
Abstract
Background:
Physical activities (PA) have been suggested to reduce the risk of Alzheimer‘s disease (AD) dementia. However, information on the neuropathological links underlying the relationship is limited.
Objective:
We investigated the role of midlife and late-life PA with in vivo AD neuropathologies in old adults without dementia.
Methods:
This study included participants from the Korean Brain Aging Study for Early Diagnosis and Prediction of Alzheimer’s disease (KBASE). The participants underwent comprehensive clinical and neuropsychological assessment, [11C] Pittsburgh Compound B positron emission tomography (PET), [18F] fluorodeoxyglucose PET, and magnetic resonance imaging. Using the multi-modal brain imaging data, in vivo AD pathologies including global amyloid deposition, AD-signature region cerebral glucose metabolism (AD-CM), and AD-signature region cortical thickness (AD-CT) were quantified. Both midlife and late-life PA of participants were measured using the Lifetime Total Physical Activity Questionnaire.
Results:
This study was performed on 260 participants without dementia (195 with normal cognitive function and 65 with mild cognitive impairment). PA of neither midlife nor late-life showed direct correspondence with any neuroimaging biomarker. However, late-life PA moderated the relationship of brain amyloid-β (Aβ) deposition with AD-CM and AD-CT. Aβ positivity had a significant negative effect on both AD-CM and AD-CT in individuals with lower late-life PA, but those with higher late-life PA did not show such results. Midlife PA did not have such a moderation effect.
Conclusion:
The findings suggest that physically active lifestyle in late-life, rather than that in midlife, may delay AD-associated cognitive decline by decreasing Aβ-induced neurodegenerative changes in old adults.
INTRODUCTION
Multiple studies have suggested that physical activities (PA) can lower the risk of Alzheimer’s disease (AD) dementia or related cognitive decline in old adults [1 –3]. Nevertheless, neuropathological mechanisms showing the role of PA in lowering the risk of AD dementia and cognitive impairment have not been established. Although several studies have investigated the effect of PA on AD biomarkers, such as amyloid-β (Aβ) deposition [4 –7], and on regional neurodegeneration [6, 8], the findings have not been consistent.
Aβ deposition is known to begin at about 10 to 15 years before the presentation of significant cognitive dysfunction [9, 10]. As clinical AD dementia usually manifests in late-life, it can be inferred that Aβ pathology commences even around midlife. Furthermore, tau pathology and AD-related neurodegeneration follow Aβ pathology after a considerable duration, and their presence closely precedes appearance of cognitive decline in late-life [9, 11]. As the pathological processes proceed sequentially from midlife to late-life, the influence of PA on different AD pathologies may vary depending on the timing of PA (midlife versus late-life); thus, this may partially explain the inconsistency of the previous findings for the neuropathological mechanisms or substrates underlying the protective effect of PA on AD dementia or related cognitive impairment. Nevertheless, the effect of PA timing, specifically, midlife PA (PAmid) and late-life PA (PAlate) separately, on in vivo AD pathologies are not clear.
In addition, a longitudinal observational study reported that late-life PA attenuated the negative associations of Aβ burden with cognitive decline and gray matter volume loss [12], suggesting the possibility of moderation by PA for the relationship between amyloid pathology and subsequent neurodegeneration. A couple of animal studies also showed that exercise inhibits Aβ-related cognitive deficit in transgenic mice [13, 14]. Given these findings, much is to be established regarding the moderating role of PA on the relationship between Aβ pathology and subsequent AD-related regional neurodegenerations.
In our study, in order to advance the understanding of the neuropathological links underlying the role of PA on AD, we first investigated the direct effect of PAmid and PAlate on in vivo AD pathologies, including Aβ deposition, AD-related brain atrophy, and AD-related cerebral hypometabolism, as measured by multiple neuroimaging modalities. Considering the sequential AD pathological process and the possibility of moderation by PA for Aβ effect on subsequent neurodegeneration, we further evaluated the moderation effect of PAmid and PAlate on the relationship of Aβ deposition with AD-related brain atrophy and hypometabolism.
METHODS
Participants
This study was part of an ongoing prospective cohort study, the Korean Brain Aging Study for Early Diagnosis and Prediction of Alzheimer‘s Disease (KBASE). The KBASE started in 2014 to identify novel AD biomarkers and to determine potential contributing factors in the lifestyle to AD pathologies [15]. The present study included 260 participants without dementia [195 with normal cognitive function (CN) and 65 with mild cognitive impairment (MCI)] aged between 61 and 90 years.
