
Abstract
Background:
Amyloid-β (Aβ) oligomers induce the overproduction of phosphorylated tau and neurodegeneration. These cascades gradually cause cognitive impairment in Alzheimer’s disease (AD). While each pathological event in AD has been studied in detail separately, the spatial and temporal relationships between pathological events in AD remain unclear.
Objective:
We demonstrated that lipid rafts function as a common platform for the pathological cascades of AD.
Methods:
Cellular and synaptosomal lipid rafts were prepared from the brains of Aβ amyloid model mice (Tg2576 mice) and double transgenic mice (Tg2576 x TgTauP301L mice) and longitudinally analyzed.
Results:
Aβ dimers, the cellular prion protein (PrPc), and Aβ dimer/PrPc complexes were detected in the lipid rafts. The levels of Fyn, the phosphorylated NR2B subunit of the N-methyl-D-aspartate receptor, glycogen synthase kinase 3β, total tau, phosphorylated tau, and tau oligomers increased with Aβ dimer accumulation in both the cellular and synaptosomal lipid rafts. Increases in the levels of these molecules were first seen at 6 months of age and corresponded with the early stages of Aβ accumulation in the amyloid model mice.
Conclusion:
Lipid rafts act as a common platform for the progression of AD pathology. The findings of this study suggest a novel therapeutic approach to AD, involving the modification of lipid raft components and the inhibition of their roles in the sequential pathological events of AD.
Keywords
INTRODUCTION
Amyloid-β (Aβ) oligomers are neurotoxic species that are generated via the central cascade leading to tauopathy and neurodegeneration in Alzheimer’s disease (AD). Aβ oligomers isolated from AD brains reduced the number of synapses, inhibited long-term potentiation (LTP), and enhanced long-term synaptic depression in rodent brains [1, 2]. Two major downstream events are speculated to occur after Aβ oligomerization. The first is a pathway involving the cellular prion protein (PrPc), Fyn kinase, and N-methyl-D-aspartate receptors (NMDARs). The PrPc, a glycosylphosphatidylinositol-anchored cell-surface glycoprotein, functions as a mediator of Aβ oligomer-induced synaptic dysfunction [3]. Aβ oligomers bind to the PrPc, and the resultant complexes activate Fyn. Activated Fyn phosphorylates the NMDAR NR2B subunit, alters calcium signaling, changes the distribution of NMDARs, and destabilizes dendritic spines [4–6]. These findings are partially supported by clinical evidence regarding the beneficial effects of the NMDAR antagonist memantine against cognitive impairment [7].
The other downstream event is tauopathy. Aβ amyloid and tau double transgenic mouse models exhibited phosphorylated tau (p-tau) accumulation [8, 9]. In addition, Aβ42 fibrils induced neurofibrillary tangle (NFT) formation in P301L tau transgenic mice, and Aβ dimers isolated from the cortices of AD patients directly increased p-tau accumulation and neuritic degeneration in primary hippocampal neurons [10, 11]. Furthermore, Aβ plaques facilitated the rapid growth of AD tau seeds into large tau aggregates and NFTs in dystrophic neurites and neuropil threads, and cortical Aβ accelerated tau propagation and neurotoxicity [12–14]. Recently, advanced clinical studies have shown that Aβ oligomer accumulation induces the overproduction of p-tau in the preclinical stage of AD [15, 16].
In 2004, we reported that Aβ dimer formation in lipid rafts is the initial event of AD pathology and is closely associated with NFT tau pathology and cognitive decline in both Tg2576 Aβ amyloidosis model mice and humans [17]. Lipid rafts are special membrane microdomains, where cholesterol and sphingolipids are assembled, and they function as crucial platforms for signal transduction pathways [18]. Aβ generation, the successive cleavage of the amyloid-β protein precursor (AβPP) through β-site and γ-site processing, Aβ oligomerization, and Aβ oligomer recruitment all take place on lipid rafts [19–24]. Fyn and the PrPc are distributed in lipid rafts, and PrPc clustering in lipid rafts activates Fyn [24–26]. Large numbers of lipid rafts are present in synapses, and they are required for synaptic transmission and plasticity [27]. However, the accumulation of the AβPP, tau, and p-tau in neuronal lipid rafts disturbed neuronal functions [28]. Furthermore, numerous receptors and cell-signaling pathways, including glutamate receptor (GluR) signaling, were found to be altered in the cortical lipid rafts of the 3xTgAD murine model [29]. In addition, it was demonstrated that Aβ induces p-tau formation through both cyclin-dependent kinase 5 (cdk5) and Fyn in lipid rafts [30]. Together, these findings suggest that Aβ oligomer accumulation; PrPc, Fyn, and NMDAR signaling; and tauopathy occur on lipid rafts. We hypothesized that lipid rafts act as a common platform for the sequential processes of AD pathology. To examine this concept, we analyzed the longitudinal changes in the levels of the cardinal molecules that accumulate in cellular and synaptosomal lipid rafts in the brains of mouse models of AD.
METHODS
Subjects
Tg2576 mice (APP + mice), which express a mutant K670N/M671L/APP695 transgene under the regulation of the hamster prion promoter on an SJL/B6 strain background [31], and their non-transgenic littermates (APP- mice) were used. The numbers of mice of each age were as follows: 4 months (APP+, n = 4; APP-, n = 4), 6 months (APP+, n = 7; APP-, n = 7), 12 months (APP+, n = 4; APP-, n = 4), and 22 months (APP+, n = 7; APP-, n = 7). TgTauP301L mice (Tau + mice) expressing 2N4R human tauP301L under the regulation of the hamster prion promoter on an FBV/N strain background [32] (n = 1 at 3, 7, 9, 12, 19, and 25 months of age) and the heterozygote F1 littermates of APP + mice that had mated with Tau + mice [9] were also used. The numbers of mice of each age were as follows: APP + Tau + mice: 12–14 months (n = 3) and 22–23 months (n = 4), APP-Tau + mice: 12–14 months (n = 3) and 22–23 months (n = 9), APP + Tau- mice: 12–14 months (n = 2) and 22–23 months (n = 4), and APP-Tau- mice: 12–14 months (n = 2) and 22–23 months (n = 6). All animal experiments followed the ARRIVE guidelines and were approved by the ethics committee of Hirosaki University (approval number: M13007-1).
