takes place at the onset of IBD. Leaky gut syndrome is a well-characterized phenomenon in other metabolic diseases similar to IBD, which also have a high probability of accompanying endotoxemia. A number of studies showed that in a mouse model with high-fat-diet-induced diabetes and in patients with type 2 diabetes, live gut bacteria and bacteria-derived LPS were translocated into the blood, where they induced metabolic endotoxemia and inflammation [175–177]. In support of this notion, it is known that patients with IBS who share the same risk factor, the bacterial translocation, are also highly susceptible to metabolic disease such as obesity-induced type 2 diabetes [144, 178–180]. These results support the idea that bacterial translocation and systemic inflammation can influence the pathologies of AD patients with loss of gut barrier function.
Blood-brain barrier disruption
In the absence of infection, levels of bacterial endotoxin in the bloodstream may be mostly attributed to the permeable gut barrier. Thus, it is hypothesized that the dysbiotic gut microbiota in AD may increase gut permeability and impair BBB integrity, allowing infectious agents and/or inflammatory factors to access the brain. The BBB, which is a component of the neurovascular unit, comprises neurons, glial cells (astrocytes, microglia, oligodendrocytes), and vascular cells (pericytes, vascular smooth muscle cells, endothelial cells) [181]. It has increasingly been recognized that BBB damage can provoke neuroinflammation and is sufficient to modify neuronal activity in many CNS diseases, including AD [182–184]. Recent neuroimaging studies in patients with early AD revealed that BBB breakdown is seen in several brain regions prior to brain atrophy or dementia [185–187]. Biomarker studies have also detected BBB disruption in AD before cognitive decline or other brain pathologies [188–190]. These studies suggested that the BBB leakage in AD might be part of a cascade leading to cognitive impairment and dementia. Thus, it is notable that the translocation of gut microbes and their derivatives to blood can cause BBB impairment and neuroinflammation, contributing to the pathogenesis of AD (Fig. 2).
The endothelial cells of the BBB separate the brain from the circulation to protect the CNS from neurotoxins in the periphery, while simultaneously playing a crucial role in transporting serum factors into the brain to maintain brain homeostasis [191]. Lipoteichoic acid and LPS, which are the major cell wall components of Gram-positive and Gram-negative bacteria, respectively, are known to induce inflammation and impair the structure and function of the BBB. Lipoteichoic acid and LPS interact directly with the endothelial cells of the BBB via toll-like receptors (TLRs; TLR2, TLR4) to trigger the release of pro-inflammatory cytokines associated with increased BBB permeability [192]. Indirectly, lipoteichoic acid and LPS activate the body’s response to blood endotoxin, resulting in elevated plasma concentrations of cytokines (TNF-α, IL-1β, and IL-6) and triggering the degradation of TJ proteins of endothelial cells to decrease the paracellular integrity of BBB [193, 194]. The disassembled BBB may allow endotoxin to cross the BBB and accelerate the aggregation of Aβ and tau within the AD brain [195]. Overall, BBB breakdown promotes the entry of blood-derived molecules and microbial agents into the CNS and is associated with neuroinflammation and neurodegeneration [196] (Fig. 2).
Braniste et al. reported that GF mice displayed increased BBB permeability compared to SPF mice with normal gut microbiota. The increased BBB permeability seen in GF mice was associated with reduced expression of the TJ proteins, occludin and claudin-5, in endothelial cells. Moreover, fecal transfer of normal gut microbiota or transplantation of SCFA-producing bacteria into GF mice reduced BBB permeability and restored the expression of TJ proteins [197]. Hoyles et al. found that propionate, an SCFA produced from dietary substrates by colonic bacteria, has beneficial effects on the BBB, alleviating the effects of deleterious inflammatory and oxidative stimuli. Propionate inhibits pathways related to non-specific microbial infections via a cluster of differentiation 14 (CD14)-dependent mechanism. It also suppresses the expression of low-density lipoprotein receptor-related protein 1 and protects the BBB from oxidative stress via nuclear factor erythroid-2-related factor 2 signaling [198]. These findings suggest that certain bacterial types can aid BBB maintenance, helping to delay AD pathology.