The CN individuals had clinical dementia rating (CDR) score [16] was 0 with no diagnosis of MCI or dementia. MCI status was assigned to individuals based on the current consensus guidelines [17] for MCI: 1) memory complaint corroborated by informant; 2) objective memory impairment; 3) preserved global cognitive function; 4) independence in functional activities; and 5) no dementia. For criterion 2), the age-, education, and sex-adjusted z-scores of < – 1.0 for at least 1 of the 4 episodic memory tests: Word List Memory, Word List Recall, Word List Recognition, and Constructional Recall test in the Korean version of Consortium to Establish a Registry for Alzheimer‘s Disease (CERAD-K) neuropsychological battery [18]. All individuals with MCI had a global CDR score of 0.5.
The exclusion criteria were as follows: 1) presence of a major psychiatric illness, including alcohol-related disorders; 2) significant neurological or medical conditions or comorbidities that could affect mental function; 3) contraindications for a magnetic resonance imaging (MRI) scan (e.g., pacemaker or claustrophobia); 4) illiteracy; 5) presence of significant visual/hearing difficulties and/or severe communication or behavioral problems that would make clinical examinations or brain scans difficult; and 6) taking an investigational drug. All participants underwent standardized clinical and neuropsychological evaluations according to the KBASE clinical assessment protocol [15], which included the CERAD-K assessment battery [18, 19]. Details regarding the KBASE study methodology, including the enrollment and assessment of participants, has been described previously [15].
This study was approved by the Institutional Review board of Seoul National University Hospital (C-1401-027-547) and SNU-SMG Boramae medical center (26-2015-60), Seoul, South Korea. All participants of this study provided written informed consent prior to participation.
Assessment of physical activity
PA of the participants were measured using the interviewer-administered Lifetime Total Physical Activity Questionnaire (LTPAQ), a tool with demonstrated reliability [20] and validity [21]. This questionnaire assesses occupational, household, and leisure activities separately throughout the respondent’s lifetime. The frequency and duration of these activities were assessed by recording the number of years, months per year, weeks per month, days per week, and hours per day that each activity was performed. The intensity of activity was assigned by the participant as sedentary, light, moderate, or heavy. A metabolic equivalent (MET) value was matched to each activity based on the Compendium of Physical Activities [22]. We calculated PAmid and PAlate as the average MET-hour/week spent on all occupational, household, and exercise/sports activities of participants between the ages of 41–60 years and over the past one year, respectively.
Assessment of potential confounders
The comorbid vascular risk factors (VRF), including hypertension, diabetes mellitus, dyslipidemia, coronary heart disease, transient ischemic attack, and stroke, were also assessed. The vascular risk score (VRS) was calculated by the number of the VRFs as a percentage [23].
Cognitive activities (CA) of participants were also measured using a 39-item expanded version of the lifetime cognitive activity scale [24] of a previous 25-item self-report questionnaire [25]. Participants were asked to report the frequency of participation in common cognitively demanding activities with few barriers, such as reading newspapers, magazines, or books; visiting a museum or library; attending a concert, play, or musical; and writing letters; and playing games, at 5 age epochs: 6, 12, 18, and 40 years and the current age. Responses for each item were recorded using a 5-point frequency scale: 5, every day or almost every day; 4, several times a week; 3, several times a month; 2, several times a year; and 1, once a year or less. Among the 39 items for CA, there were nine items each for current age (late-life) and 40 years of age (midlife). The scores of all items for both late-life and midlife were averaged to yield a CA score.
Whole blood samples were obtained for extracting genomic DNA. Apolipoprotein E ɛ4 (APOE4) genotyping was performed according to established techniques [15, 26].
Measurement of cerebral amyloid deposition
All the participants underwent simultaneous three-dimensional (3D) [11C] Pittsburgh compound B-positron emission tomography (PiB-PET), and 3D T1-weighted MRI, using the 3.0 T Biograph mMR (PET-MR) scanner (Siemens, Washington, DC, USA). Details of PiB-PET imaging acquisition and preprocessing were previously described [27].