Cellular lipid raft preparation
After the mice had been deeply anesthetized with halothane, their hemibrains were minced with a razor blade and homogenized on ice in MBS-T buffer (50 mM 2-[N-morpholino] ethanesulfonic acid [MES]; 150 mM NaCl, pH 6.5; and 1% Triton X-100) supplemented with protease inhibitors (Complete®, Roche Diagnostics, Indianapolis, IN) at a concentration of 200 mg brain tissue/ml MBS-T using 20 strokes of a Dounce homogenizer. The homogenate was sonicated and centrifuged at 2,000 g for 10 min at 4°C to remove any nuclei and cell debris. Then, sucrose density gradient (1.5 ml homogenate + 1.5 ml of 80% sucrose, 10 ml of 38% sucrose, and 3 ml of 5% sucrose) centrifugation was performed at 100,000 g for 19 h at 4°C. Starting from the top, 2 ml samples were aspirated and named fractions 1–8, with the pellet designated fraction 9 [17]. The procedure is illustrated in SupplementaryFigure 1.
Preparation of synaptosomes and synaptosomal lipid rafts
The forebrains were homogenized in 10% w/v buffer A (0.32 M sucrose; 10 mM Tris, pH 7.2; 1 mM ethylenediaminetetraacetic acid-dipotassium dihydrate (EDTA-2K); and protease inhibitors), before being centrifuged at 1,000 g for 10 min. The supernatant was transferred to a new tube and centrifuged at 20,000 g for 10 min. The pellet was transferred to a homogenizer, homogenized with 0.625 ml of buffer A, and centrifuged at 20,000 g for 10 min. The resultant pellet was then homogenized with 2 ml of buffer C (14% Ficoll; 0.32 M sucrose; 10 mM Tris, pH 7.2; and 50 mM EDTA-2K), mixed up to 6 ml in a centrifuge tube, and overlayed with 4.5 ml of buffer B (6% Ficoll; 0.32 M sucrose; 10 mM Tris, pH 7.2; and 50 mM EDTA-2K) and 2 ml of buffer A. After the mixture had been centrifuged at 99,000 g for 30 min, the layer between buffers B and C was collected. The collected layer was diluted with 1.8 ml of buffer A, before being centrifuged at 15,000 g for 15 min. The pellet was washed with 1 ml of Kreb’s solution, which consisted of 45 mM NaCl, 5 mM KCl, 1.2 mM CaCl2, 1.3 mM MgCl2·6H2O, 1.2 mM NaH2PO4·2H2O, 10 mM glucose, and 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) sodium salt at pH 7.5, containing protease inhibitors, before being resuspended in 1 ml of Kreb’s solution. This fraction was named the synaptosomefraction [33].
To separate out the synaptosomal lipid rafts, the synaptosome fraction was subjected to further sucrose density gradient (1 ml homogenate, 1 ml of 80% sucrose, 10 ml of 38% sucrose, and 3 ml of 5% sucrose) centrifugation at 100,000 g for 19 h at 4°C. The fractionation of the synaptosome fraction into fractions 1 to 9 was carried out as described in the previous section. The procedure is illustrated in Supplementary Figure 1.
Antibodies
The following antibodies were used: Anti-flotillin antibodies (flotillin was used as a lipid raft marker) (IBL, Fujioka, Gunma, Japan); anti-postsynaptic density protein-95 (PSD95) antibodies (PSD95 was used as a postsynaptic density marker) (Thermo Fisher, Waltham, MA); anti-synaptophysin antibodies (synaptophysin was used as a presynaptic marker) (Dako); anti-Fyn antibodies (sc-434, Santa Cruz Biotechnology, Dallas, TX); anti-glycogen synthase kinase 3β (GSK3β) antibodies [34]; 6E10 antibodies against Aβ1-6 (BioLegend, San Diego, CA); 4G8 antibodies against Aβ17–24 (BioLegend); 82E1 antibodies against Aβ1-16 (IBL, Fujioka, Japan); 72D9 antibodies, which specifically bind to Aβ oligomers [35]; 6D11 antibodies against PrPc 93–109 (BioLegend); and C-20 antibodies against an antigen located near the C-terminus of the PrPc (sc-7693, Santa Cruz Biotechnology). In addition, the following anti-tau antibodies were used: TAU-5 antibodies against mouse total tau (t-tau) (Thermo Fisher); PHF-1 antibodies against tau that was phosphorylated at the Ser396/Ser404 sites (a gift from Peter Davies); and pSer396Tau antibodies against tau that was phosphorylated at the Ser396 site (Santa-Cruz Biotechnology). Anti-NMDAR NR2A (Santa Cruz Biotechnology), anti-NMDAR NR2B (Santa Cruz Biotechnology), and anti-phospho-NMDAR 2B pTyr1472 (p-NR2B) (Thermo Fisher) antibodies were used as anti-NMDAR antibodies. The antibodies used are listed in the SupplementaryTable 1.
Western blotting
The samples were separated on 4–12% NuPAGE Bis-Tris gel (Thermo Fisher) and then transferred to polyvinylidene difluoride membranes at 100 V for 1.5 h. The membranes were labeled with the primary antibodies overnight at 4°C, before being incubated with horseradish peroxidase-linked secondary antibodies (Amersham Biosciences, Buckingham, UK) for 1 h. The resultant signals were detected using the SuperSignal substrate (Pierce, Rockford, IL) or an enhanced chemiluminescence detection system (Amersham, Buckinghamshire, UK) in a LuminoImage analyzer (LAS 1000-mini, Fujifilm, Tokyo, Japan). The signal intensities of proteins were quantified using a LuminoImage analyzer (LAS 1000-mini, Fuji Film, Tokyo, Japan). Each band was analyzed using Image-J (NIH).
Aβ oligomer and PrPc binding assay
Synthetic Aβ42 (Sigma-Aldrich, St. Louis, MO; 50–200 pmol) was incubated overnight at 4°C with or without the lipid raft fraction from the brains of 22-month-old APP- mice, immunoprecipitated with C-20 or 6E10 antibodies, and detected with 82E1 antibodies. To detect Aβ oligomer/PrPc complexes in lipid rafts, all of the fractions obtained from the brains of 23-month-old APP + mice (from 75μg of brain tissue) through sucrose density gradient centrifugation were immunoprecipitated with C-20 antibodies and detected with 4G8 antibodies, or immunoprecipitated with 82E1 antibodies and detected with 6D11 antibodies.