Neuroinflammation
Microglia are the primary resident macrophages in the CNS and are considered to be immune sentinels that are responsible for initiating an innate immune response upon receipt of diverse damage signals in the brain [199]. Under normal conditions, homeostatic microglia (resting microglia) continuously survey their local environment via highly motile processes. In response to brain injuries or immunological stimuli, microglia are capable of rapidly transitioning to an activated state characterized by alterations in their proliferation rate, morphology, and phagocytic activity, and thereafter scavenge the CNS for damaged neurons, infectious agents, and/or foreign substances [200]. Microglia are equipped with a wide range of membrane receptors that enable them to sense alterations in the local environment, and activated microglia produce the inflammatory factors, including cytokines and chemokines, which enable them to fulfill their immune functions [201]. However, prolonged microglial activation leads to the excessive release of pro-inflammatory cytokines and reactive oxygen species, resulting in detrimental consequences for neurons and other glial cells in the brain. Indeed, accumulating evidence indicates that a chronic microglial activation status is implicated in the pathology of neurodegenerative diseases [202], and uncontrolled inflammatory responses and microglial dysfunction due to persistent microglial activation were revealed as major contributors to AD pathology [203].
Due to microbial dysbiosis and the resulting increased gut permeability, certain pro-inflammatory cytokines that are elevated in plasma can cross the BBB and likely contribute to the neuroinflammation initiated by microglia. The peripheral inflammatory cytokines that are used as diagnostic biomarkers for AD [204] could be reflecting gut dysbiosis. The loss of BBB integrity following increased gut permeability in AD may allow bacterial components and metabolites to enter the CNS and trigger aberrant microglial activation [205]. Based on recent studies showing that microglia activation is profoundly affected by gut inflammation and bacteria-derived materials, the imbalanced gut microbiota composition and high gut permeability underlying these symptoms can be suggested as major risk factors for AD (Fig. 2).
Gut inflammation. In mice that received intragastric administration of LPS and ethanol, increased levels of BBB-permeable pro-inflammatory cytokines (TNF-α, MCP-1, IL-1β) in the liver and serum were found to induce microglia activation, leading to morphological changes with reduced neurogenesis [206]. The gut inflammation induced by intracolonic administration of 2, 4, 6-trinitrobenzene sulfonic acid was found to increase hippocampal susceptibility to TNF-α-mediated neuronal excitability arising from microglial activation [207]. 3xTg-AD mice (an AD mouse model) were reported to be susceptible to gut infection, which can trigger inflammation, resulting in exaggerated microglial activation [208]. A systemic immune challenge was shown to predispose WT mice to develop age-associated AD-like phenotypes and cognitive decline [209]. Several studies have thoroughly documented the neuroinflammation, amyloidogenesis, tau phosphorylation, and memory impairment that arises following LPS-induced systemic inflammation in AD model mice [115, 210]. Tejera et al. investigated microglial changes in APP/PS1 mice following peripheral immune challenge with intraperitoneal LPS injection. The microglia exposed to peripheral LPS challenge were particularly poor in their Aβ clearance capacity. The impaired phenotype of microglia was restored by NLRP3 knockout, indicating that the NLRP3 inflammasome is deeply involved in the microglial activation that occurs during systemic inflammation [211]. These results suggest that peripheral inflammation contributes to the progression of AD. This hypothesis is supported by clinical studies showing that both acute and chronic systemic inflammation are followed by increased cognitive decline in AD [212]. Thus, systemic inflammation due to microbial dysbiosis can affect microglia in AD, further exacerbating neuroinflammation and accelerating the progression of AD.