The automated anatomical labeling algorithm [28] and a region-combining method [29] were applied to determine the regions of interest (ROIs) to measure global amyloid retention level in the frontal, lateral parietal, precuneus/posterior cingulate precuneus (PC/PCC), and lateral temporal regions. The standardized uptake value ratios (SUVR) were generated by the mean value for all voxels within the ROIs by the mean cerebellar uptake value in the same image [28, 29]. Participants were classified as Aβ positive if the SUVR value was > 1.4.
Measurement of AD-signature neurodegeneration
Participants also underwent fluorodeoxyglucose (FDG)-PET imaging using the PET-MR. Details of FDG-PET imaging acquisition and preprocessing have been established previously [27]. The AD-signature FDG-PET ROIs included the angular gyrus, posterior cingulate cortex, and inferior temporal gyrus which were known to be sensitive to change associated with AD [30]. The AD-signature cerebral glucose metabolism (AD-CM) was defined as the voxel-weighted mean SUVR extracted from the AD-signature FDG ROIs [30].
All T1-weighted images were acquired in the sagittal orientation using the PET-MR machine. Details of MRI imaging acquisition and preprocessing have been established previously [27]. The AD-signature cortical thickness (AD-CT) was defined as the mean cortical thickness values extracted from AD-signature regions including the medial temporal (entorhinal and parahippocampal) area, middle temporal gyrus, angular gyrus, and PC/PCC [31, 32].
Statistical analysis
A multiple linear regression analysis was conducted to examine the association of PAmid (or PAlate) (independent variable) with each of global Aβ retention, AD-CM, and AD-CT (dependent variables) controlling for age, sex, years of education, APOE4 carrier status, VRS, CA score, and clinical diagnosis (CN versus MCI). A logistic regression analysis was also performed to investigate the association between each PAmid (or PAlate) (independent variable) and Aβ positivity (dependent variable) controlling for the same covariates. To evaluate the moderation effect of PAmid (or PAlate) on the relationship between Aβ positivity and AD-CM or AD-CT, general linear model (GLM) analyses were conducted including PAmid (or PAlate)×Aβ positivity interaction term with PAmid (or PAlate) and Aβ positivity as independent variables, AD-CM or AD-CT as dependent variables, and the same covariates used in above analyses. If there was a significant PA×Aβ positivity interaction effect, further subgroup analyses were performed for each of the lower PA (median and below median value of PA) and higher PA (above median value of PA) group. All statistical analyses were conducted using SPSS software (ver. 25.0 for windows; SPSS Inc., Chicago, IL) and a p-value < 0.05 was considered statistically significant.
RESULTS
Demographics
The characteristics of the participants are summarized in Table 1. The mean age and education of all participants were 72.3 (SD 6.3) and 11.1 (SD 4.8), respectively, and 55.8% of them was female. In regard of cognitive function, mean MMSE and CERAD total score were 25.5 (SD 3.4) and 72.4 (SD 15.5), respectively.
Demographic data
SD, standard deviation; CN, cognitive normal; MCI, mild cognitive impairment; VRS, vascular risk score; APOE4, apolipoprotein E ɛ4; MMSE, Mini-Mental Status Examination; CERAD, Consortium to Establish a Registry for Alzheimer’s Disease; LTPAQ, lifetime total physical activity questionnaire; MET, metabolic equivalent; Aβ, amyloid-β; SUVR, standardized uptake value ratio; AD-CM, Alzheimer’s disease signature region cerebral glucose metabolism; AD-CT, Alzheimer’s disease signature cortical thickness.
Association between PA and AD biomarkers
Multiple linear regression analyses revealed that neither PAmid nor PAlate affected global Aβ retention (p = 0.386 in PAmid, p = 0.819 in PAlate, respectively), AD-CM (p = 0.649 in PAmid, p = 0.427 in PAlate, respectively), and AD-CT (p = 0.889 in PAmid, p = 0.145 in PAlate, respectively). Further, logistic regression analysis did not show any significant association between PAmid (p = 0.226) or PAlate (p = 0.292) and Aβ positivity (Table 2).
Logistic and linear regression analyses for the relationship of physical activity with AD biomarkers
Adjusted for age, sex, education, apolipoprotein E ɛ4, vascular risk score, cognitive activity score, and clinical diagnosis. AD, Alzheimer’s disease; OR, odds ratio; CI, confidence interval; Aβ, amyloid-β; PA, physical activity; AD-CM, Alzheimer’s disease signature region cerebral glucose metabolism; AD-CT, Alzheimer’s disease signature cortical thickness.
