
Abstract
The brain-derived neurotropic growth factor (BDNF) gene has been linked to dementia, inflammation, and Apolipoprotein E (APOE) ɛ4 status. We used cerebrospinal fluid (CSF) amyloid-β (Aβ)42 and phosphorylated tau (p-tau) to investigate associations with BDNF polymorphisms and modifications by APOE ɛ4 or inflammation in a memory clinic population (n = 114; subjective cognitive decline, mild cognitive impairment, Alzheimer’s disease). We found distinct pathways to Alzheimer’s disease pathology: Val-Met displayed lower CSF-Aβ42 in APOE ɛ4+ carriers, independent of p-tau, while Val-Val displayed greater p-tau at higher IL-6 and sub-threshold Aβ42. This may contribute to resolving some inconsistencies in the BDNF literature and provide possible inroads to specific Aβ and tau interventions depending on BDNF polymorphism.
Keywords
INTRODUCTION
Brain-derived neurotropic growth factor (BDNF), a protein encoded by the BDNF gene, plays an important role in neuronal maintenance, neuronal survival, neurotransmitter regulation, and in long term potentiation and plasticity, especially in the hippocampus, a structure that plays a critical role in memory formation and Alzheimer’s disease (AD) [1–3]. The single nucleotide polymorphisms (SNP) of BDNF Val66Met may be a genetic risk factor for dementia. Previous research has linked BDNF SNPs with AD and observed worse cognitive performance in Met allele carriers diagnosed with autosomal dominantly inherited AD, caused by mutations in the amyloid precursor protein, presenilin-1, and presenilin-2 [4]. Likewise, in a large middle-aged cognitively healthy cohort enriched for AD risk, carriage of the Met-allele was associated with steeper decline in episodic memory and executive function [5].
However, so far studies linking BDNF to AD pathophysiology remain inconclusive. Previous studies have shown an association of lower serum BDNF to widespread Aβ42 in PET studies [6] and faster disease progression in Met carriers, and this effect is magnified by the presence of an Apolipoprotein E (APOE) ɛ4 allele [7, 8]. On the other hand, clinically healthy Val-Val individuals showed lower hippocampal volume compared to heterozygotes and with a dose-response effect with the number of APOE ɛ4 alleles [9]. Older meta-analyses in both healthy and neuropsychiatric populations found no effects of Val66Met on brain volume. It should be noted that the role of amyloid-β (Aβ) positivity was not considered in the meta-analyses, and one recurring observation across several recent studies is that Aβ42 interacts with Met carriership on cognitive decline and hippocampal volume [10, 11].
We hypothesize that these inconsistencies may also be partly due to variability in immune responses. Higher immune response in Met carriers was associated with higher depression symptoms within the cognitive dimension [12]. Interleukin 6 (IL-6) is a pleiotropic cytokine with both pro- and anti-inflammatory actions [13] that in animal models also plays a role in memory [14] and neuronal differentiation and maintenance [15]. Plasma IL-6 has been associated with chronic neuroinflammation, is elevated in AD [16], plays a central role in the activation of microglia, negatively affects the clearance system of Aβ42 [17, 18], and modulates the relationship between AD pathology and vascular/blood brain barrier damage [19]. IL-6 was shown in vitro to have a reciprocal relation supporting neuronal survival in combination with endogenous BDNF [20]. Animal models showed that chronically increasing IL-6 markedly decreases neurogenesis [21].
Inflammation is associated with, and possibly exacerbates, AD pathology, and based on the above observations, we posit that BDNF Val66Met may moderate this association [19, 22–24]. Thus, we sought to examine the relationship of the BDNF polymorphism with Aβ42 versus phosphorylated tau (p-tau), and whether these associations were modified by APOE ɛ4, or inflammation (specifically IL-6) in a memory clinic population.
MATERIALS AND METHODS
A convenience sample of 114) subjects was recruited from the memory clinic of the Maastricht University Medical Center (MUMC) for this cross-sectional study. This group includes individuals diagnosed with subjective cognitive decline (SCD), mild cognitive impairment (MCI), and AD dementia. Diagnoses were made by experienced physicians based on the Petersen core clinical criteria for MCI [25] and AD dementia. Criteria for SCD included self-reported presence of subjective cognitive complaints and endorsing the question “Do you think your memory is becoming worse” and absence of impairments on cognitive tests (defined as a score below –1.5 SD of the age-, sex-, and education-adjusted mean) [26]. Exclusion criteria were major neurological disease, clinical diagnosis of other neurodegenerative disorders (e.g., frontotemporal dementia), recent transient ischemic attack or cerebrovascular accident (< 2 years), history of psychiatric disorders, and alcohol or drug abuse. All patients provided informed written consent and the study protocols were approved by the Medical Ethics Committee of the MUMC in confirmation with the declaration of Helsinki.