Bacterial LPS and gut microbiota-secreted amyloids. Bacterial LPS and gut microbiota-secreted amyloids are also known to directly contribute to the microglial inflammatory response. LPS binds to microglial receptors (TLR2, TLR4, and CD14) and activates the signaling of the myeloid differentiation primary response gene 88-dependent nuclear factor kappa B (NF-κB). The NF-κB signaling pathway can evoke inflammatory responses through a cascade of cytokines and chemokines [213]. These inflammatory mediators eventually act on components of AD pathogenesis, including Aβ production/deposition and tau phosphorylation. For example, the inflammatory cytokines, IL-1β, IL-6, IL-18, TNF-α, and interferon-gamma, increase the synthesis and processing of APP in astrocytes and/or neurons. Most inflammatory cytokines are linked to the activation of several tau kinases in the brain, including cyclin dependent kinase 5, glycogen synthase kinase-3β, and p-38 mitogen-activated protein kinase, to provoke abnormal tau phosphorylation at AD-associated residues [214]. Hence, the neuroinflammatory processes triggered by bacterial LPS can contribute to the formation of both Aβ plaques and NFTs and can perpetuate as a vicious cycle, thereby playing active roles in AD progression.
In addition to LPS, amyloids produced by some bacterial species can easily pass through a compromised gastrointestinal tract or BBB and accumulate in the AD brain [215]. Bacterial amyloids are also recognized as PAMPs by TLR1/2 and CD14 to induce the innate immune response through NF-κB signaling, thereby eliciting the production of pro-inflammatory chemokines and cytokines [216]. Therefore, it has been suggested that microglial activation by bacterial amyloids in the brain may alter the misfolded protein load in neurodegenerative diseases. Chen et al. verified this hypothesis by comparing rats exposed to curli-producing bacteria or curli-deficient bacteria. Compared to rats exposed to mutant bacteria unable to synthesize curli, rats exposed to curli-producing bacteria displayed increased neuronal α-syn deposition in both gut and brain, along with enhanced inflammation indicated by microgliosis and astrogliosis [217]. In the AD brain, both LPS and microbial amyloids can alter microglial function through NF-kB-mediated pathways. NF-kB-dependent microRNA-34a (miRNA-34a) can downregulate a triggering receptor expressed on myeloid cells 2 that is directly involved in Aβ peptide sensing, phagocytosis, and clearance in microglia [218]. LPS-induced NF-kB stimulates miRNA-146a and miRNA-155; these microRNAs potently modulate complement factor H (CFH), which regulates the alternative pathway of complement activation in innate immunity [219]. Given the findings that systemic CFH deficiency contributes to excessive activation of the complement pathways associated with autoimmune, host tissue damage, and persistent/chronic inflammatory responses [220, 221], it is reasonable to assume that NF-kB stimulating microRNAs may act on microglia-mediated inflammation.
Microbe-derived metabolites. Microbe-derived metabolites also contribute to microglial activation. SCFAs are the major end products of bacterial fermentation in the colon [222]. It has been well described that supplementation of GF WT mice with SCFAs reversed the phenotype and functions of stunted microglia through other peripheral myeloid cells that express free fatty acid receptor 2 [26]. However, since there is evidence that SCFAs can cross the BBB [223], the possible direct effect of SCFAs on microglia cannot be excluded. A recent study found that SCFA supplementation of GF APP/PS1 mice altered the microglial transcriptomic profile and caused apolipoprotein E upregulation in microglia, but not astrocytes, to promote Aβ deposition [224]. The results underscore that microbiota-derived SCFAs can be critical mediators along the GBA and potentially increase Aβ plaque load via altering microglia. Alteration of the gut microbiota was found to elevate the levels of phenylalanine and isoleucine in the periphery in 5XFAD mice to provoke pro-inflammatory Th1 cell infiltration to the brain with microglia activation. GV-971, a sodium oligomannate that inhibits phenylalanine and isoleucine accumulation, was found to attenuate neuroinflammation and reverse cognitive impairment, and was accordingly proposed as a treatment for AD pathology caused by gut dysbiosis [100]. Another study provided evidence that the butyrate-producing probiotic bacterium, Clostridium butyricum (CB), can attenuate microglia-mediated neuroinflammation via its metabolites [225]. Together, these studies show that microbial-related products can mediate microglia activity and may serve as potential therapeutic targets acting through the GBA in AD.