Moderation of PA on the relationship between Aβ deposition and neurodegeneration
GLM analysis showed that PAlate significantly moderated the relationship between Aβ positivity and AD-CT (p = 0.004) (Table 3). PAlate attenuated the negative influence of Aβ positivity on AD-CT. Although not statistically significant, PAlate also appears to reduce the negative association between Aβ positivity and AD-CM (p = 0.060). Subsequent subgroup analyses demonstrated that Aβ positivity was associated with lower AD-CT (p = 0.028) or AD-CM (p = 0.009) in lower PAlate group, whereas it did not have any significant relationship with those markers (p = 0.219 in AD-CT, p = 0.086 in AD-CM, respectively) in higher PAlate group (Table 4 and Fig. 1A, B). As described in statistical analysis section, the participants were divided into the lower PAlate and higher PAlate subgroup by the median value (55.58 MET*hour/week) of the PAlate. In contrast, PAmid did not show such moderation effect on the relationship of Aβ positivity with AD-CT (p = 0.267) and AD-CM (p = 0.553).
Moderation of physical activity on the relationship between amyloid positivity and AD-related neurodegeneration
Adjusted for age, sex, education, apolipoprotein E ɛ4, vascular risk score, cognitive activity score, and clinical diagnosis. AD, Alzheimer’s disease; PA, physical activity; CI, confidence interval; AD-CM, Alzheimer’s disease signature region cerebral glucose metabolism; Aβ, amyloid-β; AD-CT, Alzheimer’s disease signature cortical thickness.
Subgroup analysis by late-life physical activity intensity between amyloid positivity and AD-related neurodegeneration
Adjusted for age, sex, education, apolipoprotein E ɛ4, vascular risk score, cognitive activity score, and clinical diagnosis. Lower late-life PA group (n = 130) consisted of participants who performed median and below median values of late-life PA whereas higher late-life PA group (n = 130) consisted of the ones who performed above median value of PA. AD, Alzheimer’s disease; CI, confidence interval; PA, physical activity; AD-CM, Alzheimer’s disease signature region cerebral glucose metabolism; Aβ, amyloid-β; AD-CT, Alzheimer’s disease signature cortical thickness.
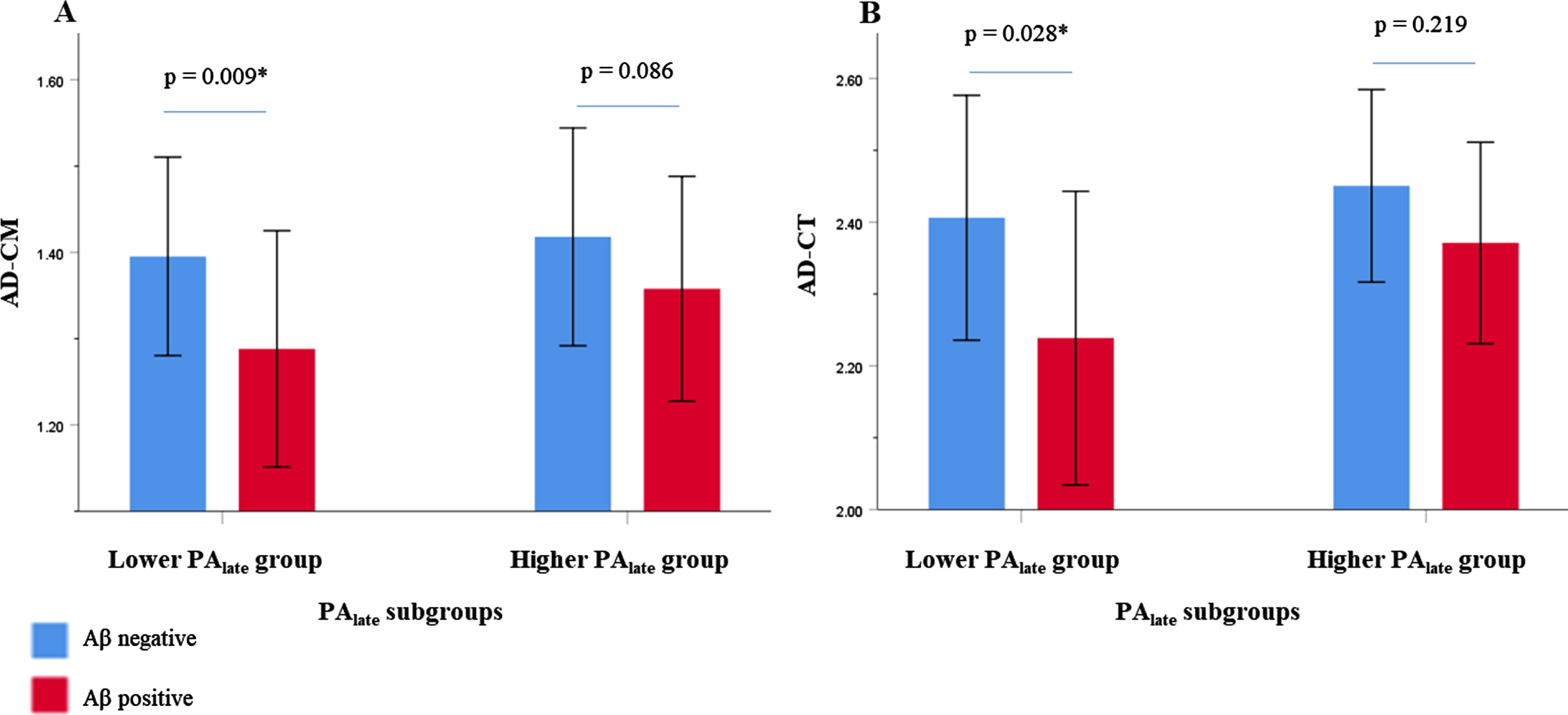
Relationship between Aβ positivity and AD-CM (A) or AD-CT (B) in late-life PA subgroup by intensity. Error bars represent standard deviation. Lower PAlate group (n = 130) consisted of participants who performed median and below median values of PAlate whereas higher PAlate group (n = 130) consisted of the ones who performed above median value of PA. Multiple linear regression analysis was performed after adjusting for age, sex, education, apolipoprotein E ɛ4, vascular risk score, cognitive activity score, and clinical diagnosis. Aβ, amyloid-β; AD-CM, Alzheimer’s disease signature region cerebral glucose metabolism; AD-CT, Alzheimer’s disease signature cortical thickness; PA, physical activity; PAlate, late-life physical activity.