CSF and blood analyses
CSF was collected via a lumbar puncture in the L3 to L5 vertebral interspaces, centrifuged, aliquoted, and stored at –80°C in polypropylene tubes. Biochemical analysis of CSF Aβ42, total tau, and p-tau181p (Innotest ELISA, Innogenetics, Ghent, Belgium) was done following standardized protocol and blinded to diagnostic information. APOE ɛ4 and BDNF SNP genotyping was determined on genomic DNA using polymerase chain reaction and restriction fragment length polymorphism at the MUMC. BDNF allele distribution did not deviate from the Hardy-Weinberg equilibrium [27]. Met-Met homozygotes (n = 3) in this sample were pooled with the Val-Met. Cytokine interleukin IL-6 was analyzed from serum using multiplex BD cytometric bead array (BD-biosciences, Franklin Lakes, NJ, USA). Samples were collected between 2011 and 2017 and stored at –80°C in a biobank during this time (aliquoted to avoid freeze thaw cycles). Prolonged storage, even at –80°C, can differentially affect the level of cytokines [28] and therefore we linearly adjusted for storage times calculated from the day of entry in the biobank to the date of analysis [19].
Statistical analyses
Statistical analyses were conducted in R version 3.6.2 (http://www.Rproject.org). Demographics are provided in Table 1. For each biomarker (Aβ42, Tau, p-tau) we set up the following hierarchical multivariable regression models: 1) differences between BDNF Val-Val and Val-Met for each biomarker; 2) interaction between the BDNF and APOE ɛ4 genotype; 3) interaction between the BDNF genotype and IL-6 levels (as continuous variable). Each model was adjusted for age, sex, APOE ɛ4 and storage time (and diagnosis in a separate step); and 4) interaction models additionally adjusted for either Aβ42 or p-tau to determine whether the effect was independent of the other pathology. Post hoc, we also interacted BDNF genotype by IL-6 by Aβ42 on p-tau and performed Johnson-Neyman analyses [29, 30] using simple slopes to determine at which range of pathology the association was significant. To ensure effects where robust, linear models were bootstrapped (30,000 replications) and p-values were derived from the resulting distribution. In addition, FDR correction at alpha < 0.05 was implemented to adjust for multiple comparisons.
Demographics stratified by BDNF genotype
Demographics stratified by BDNF genotype
Education is measured using an evaluation system based on Verhage (1964) and the Standard Classification of Education of the Dutch Central Bureau of Statistics (CBS, 2014). It is equivalent to the International Standard Classification of Education (UNESCO, 1997). †χ2 -test for dichotomous variables, t-test for continuous variables, ‡Fisher Exact Test for count data; # #Aβ42 CSF positive clinical cutoff at < 500.
Demographics stratified by BDNF allele are listed in Table 1. Our sample had a mean age of 62.97 (SD = 9.04) years, 71% of the participants were male, 46.6% of Val-Met, and 49.2% Val-Val carried at least one APOE ɛ4 allele (no difference p = 0.935) There was no difference in demographics between BDNF Val-Val and Val-Met carriers (Table 1).
BNDF-Val66Met effects (covaried for APOE ɛ4, age, and sex)
There is no direct association between BDNF and Aβ42 (t109 = 0.22, pFDR = 0.822) or total tau (t109 = 1.12, pFDR0.265). However, BDNF Val-Val carriers exhibited higher p-tau values (t109 = 2.04, pFDR = 0.043). There is a trend level association between BNDF and p-tau (t109 = 1.87, pFDR = 0.08) when Aβ42 is added as a covariate (t109 = 2.04, p = 0.04 pFDR = 0.08), suggesting shared variance in the relation of p-tau and Aβ42. There are no differences in mean IL-6 level in the Val-Met group (1.430) and in the Val-Val group (1.352) (t99 = 0.46211, p = 0.645). There are no differences in IL-6 levels for APOE ɛ4 groups (APOE ɛ4–1.50/APOE ɛ4+ 1.25), (t112 = 1.47, p = 0.14). The differences of CSF Aβ42 in APOE ɛ4 groups were significant (APOE ɛ4–1049/APOE ɛ4+ 787), (t112 = 4.15, p≤0.0001).
BDNF by APOE ɛ4 interaction (covaried for age and sex)
When looking at the interaction of BDNF and APOE ɛ4, we found lower CSF Aβ42 in Val-Met carriers who also carried the APOE ɛ4 allele (t108 = 2.19, p = 0.03 (pFDR = 0.03), Fig. 1C), compared to Val-Val carriers with or without APOE ɛ4 carriage. When adding p-tau as covariate, the interaction be-tween BDNF and APOE ɛ4 remained significant (t107 = 2.62, p = 0.009 (pFDR = 0.018)), indicating that this effect was independent of p-tau. If the sample was post hoc restricted to MCI and SCD, the interaction between BDNF and APOE ɛ4 was not significant (t85 = 1.83, pFDR = 0.11). The interaction between BDNF and APOE ɛ4 was not significant for t-tau (t108 = 1.09, p = 0.275) or p-tau (t108 = 0.648 pFDR = 0.56).