Double-labeling immunofluorescence
Frozen 8-μm-thick sections were cut from the brains of the APP + mice. C-20 and 72D9 antibodies were used as primary antibodies. Secondary antibodies conjugated with Alexa Fluor 594 (Thermo Fisher Scientific) to produce red signals and Alexa Fluor 488 (Thermo Fisher) to produce green signals were used for the double immunofluorescent labeling.
Statistical analysis
Data are expressed as the mean±standard deviation (SD). The unpaired t-test was used for comparisons between two groups. The Prism 9 software (GraphPad, La Jolla, CA) was used for all statistical analyses. p-values of < 0.05 were considered to be significant for all statisticaltests.
RESULTS
Detection of Aβ oligomer/PrPc complexes in cellular lipid rafts
All sucrose gradient fractions from the brains of the APP- and APP + mice were analyzed by Western blotting. The strongest flotillin band was found in fraction 2, indicating that the lipid rafts had migrated into fraction 2. The PrPc was mainly recovered from fraction 2 in both the APP- and APP + mice. Aβ monomers and dimers were detected in fraction 2 and the pellet from the APP + mice (Fig. 1A). Fifty to 200 pmol of synthetic Aβ42 peptides was added to fraction 2 from the APP- mice and immunoprecipitated with C-20 antibodies and labeled with 82E1 antibodies, which showed that the formation of complexes between the PrPc and Aβ dimers/monomers occurred in the lipid rafts (Fig. 1B). Then, all of the fractions obtained from the APP + mice by sucrose density gradient centrifugation were immunoprecipitated using C-20 antibodies and labeled with 4G8 or 6D11 antibodies. Complexes involving the PrPc and Aβ dimers/monomers were present in the lipid rafts, and they accumulated in the pellet (Fig. 1C). A differently configured experiment involving immunoprecipitation by 82E1 antibodies and detection by 6D11 or 4G8 antibodies also confirmed the presence of complexes between the PrPc and Aβ dimers/monomers in the lipid rafts (Fig. 1D). Histological examinations showed the presence of Aβ oligomer/PrPc complexes in the APP + mouse brains from the early (12 months) to advanced stages (23 months) of Aβ deposition (Fig. 1E, arrows).
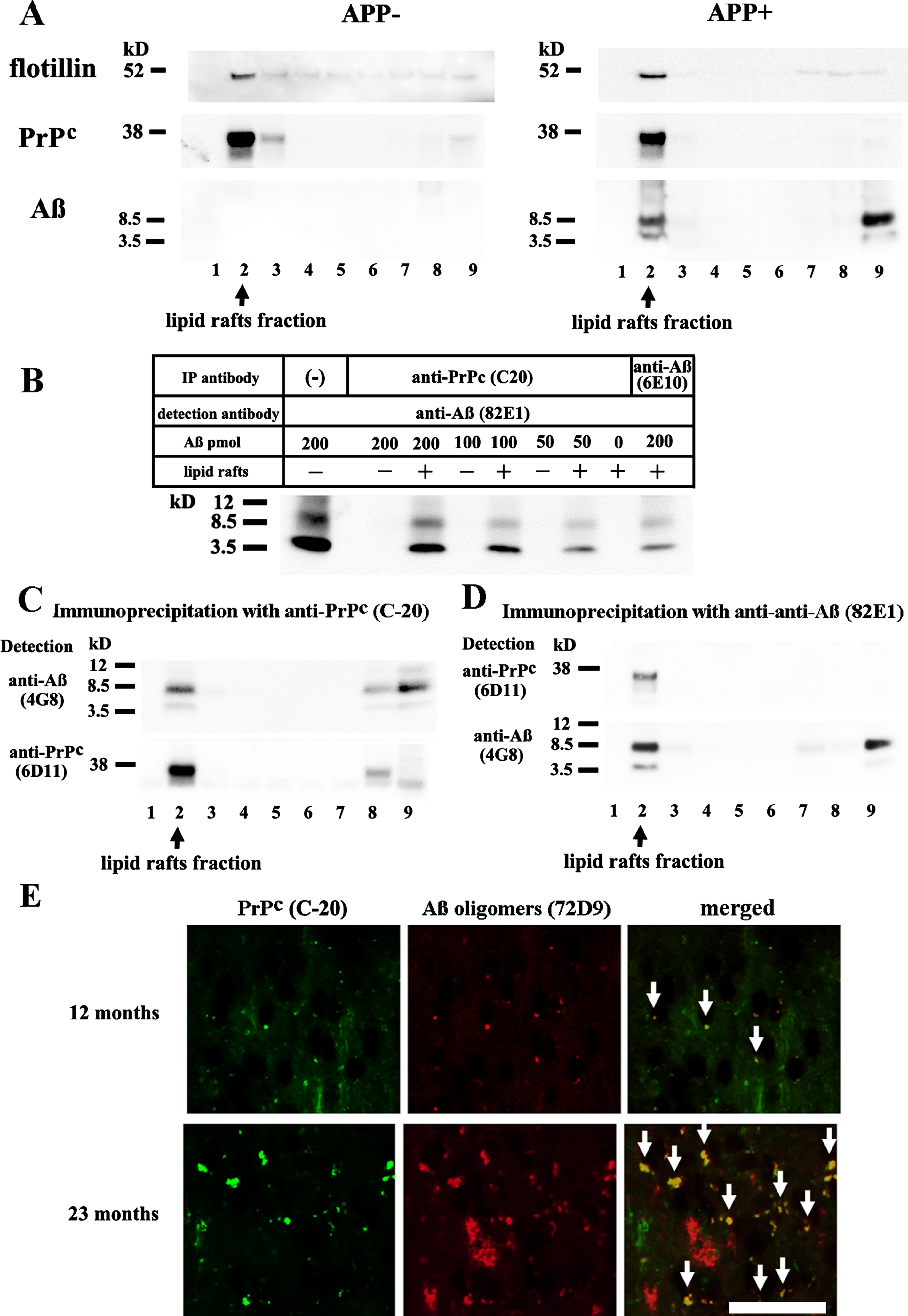
Presence of Aβ dimer/PrPc complexes in lipid rafts. A) All of the fractions obtained from the brains of the 22-month-old APP- and APP + mice through sucrose density gradient centrifugation were subjected to Western blot analysis. Flotillin was mainly detected in fraction 2. 6D11 antibodies labeled PrPc in fraction 2 in both the APP- and APP + mice. 82E1 antibodies labeled Aβ monomers and dimers in fraction 2 and the pellet in the APP + mice. B) Fifty to 200 pmol of synthetic Aβ42 peptides was added to fraction 2 from the 22-month-old APP- mice. Immunoprecipitation was performed with C-20 or 6E10 antibodies, and detection was carried out with 82E1 antibodies. The left column shows the results obtained after the direct application of oligomerized Aβ42 as a control. Complexes between the PrPc and Aβ dimers/monomers were identified. C) All of the sucrose gradient fractions from the 22-month-old APP + mice were immunoprecipitated with C-20 antibodies and detected with 4G8 or 6D11 antibodies. Complexes between the PrPc and Aβ dimers/monomers were detected in the lipid rafts, and they accumulated in the pellet fraction. D) Inverted immunoprecipitation experiments (immunoprecipitated with 82E1 antibodies and detected with 6D11 or 4G8 antibodies) confirmed the presence of complexes between the PrPc and Aβ dimers/monomers in the lipid rafts. The PrPc was not detected in the pellet fraction, suggesting that the levels of these complexes were much lower in the pellet. E) Frozen sections from the cerebral cortices of the 12-month-old and 22-month-old APP + mice were subjected to double immunofluorescent labeling. C-20 staining produced green signals, and 72D9 staining produced red signals. Yellow fluorescence, indicating the colocalization of Aβ oligomers and the PrPc, was detected in the brains of both the 12-month-old and 22-month-old APP + mice (arrow). The scale bar represents 100μm.