When global Aβ retention was used as a marker for Aβ deposition in the analysis, the results for the moderation effect of Aβ were very similar (Supplementary Table 1). In addition, when we controlled cognitive function or performance (i.e., Mini-Mental Status Examination or CDR sum of boxes score) instead of clinical diagnosis, the results did not change (Supplementary Tables 2 and 3).
DISCUSSION
In the current study, we found that PAlate, but not PAmid, moderated the relationship between Aβ deposition and AD-related neurodegeneration, while neither PAmid nor PAlate directly affected any in vivo AD pathological markers. More specifically, higher Aβ deposition was observed with greater AD-related neurodegeneration in the lower PAlate group, but not in the higher PAlate group.
In line with our findings, several previous studies reported no direct association between PAmid or PAlate and AD pathological markers, such as Aβ deposition, cerebral glucose metabolism, and hippocampal volume [5 , 33–35]. Recent intervention studies also did not demonstrate any protective effect of PA on amyloid deposition and whole brain or hippocampal volume in non-demented or CN participants [36 –38], while a couple of studies reported the association between PAmid or PAlate with Aβ deposition [4, 7]. Some studies also indicated that PA may relate to relatively preserved brain or hippocampal volume in non-demented old adults [39 –41]. However, they simply evaluated the relationship between PA and brain volume, while the influence on Aβ deposition was relatively unknown.
We found that a higher PAlate attenuated the relationship between amyloid deposition and neurodegeneration. This finding is very similar to a previous report which showed that PAlate attenuated the relationship of Aβ burden with gray matter volume loss [12]. It is not easy to provide accurate mechanism underlying such moderation effect of PAlate, the following explanations may help to understand the effect of PAlate. First, PA is known to decrease insulin resistance [42]. Increased insulin resistance has been observed with a higher phosphorylated and total tau level in cerebrospinal fluid (CSF) [43] and a decreased gray matter volume in AD-related brain regions [44]. Insulin itself was also reported to attenuate Aβ-induced neurotoxicity [45]. Thus, a higher PAlate may reduce Aβ-induced neurodegeneration by decreasing insulin resistance or facilitating insulin effect in the brain. Second, PA may stimulate the release of neurotransmitters such as norepinephrine and dopamine [46], which have been suggested to have protective effects against AD pathological changes [47, 48]. In mice, norepinephrine stimulation of microglia was reported to suppress Aβ-induced cytokine and chemokine production [47]. Third, PA may modulate neuroinflammation related to AD neuropathologies through upregulation of anti-inflammatory cytokines and inhibition of microglial activation [49]. Microglial cells with lipopolysaccharide activate inflammatory cytokines when exposed to Aβ that can cause neuronal damages [50]. In the present study, PAlate showed a significant moderating role even after adjusting for vascular risk factors. This finding indicates that the effect of PAlate is not only by lowering the vascular risks.