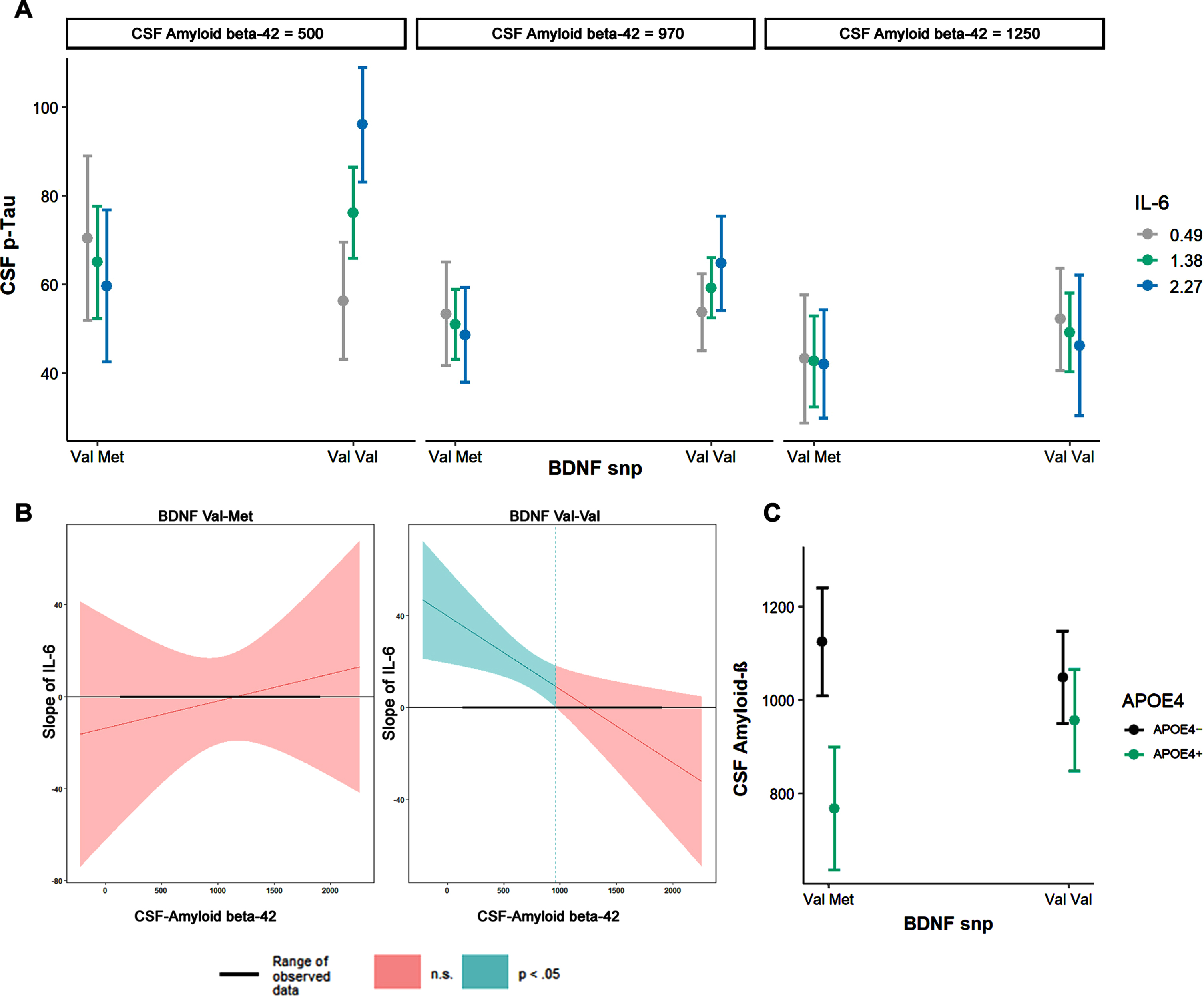
A) BDNF polymorphism SNP show different associations between IL-6 and CSF p-tau, dependent on Aβ42 (continuous variable, plotted marginal effects at mean±1SD). B) Johnson-Neyman plot showing the level of CSF Aβ42 (< 963 pg/ml) where the interaction BDNF*IL6*Aβ42 becomes significant. Clinical threshold for Aβ42 is 500 pg/ml and p-tau (85 pg/ml). C) CSF Aβ42 levels differ among BDNF by APOE ɛ4 SNPs.