Increased Fyn and p-NR2B levels in cellular lipid rafts
Fyn and the NMDAR subunits NR2A and NR2B were detected in fractions 2, 3, 7, and 8 and the pellet in both the APP- and APP + mice. Western blots of all sucrose gradient fractions were performed three times using different mouse samples in each experiment. However, a sample size of 3 is not adequate for statistical analyses. Therefore, we visually compared the mean values for the obtained data. The Fyn levels in fraction 2 from the APP + mice were greater than those seen in fraction 2 from the APP- mice. The amounts of NR2A and NR2B did not differ between the APP- and the APP + mice. P-NR2B was mainly detected in fraction 2 in both types of mice, but higher levels were observed in the APP + mice. These findings indicate that the presence of an increased amount of Fyn facilitates the phosphorylation of NR2B in the lipid rafts of Aβ amyloid model mice(Fig. 2A, B).
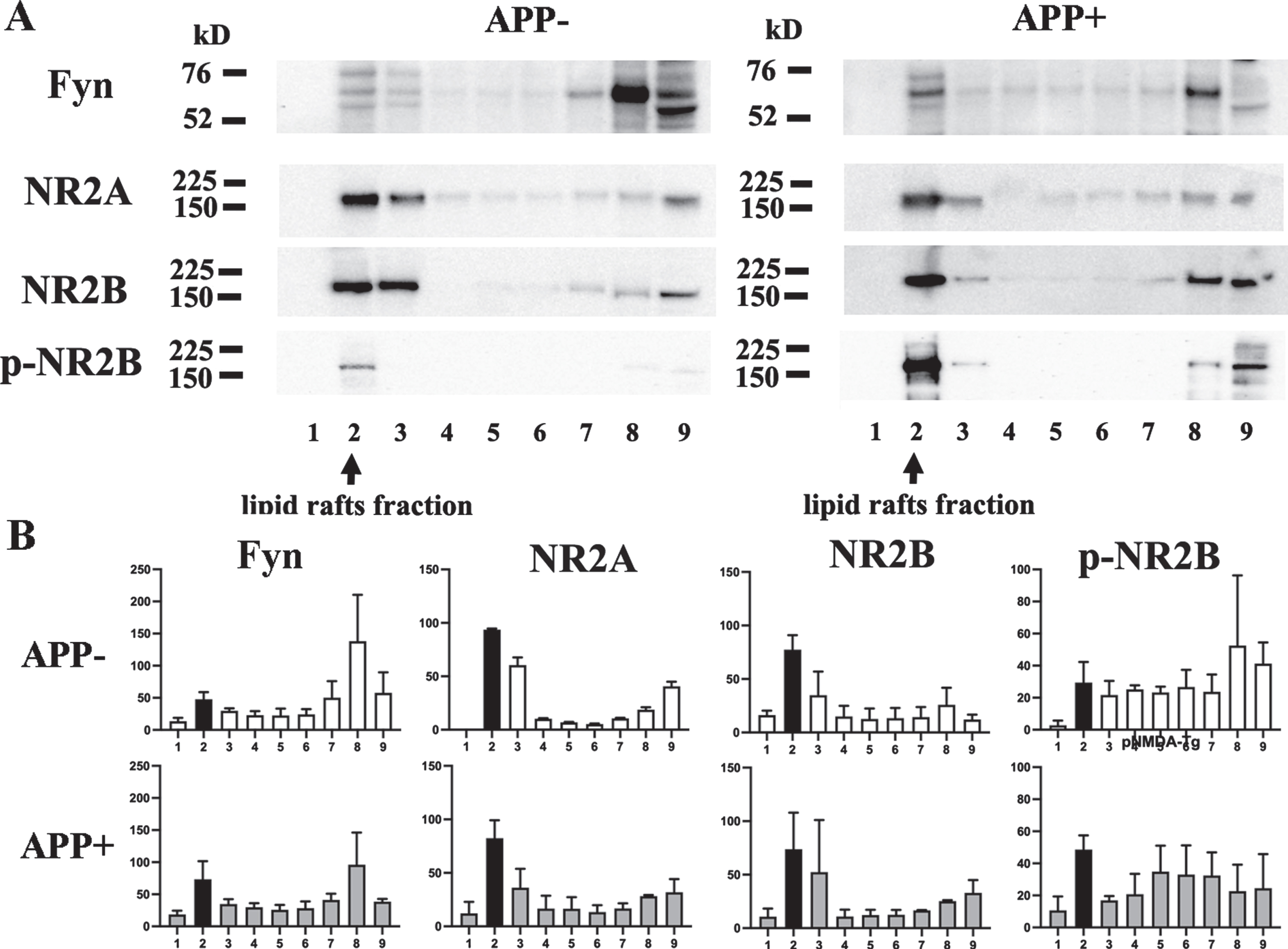
Increased levels of Fyn and p-NR2B in lipid rafts. A) The sucrose gradient fractions from the brains of the 22-month-old APP + and APP- mice were subjected to western blotting. B) Semi-quantitative analysis of the western blot bands was performed. Fraction 2 was shown in black. Fyn, NR2A, and NR2B were present in fractions 2 and 3, Triton-soluble fractions 7 and 8, and the pellet. The majority of p-NR2B was detected in fraction 2. Increased levels of Fyn and p-NR2B were observed in fraction 2 from the APP + mice. The amounts of NR2A and NR2B detected did not differ between the APP- and APP + mice.