In contrast to PAlate, PAmid did not show a moderating effect on the relationship between Aβ burden and AD neurodegeneration biomarkers. This result may be due to the sequential pathological events toward AD. In other words, sufficient “upstream” pathology of Aβ accumulation precedes “downstream” pathology of abnormal tau, neuronal dysfunction, glial activation, neuronal loss, and atrophy [11]. Aβ start to deposit commonly from middle age or early stage of late life and AD-related neurodegeneration develops mainly in late life period after Aβ accumulation to some degree [9 –11]. Hence, the protective role of PA which attenuates the negative effect of Aβ on “downstream” pathology of neurodegeneration could be observed more easily in late-life period when Aβ pathology accumulate sufficiently than in midlife. The null finding for the effect of PAmid on AD pathologies appears contrast to the results from some previous studies which suggested that PAmid may decrease the risk of dementia inlate-life [51]. However, the Whitehall Study reporting on the 28-year follow-up of 10,308 people revealed that moderate-to-vigorous physical activity lowered dementia risk over 10, but not 28 years [52]. In addition, a recent individual-level meta-analysis of 19 observational studies including 404,840 participants’ data (mean baseline age 45.5 years; mean follow-up duration 14.9 years) reported that there was no increase in overall and AD dementia risk in those who were physically inactive in the 10–15 year period before diagnosis except in those with comorbid cardio-metabolic disease, whereas an increased incidence of dementia was found in those who were physically inactive in the 10-year period before diagnosis [53]. These findings are generally consistent with our results showing the moderation effect of PAlate but not that of PAmid.
It is a strong point of the present study that we evaluated the relationships of both PAlate and PAmid with in vivo AD pathologies using multi-modal brain imaging in a relatively large number of participants. Furthermore, it is a novel finding that PA had a differential moderation effect on the relationship of Aβ deposition with AD-CT and AD-CM according to the age when PA were performed.
Nevertheless, a few limitations exist. First, as we investigated the effects of PA cross-sectionally, it is difficult to infer the causal relationship between the variables and we could not fully exclude the possibility of reverse causality (i.e., the possibility that brain change itself may cause decreased PA in late-life). Therefore, further longitudinal studies need to be conducted to confirm the actual influence of PA on AD pathological changes. Second, we used self-reported questionnaire to measure the level of PA depending on the recall of the participants. Although the questionnaire was proved to have sufficient validity and reliability, some concerns about the possibility of recall bias exist as individuals with MCI were included. However, although individuals with MCI have issues with their recent memory, their remote memory is very well-preserved [54]. Therefore, it is unlikely that individuals with MCI reported their PA history more erroneously, because the self-report for PA mainly depends on remote memory rather than recent memory. Nevertheless, given time interval between recall and PA, detailed recall of PAmid might have lower reliability than that of PAlate. Third, we did not evaluate tau pathology, despite its prime role in AD. Further studies including tau PET or CSF tau level areneeded.
Conclusion
In conclusion, our results indicate that physically active lifestyle in late-life, rather than that in midlife, may delay AD-associated cognitive decline by decreasing the Aβ-induced neurodegenerative changes in old adults.
Footnotes
ACKNOWLEDGMENTS
This study was supported by a grant from the Ministry of Science and ICT, Republic of Korea (grant No: NRF-2014M3C7A1046042), a grant from the Ministry of Health & Welfare, Republic of Korea (HI18C0630 & HI19C0149), a grant from the Seoul National University Hospital, Republic of Korea (No. 3020200030), and a grant from the National Institute of Aging, United States of America (U01AG072177). The funding source had no role in the study design, data collection, data analysis, data interpretation, writing of the manuscript, or decision to submit it for publication.