The interaction between BDNF and IL-6 showed no significant associations with Aβ42 (t106 = 0.74, pFDR = 0.45) and t-tau (t106 = 1.78, pFDR = 0.07). Higher p-tau was associated with BDNF Val-Val and higher IL-6 (t106 = 2.957, pFDR = 0.01). When Aβ42 was added as a covariate, this association did not change (t106 = 2.79, pFDR = 0.02); likewise adding diagnosis as a covariate did not change results (t96 = –3.037 pFDR = 0.02).
The 3-way interaction of BDNF, IL-6, and Aβ42 was significant (t102 = –3.00, p = 0.0033, pFDR = 0.01, adjusted R-square: 0.4825): Val-Val carriers with lower CSF Aβ42 and higher IL-6 levels (Fig. 1A) exhibited higher p-tau levels. The Johnson-Neyman interval revealed that this three-way interaction became significant at Aβ42 < 962.96 pg/ml (pFDR < 0.05, t = 2.30; Fig. 1B)
DISCUSSION
In this study we found distinct relationships of BDNF polymorphism to either Aβ42 or p-tau. We observed that individuals with Val-Met genotype displayed lower CSF Aβ42 when also carrying at least one APOE ɛ4 allele, independent of p-tau. However, Val-Val individuals exhibited higher p-tau under higher levels of IL-6, and subthreshold levels of Aβ42 (< 963 pg/ml; clinical threshold for Aβ42 = 500). BDNF plays a crucial role in synaptic plasticity and long-term potentiation in the hippocampus and therefore SNPs of the BNDF gene have been of interest for dementia research. These findings could provide new inroads to specific interventions for Aβ42 and p-tau depending on BDNF polymorphism and immune response.
While only few studies examined the effect of BDNF on AD pathology, the literature is consistent in that the relationship between BDNF and memory is dependent on amyloid, but potential effect-modification by APOE is less clear. In in Aβ- SCD participants, no increased risk of dementia was found, but combined Aβ+ and Val66Met+ did cognitively worse than Aβ+ only [8], with similar findings in unimpaired but increased AD risk [11] suggesting a synergistic effect of Met+ and Aβ. However, no moderating effect of APOE ɛ4 was found on cognition. In the AIBL study, Aβ+ APOE ɛ4+ and Val66Met+ had worse memory performance [7] but interaction effects of APOE by BNDF on Aβ are not described. Two studies took a similar approach as ours and examined the relationship between BDNF Val66Met on AD pathology in cognitively normal older individuals [31, 32] and reported higher PET Aβ42 in APOE ɛ4+ in Val66Met+ [32]. In a combined autosomal dominant AD group, lower CSF Aβ42 in APOE ɛ4+ participants but no interaction with BDNF was found [2]; however, Val66Met+ was associated with hippocampus-frontal connectivity in autosomal dominant AD and AD and stronger associations with Aβ in sporadic AD but not in SCD [33]. Reasons underlying these differences between sporadic AD, preclinical autosomal dominant AD, and SCD might be related to different ages of onset and disease progression.
It is increasingly accepted that inflammation plays an important role in linking concurrent pathologies in AD dementia [19, 35]. IL-6 is a pleiotropic cytokine that has both pro- and anti-inflammatory effects, plays an important part in the innate immune system, can induce hyperphosphorylation of tau in animal models [36], and can interfere with Aβ42 clearance [17, 37]. Inflammation and microglial activation lead to production of multiple neurotrophic factors, including BDNF. Sustained immune activation, however, strongly reduces the generation of these neurotropic factors possibly interfering with neuroplasticity [38].
This study has some limitations. This is a me-dium sized cross-sectional convenience sample from a referral-only university hospital memory clinic in northern Europe. The Val66Met prevalence is extremely dependent (0 to > 70%) on genetic background [39] and the prevalence of the Val66Met polymorphism is approximately 20% in Europe. The locale should be considered when comparing these results with studies conducted in a different genetic background. Both IL-6 and BDNF are not specific to any single disease complicating the interpretation of associations in various populations. Larger studies are needed to better model interactions between pathology, genetics, and the innate immune system. We tentatively hypothesize based on this data that trials targeting AD pathology (both Aβ and p-tau), especially using monoclonal antibodies, may have to consider APOE status, BDNF polymorphisms as well as innate immune system activation. Substantially larger cohorts are needed in other to be able to investigate the downstream effects of BDNF on cognition.
Conclusion
Both BDNF genotypes are associated with AD pathology, depending on the interaction with either APOE ɛ4 or inflammation. Elevated p-tau was associated with BDNF Val-Val carriers with elevated IL-6, with a dose response to Aβ42 and starting at subthreshold levels, while lower CSF-Aβ42 was observed in BDNF Val-Met/APOE ɛ4 carriers, independent of p-tau.