Increased levels of phosphorylated and oligomerized tau in cellular lipid rafts
GSK3β, t-tau, p-tau (pSer396/pSer404), and p-tau (pSer396) were detected in fractions 2, 7, and 8, and the pellet in both the APP- and APP + mice. In the lipid raft fraction, the levels of GSK3β and t-tau monomers (blue arrow), dimers (green arrow), and high-molecular-weight oligomers (red arrow) were higher in the APP + mice than in the APP- mice. The levels of p-tau (pSer396/pSer404) and p-tau (pSer396) monomers (blue arrow), dimers (green arrow), and high-molecular-weight oligomers (red arrow) were also increased in the APP + mice (Fig. 3A, D).
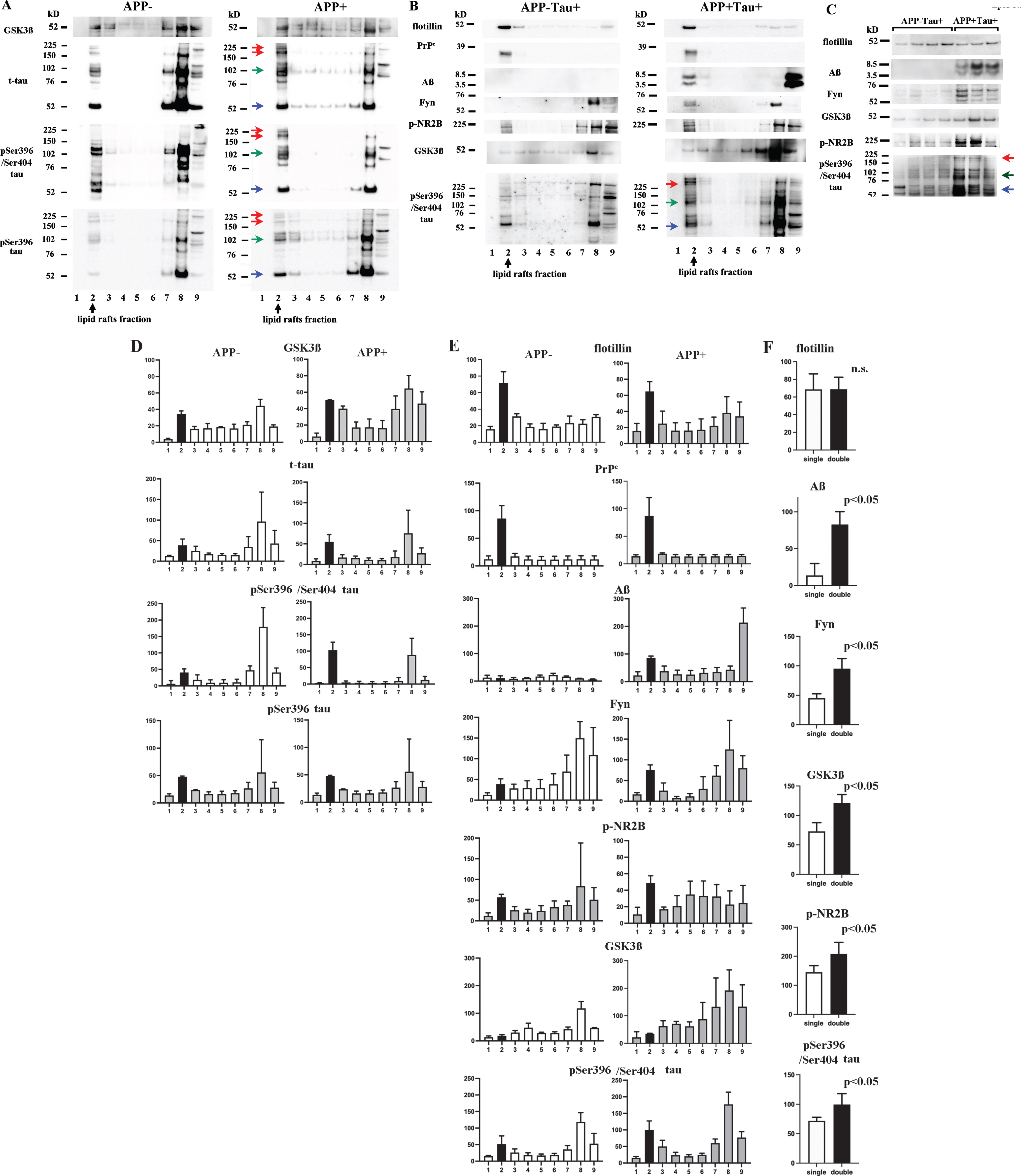
Levels and oligomerization of tau increased with Aβ dimer accumulation in lipid rafts. The sucrose gradient fractions from the 22-month-old APP- and APP + mice (A) and the 22-month-old APP-Tau + and APP + Tau + mice (B, C) were subjected to western blotting. D, E, and F show the results of semi-quantitative analysis of the western blots shown in A, B, and C, respectively. Fraction 2 was shown in black. A, D) GSK3β, t-tau, p-tau (pSer396/pSer404), and p-tau (pSer396) were detected in fractions 2, 7, and 8, and the pellet. The levels of GSK3β and monomers (blue arrow), dimers (green arrow), and high-molecular-weight oligomers (red arrow) of t-tau and p-tau were increased in the lipid rafts from the APP + mice. B, E) Aβ dimers and monomers were observed in fraction 2 and the pellet from the APP + Tau + mice. The levels of GSK3β and monomers (blue arrow), dimers (green arrow), and high-molecular-weight oligomers (red arrow) of p-tau (pSer396/pSer404) were higher in fraction 2 from the APP + Tau + mice than in fraction 2 from the APP-Tau + mice. C, F) A comparison of fraction 2 between the APP-Tau + (n = 7) and APP + Tau + (n = 4) mice revealed that in the APP + Tau + mice the cellular lipid raft levels of GSK3β and monomers (blue arrow), dimers (green arrow), and high-molecular-weight oligomers (red arrow) of p-tau (pSer396/pSer404) increased with the accumulation of Aβ monomers and dimers. The levels of Aβ, Fyn, GSK3β, p-NR2B, and p-tau (pSer396/pSer404) were significantly higher in the APP + mice than in the APP- mice (p < 0.05).