CONCLUSION
In summary, an imbalanced gut microbial composition in AD negatively impacts the properties and integrity of the gut barrier, as well as gut permeability. Gut dysbiosis and high gut permeability perturb gut immunity, thereby causing persistent inflammatory responses. A highly permeable gut also allows bacteria-derived molecules and inflammatory mediators to translocate into the bloodstream and the brain. Bacteria-derived substances such as amyloids and LPS cross the permeable BBB to promote amyloid aggregation and microglia activation in the brain. The persistent microglial activation driven by peripheral inflammation results in a blunted phagocytic activity and chronic inflammatory response. This neuroinflammation increases Aβ accumulation and facilitates tau phosphorylation. Thus, the imbalance in the gut microbiota can be proposed as a hidden player in triggering the neurodegeneration of AD (Fig. 3).

Graphical summary and table of the mechanisms of the gut-brain axis in AD. LPS, lipopolysaccharide; AD, Alzheimer’s disease; Aβ, amyloid-beta; SCFAs, short chain fatty acids; TJs, tight junctions; AJs, adherens junctions; LCN2, lipocalin 2; COX, cyclooxygenase; NLRP3, NOD-like receptor pyrin domain-containing protein 3; BBB, blood-brain barrier.
The extensive evidence presented in this review for possible mechanisms by which gut dysbiosis accelerates the onset of AD raises several points that need to be addressed. First of all, since the stages in which AD patients are in at the time of sampling are not specified in most studies, the relationship between gut microbes and different stages of AD is still unclear. One study shows that pro-inflammatory bacteria such as Enterobacteriales of the phylum Proteobacteria are progressively enriched from healthy control to mild cognitive impairment to AD, and the changes are significantly correlated with the clinical severity [131]. Another study involving 3xTG-AD mice suggests that gut microbiota alterations are already present in a pre-clinical stage of 3 and 5 months, at which no robust pathology is observed [83]. These studies collectively point out the importance of taking different AD stages into account. Chronologically ordering the alterations in gut microbiota and the concomitant AD lesions will further clarify the cause-and-effect relationship between AD pathogenesis and dysbiosis of gut microbiota. Furthermore, a longitudinal study spanning the course of disease progression or a cross-sectional study using strictly characterized cohorts of various disease stages will help broaden our understanding of it.
It is worth envisaging the possibility that gut microbiota alterations are the consequences of AD brain pathology rather than vice versa. Transgenic mouse models are driven by powerful transgenes that overexpress representative pathological proteins. The very fact that these mouse models of AD, which are designed to have pathological proteins overexpressed only in the brains, show alterations in gut microbiota indicates that the brain pathology can be a preceding event. Thus, a study on the link between AD and gut microbiota should always consider the interconnectivity and bidirectionality of GBA and retain a holistic viewpoint.
Lastly, the limitations of animal models of AD must always be kept in mind. AD animal models retain poor genetic diversity, cannot fully recapitulate physiologically-relevant disease phenotypes, and carry gene mutations for familial AD cases, representing only a minor portion of the overall patient population [226–228]. As failed clinical trials have told from the past, a successful amelioration of pathology in animal models does not necessarily mean that the treatments will work the same way in human physiology. Bacterial species that are beneficial in animals could potentially have no apparent effect or even be detrimental in humans.
In conclusion, the alteration of gut microbiota composition (gut dysbiosis) plays a critical role in the progression of AD. Multiple animal experiments focused on manipulating the gut microbiota in ways that have benefits in AD are paving the way for developing new therapeutic targets. There are still limitations to the translation of animal research results to patients, such as those arising from complications related to age, sex, genetic factors, and environmental factors. However, in the years to come, accumulating experimental evidence obtained through epidemiologic studies will provide a solid ground on which human clinical trials can stand. Given the growing body of evidence linking gut dysbiosis and AD pathogenesis, researchers should pay more attention to curing AD by modulating the gut microbiome. Improving a healthy gut environment via restoring balance to the gut microbiota composition could eventually be a breakthrough strategy for the treatment of AD.
Footnotes
ACKNOWLEDGMENTS
This research was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) and Korea Dementia Research Center (KDRC), funded by the Ministry of Health & Welfare and Ministry of Science and ICT, Republic of Korea (grant number: HU20C0187) to I. Mook-Jung. The work was also supported by a grant from the National Research Foundation of Korea (NRF-2020R1A6A3A1306860612) to H. Choi. All figures were created with the confirmation of publication and licensing rights from BioRender.