Enhanced phosphorylation and oligomerization of tau in the cellular lipid rafts of double transgenic mice
Aβ dimers and monomers were observed in fraction 2 in the APP + Tau + mice. In a comparison of lipid rafts between the APP-Tau + and APP + Tau + mice, it was found that the levels of GSK3β and p-tau (pSer396/pSer404) monomers (blue arrow), dimers (green arrow), and high-molecular-weight oligomers (red arrow) were increased in the APP + Tau + mice (Fig. 3B, 3E). A semi-quantitative western blotting-based comparison between the APP-Tau + and APP + Tau + mice revealed identical findings (Fig. 3C). The levels of Aβ, Fyn, GSK3β, p-NR2B, and pSer396/Ser404 tau in the lipid rafts were significantly higher in the APP + Tau + mice (n = 4) than in the APP-Tau + mice (n = 7) (Fig. 3F). Thus, Aβ oligomers enhanced the levels, hyperphosphorylation, and oligomerization of tau in cellular lipid rafts.
Increased levels of Fyn and p-NR2B and enhanced tauopathy in synaptosomal lipid rafts
PSD95 was recovered from the synaptosome fraction between buffer B and C in both the APP + and APP- mice. Synaptophysin, the PrPc, Fyn, p-tau (pSer396/pSer404), and p-tau (pSer396) were also detected in the synaptosome fraction in both the APP + and APP- mice. A large amount of the Aβ dimer and a smaller amount of the Aβ monomer were recovered from the synaptosome fraction in the APP + mice. The synaptosome fraction from the APP + mice contained greater amounts of the PrPc, Fyn, p-tau (pSer396/pSer404), and p-tau (pSer396) than that from the APP- mice (Fig. 4A).
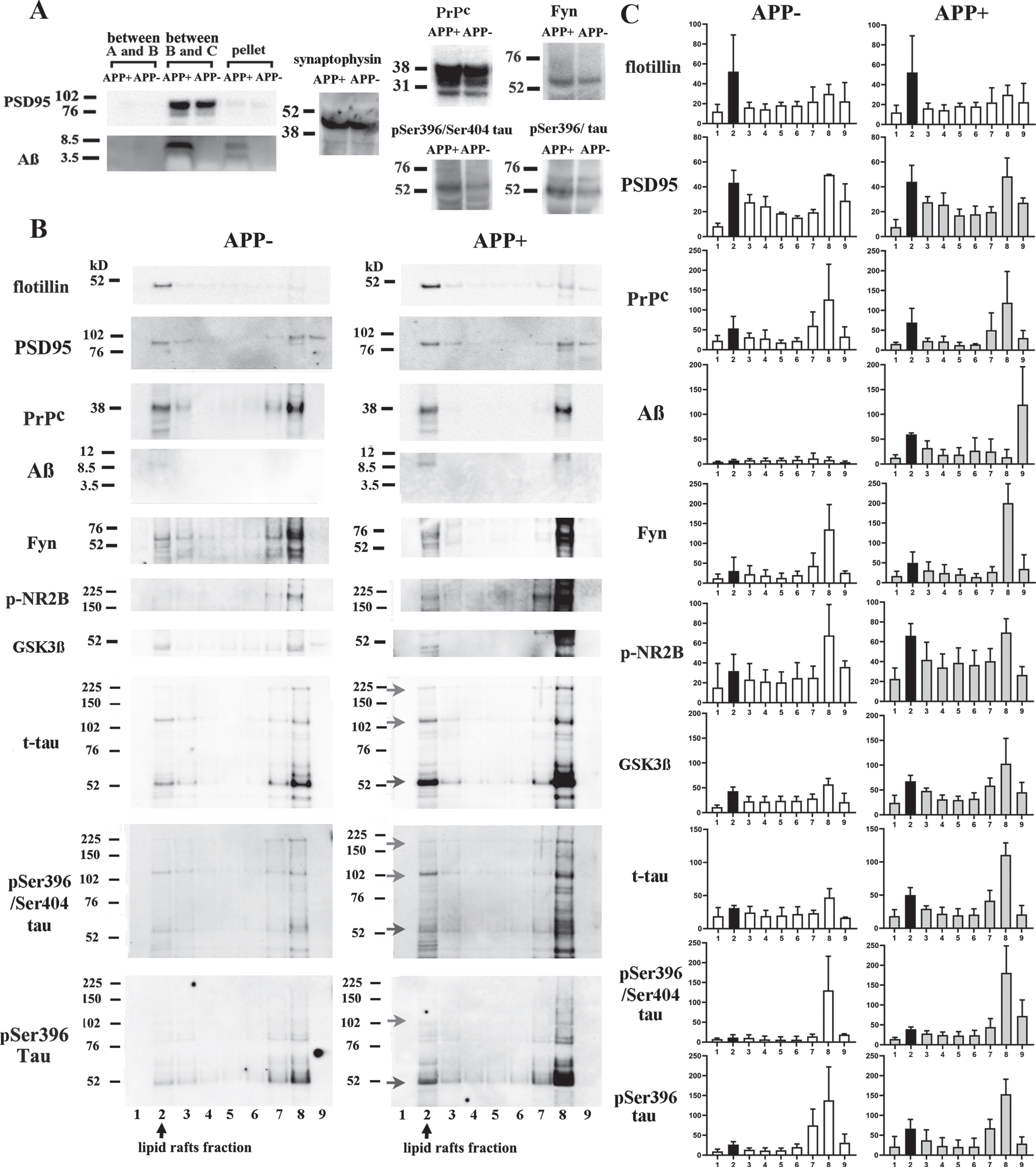
Levels of Fyn, p-NR2B, and p-tau oligomers increased with Aβ dimer accumulation in synaptosomal lipid rafts. A) The synaptosomes of the 12-month-old APP + and APP- mice were extracted from the fraction between buffers B and C, and identified based on their expression of PSD95. A large amount of Aβ dimers and a smaller amount of Aβ monomers were recovered from the synaptosomes of the APP + mice. The synaptosomes of the APP + mice contained larger amounts of the PrPc, Fyn, p-tau (pSer396/pSer404), and p-tau (pSer396) than those of the APP- mice. The amount of synaptosomal synaptophysin did not differ between the APP+ and APP- mice. B) The synaptosomes were subjected to sucrose density gradient centrifugation to separate out the lipid rafts. C shows the results of semi-quantitative analysis of the fractions shown in B. Fraction 2 was shown in black. Flotillin was mainly present in fraction 2. PSD95, the PrPc, Fyn, p-NR2B, GSK3β, t-tau, p-tau (pSer396/pSer404), and p-tau (pSer396) were recovered from fractions 2, 7, and 8. Aβ dimers were detected in fraction 2 in the APP + mice. Increased levels of Fyn, p-NR2B, and GSK3β were also observed in this fraction. Monomers (blue arrow), dimers (green arrow), and high-molecular-weight oligomers (red arrow) of t-tau and p-tau (pSer396/pSer404) and p-tau (pSer396) were detected in the synaptosomal lipid rafts from the APP + mice.
The synaptosome fraction was separated further using sucrose density gradient centrifugation to obtain synaptosomal lipid rafts. Flotillin migrated into fraction 2 as well as the cellular lipid rafts fraction. PSD95, the PrPc, and Fyn and low levels of GSK3β, t-tau, and p-tau were detected in the synaptosomal lipid rafts from the APP- mice. The synaptosomal lipid raft levels of Aβ dimers, p-NR2B, and GSK3β and monomers (blue arrow), dimers (green arrow), and high-molecular-weight oligomers (red arrow) of t-tau and p-tau were higher in the APP + mice than in the APP- mice (Fig. 4B, 4C). Thus, the levels of Fyn and p-NR2B and the levels, phosphorylation, and aggregation of tau in the synaptosomal lipid rafts increased with Aβ dimer accumulation.
Age-dependent increases in the levels of Fyn and p-NR2B and enhanced tauopathy in synaptosomal lipid rafts with Aβ dimer accumulation
To examine the sequential alterations that occur in the synaptosomal lipid rafts of AD model mice, we longitudinally examined the levels of various AD-related molecules in the brains of 4-, 6-, and 12-month-old APP- and APP + mice. In the APP + mice, Aβ dimers first appeared at 6 months of age, and their levels then increased age-dependently. The levels of Fyn and p-NR2B and p-tau (pSer396/pSer404) monomers (blue arrow), dimers (green arrow), and high-molecular-weight oligomers (red arrow) rose in parallel with the increase in the amount of Aβ dimers in the synaptic lipid rafts of the APP + mice (Fig. 5). These findings suggest that in the synaptic lipid rafts of AD model mice Fyn and NMDR activation and the production, phosphorylation, and oligomerization of tau were continuously enhanced from 6 months of age as the accumulation of Aβ dimers progressed.

Age-dependent increases in the levels of Fyn, p-NR2B, and p-tau (pSer396/pSer404) with Aβ dimer accumulation in synaptosomal lipid rafts. All of the synaptosome fractions obtained from the 4-, 6-, and 12-month-old APP- and APP + mice were subjected to western blot analysis. The results of semi-quantitative analysis of the fractions are shown. Fraction 2 was shown in black. In the APP + mice, Aβ dimers appeared in the synaptosomal lipid rafts at 6 months of age, and the level of Aβ dimers had increased at 12 months. The levels of Fyn and p-NR2B and monomers (blue arrow), dimers (green arrow), and high-molecular-weight oligomers (red arrow) of p-tau (pSer396/pSer404) in the synaptosomal lipid rafts increased age-dependently from 6 to 12 months of age in the APP + mice.
DISCUSSION
The sequential separation of brain components revealed that Aβ monomers and dimers and the PrPc were predominantly detected in lipid rafts. Immunoprecipitation experiments showed Aβ dimer/PrPc complex formation in the lipid rafts from mouse brains, and histological examinations showed the colocalization of Aβ oligomers and the PrPc in the brains of the APP + mice. Although a small amount of the Aβ dimer/PrPc complexes moved to the pellet fraction, these findings indicate that lipid rafts are the major platform for Aβ oligomerization and the formation of Aβ oligomer/PrPc complexes. The PrPc, which functions as an Aβ oligomer receptor, is predominantly found in lipid rafts, where the conversion of the PrPc to the scrapie isoform of the prion protein (PrPsc) also occurs [3, 36–40]. In a previous study, the genetic ablation of the PrPc and treatment with anti-PrPc antibodies both restored Aβ oligomer-induced suppression of hippocampal LTP [3]. These findings support our suggestion that lipid rafts are the major platform for Aβ oligomer/PrPc complex formation.
Aβ oligomer/PrPc complexes activate Fyn via metabotropic GluR 5 (mGluR5) [41]. Cholesterol depletion removes the PrPc and Fyn from lipid rafts and reduces the binding of Aβ oligomers to cells and Fyn activation [26]. In our study, Fyn, NR2A, NR2B, and p-NR2B were detected in lipid rafts. In addition, the levels of Fyn and p-NR2B were increased in the lipid rafts of the APP+ mice, but no such increases were seen in the levels of NR2A or NR2B. Um et al. showed that Aβ oligomers increased the levels of p-NR2B and induced calcium signaling and cell toxicity in primary cultured cortical neurons [4]. Kaufman demonstrated that Aβ oligomers caused increases in the levels of p-NR2B in mouse hippocampal slices and that the administration of the Fyn inhibitor AZD0530 blocked the Aβ oligomer-induced increase in the level of p-NR2B [42]. Our experiments support the idea that Aβ oligomer/PrPc complexes increase the levels of p-NR2B in the lipid rafts of animal models of AD. Thus, lipid rafts act as a platform for the Fyn-NMDAR signaling induced by Aβ oligomers.
In the present study, t-tau, p-tau, tau oligomers, and GSK3β were found in lipid rafts, and the lipid raft levels of these molecules were higher in the APP + mice than in the APP- mice. Since APP+mice do not develop marked tauopathy, we performed further experiments in APP+Tau+double transgenic mice, which exhibit both Aβ amyloidosis and NFTs. As a result, we obtained essentially identical findings. These two different experiments strongly indicate that the production, phosphorylation, and oligomerization of tau occur in lipid rafts and that they occur in parallel with Aβ oligomer/PrPc complex accumulation. We previously showed that the accumulation of Aβ dimers and p-tau in lipid rafts is an early event in mild cognitive impairment (MCI) due to AD [17]. Recent clinical studies have indicated that Aβ induces the overproduction and phosphorylation of tau long before MCI occurs [15, 16]. Activated Fyn causes the overproduction of p-tau [43], tyrosine kinase 2 (Pyk2) [44], and GSK3β [45]. GSK3β phosphorylates tau [46], and activated GSK3β is recruited into lipid rafts [47]. In a previous study, Aβ treatment increased the levels of p-tau (pTyr18), p-tau (pSer396/pSer404), and cdk5 in lipid rafts [30]. Although we only investigated tau phosphorylation at the Ser396 and Ser404 sites, the levels of p-tau (pSer396/pSer404) and GSK3β were clearly increased in lipid rafts. Tau oligomers are toxic species and spread in a similar manner to prions [48]. They are generated on membranes by negatively charged phospholipids [49], which are abundant in lipid rafts [50]. Tau oligomer neurotoxicity is mediated by their binding to the PrPc [51]. In a previous study, the oligomerization of tau was blocked when Fyn was knocked out. Conversely, the level of p-tau was upregulated by Fyn [52]. In our study, the level of tau oligomers was actually increased in lipid rafts. Together, these findings indicate that lipid rafts function as a major platform that links Aβ amyloidosis to the overproduction of p-tau species and tau oligomers.
Synapses are the main target of Aβ oligomer neurotoxicity. Lipid rafts are abundant in synapses and play an important role in synaptic transmission [27]. For example, NMDARs translocate from post-synaptic non-raft membranes to post-synaptic lipid rafts during spatial memory formation [53]. In rat brain synaptosomes, 91±3% of Fyn, 52±3% of NR2A, and 58±4% of NR2B was localized in lipid rafts, and the amount of p-NR2B present in synaptic lipid rafts was increased by ischemia [54]. In addition, Fyn transgenic mice had increased levels of p-NR2B and p-tau in their synaptosomes [55]. Our results confirmed the presence of Aβ oligomer/PrPc complexes, activated Fyn, and p-NR2B and p-tau/tau oligomers in both synaptic and cellular lipid rafts.
Synaptic tau seeding precedes tau pathology in the AD brain, and sodium dodecyl sulfate (SDS)-stable tau oligomers were found to localize in the pre- and postsynaptic synaptosomes of both AD patients and non-demented controls [56, 57]. Henkins et al. showed that the SDS-stable p-tau oligomers in the synaptosomes of AD patients were tau dimers and trimers [58]. In the present study, although the absolute amounts and relative proportions of these molecules were low and varied, Aβ-induced Fyn-NMDAR signaling and tauopathy were observed in the synaptosomal lipid rafts.
Our longitudinal study of 4-, 6-, and 12-month-old APP + mice showed that Aβ dimer accumulation, Fyn-NMDAR activation, and tauopathy are first seen in synaptic lipid rafts as early as 6 months of age and then advance with aging. In APP + mice, Aβ dimer-induced memory disturbance emerged from 6 months of age [17, 59]. A recent cerebrospinal fluid biomarker study by the Dominantly Inherited AD Network (DIAN) revealed that Aβ accumulation induced the overproduction of p-tau at the Thr217 and Thr181 sites as early as two decades before the development of cognitive impairment and aggregated tau pathology [16]. Since we had not finished characterizing appropriate antibodies for p-tau at the Thr217 and Thr181 sites, we could not examine these findings or the timing of the appearance of various p-tau sites in this study.
We found Aβ, p-tau, Fyn, p-NR2B, and GSK3β in fractions 7–9 than in fraction 2 (Figs. 3A, 5). However, increased levels of these proteins were seen in the lipid raft fraction in the APP + mice and APP + Tau + mice, with the exception of p-tau (pSer396) in the experiment shown in Fig. 3A, GSK3β and p-tau (pSer396/pSer404) in the experiment shown in Fig. 3B, and p-NR2B and GSK3β in the experiment shown in Fig. 4B. The time course experiment also showed greater increases in the levels of Fyn, p-NR2B, and p-tau (pSer396/pSer404) in the lipid raft fraction than in fractions 7–9. Only 0.4–0.8% of plasma membrane molecules are found in lipid rafts [60]. Fractions 7–8 contain Triton-X-soluble proteins from the plasma membrane, cytoplasm, and organelles. Large amounts of soluble proteins are found in fractions 7–8 from most cells. Fraction 9 is a pellet that precipitates on the bottom of the centrifuge tube and contains aggregated proteins, such as Aβ fibrils, and cell debris. Thus, high concentrations of these proteins are found in lipid rafts. Therefore, we suggest that these proteins in lipid rafts may be more important for the pathogenesis of AD than those found in fractions 7–9. The changes in the levels of proteins found in Triton-soluble fractions will be analyzed in subsequent studies.
We confirmed that Aβ oligomer/PrPc complex formation and the associated downstream events emerge at an early stage in synaptic lipid rafts, which function as a common platform for AD pathology in the brains of AD model mice. Aβ is generated on the lipid rafts [20–22]. Aβ accumulation is regulated by the composition of lipid rafts [61]. Changes in the composition of the lipid rafts in the brain occur during aging [62] and are seen from the earliest stages of AD [63]. In addition, the induction of cholesterol accumulation by Nef protein increased the amounts of tau in lipid rafts [28]. Based on these findings, the modification of lipid raft components and the inhibition of their roles in the sequential events of AD pathology may be a promising novel approach to the development of disease-modifying therapies for AD.
Conclusions
Our study showed that lipid rafts act as a common platform for the progression of AD pathology. Lipid rafts function as a common platform that links sequential events in AD pathology.
Footnotes
ACKNOWLEDGMENTS
We thank Sakiko Narita, Kaori Haga, Sachiyo Ichinohe, and Sachiyo Kasai for providing research assistance; Dr. Peter Davies for providing the PHF-1 antibody, Dr. Etsuro Matsubara for providing the 72D9 antibody; and Dr. Koichi Ishiguro for providing the anti-GSK3β antibody.
The study was supported by Grants-in-Aid for Scientific Research (C) (18K07385 to M.S. and 19K07989 to T.K.) from the Ministry of Education, Science, and Culture of Japan.