
Abstract
Background:
Synaptic abnormalities in synaptic proteins are the initial hallmarks of Alzheimer’s disease (AD). The higher level of palmitoylation of synaptic proteins was closely associated with amyloid-β (Aβ) in AD. Cattle encephalon glycoside and ignotin (CEGI) have been shown to act as multitarget neurotrophic agents in APPswe/PS1dE9 (APP/PS1) transgenic AD mice. However, it is not clear whether CEGI can influence Aβ deposition or whether it does so by the regulation of protein palmitoylation and expression of synaptic proteins in transgenic AD mice.
Objective:
In this study, we investigated the roles of CEGI in modulating postsynaptic density protein 95 (PSD-95) palmitoylation, Aβ pathologies, and expression of synaptic-associated proteins in APP/PS1 mice.
Methods:
Five-month-old APP/PS1 mice were treated intraperitoneally with 6.6 mL/kg of CEGI for 6 weeks. At the end of the treatment period, APP/PS1 mice were subjected to Morris water maze to test their cognitive functions. Acyl-biotinyl exchange (ABE) for PSD-95 palmitoylation, immunofluorescent staining for expression of PSD-95, N-methyl-D-aspartic acid receptor subunit 2B (NR2B), and synaptotagmin 1 (SYT1) were assessed in mouse brain sections.
Results:
CEGI treatment in APP/PS1 mice significantly reduced Aβ deposition, relieved memory deficits, and decreased PSD-95 palmitoylation while markedly increasing the expression of PSD-95, NR2B, and SYT1 in the frontal cortex. There was a significant correlation between Aβ expression and PSD-95 palmitoylation in APP/PS1 mice.
Conclusion:
Our findings demonstrate that CEGI improved AD-like neuropathology, possibly by inhibiting PSD-95 palmitoylation, improving learning memory, and enhancing expression of synaptic-associated proteins, representing a potential therapy for AD treatment.
INTRODUCTION
Synaptic loss and abnormalities in synaptic proteins are initial hallmarks of Alzheimer’s disease (AD) [1, 2]. Compared with amyloid plaques, neurofibrillary tangles, and neuronal loss, region-specific decreases in synaptic proteins may be more closely related with cognitive deficits in AD [3]. Moreover, previous studies have demonstrated that both presynaptic and postsynaptic proteins are fundamental to normal synaptic function and that their downregulations are correlated with cognitive deficits in AD [4–6].
Compared to that of postsynaptic markers, presynaptic markers (e.g., synaptophysin) have been more commonly investigated in the context of AD. Postsynaptic density protein 95 (PSD-95), a prominent postsynaptic-scaffolding protein, is the most plentiful protein in the postsynaptic density [7] and plays a pivotal role in synaptic stability and function [8] via its trafficking, recruitment, and scaffolding of N-methyl-D-aspartic acid receptors (NMDARs) and α-amino-3-hydroxy-5-methyl-4-isoxazole-proprionic acid receptors (AMPARs) to the postsynaptic membrane; additionally, PSD-95 induces restructuring of presynaptic terminals, as well as modulates the trafficking and synaptic localization of NMDAR subunit 2B (NR2B) during development [9, 10]. Moreover, PSD-95 may directly interact with amyloid-β (Aβ) to modulate AD-implicated synaptic pathology, and it has been demonstrated that PSD-95 levels are decreased in pathological regions within the brains of AD patients [11].
In recent years, many studies have elucidated multiple influences of palmitoylation on pathophysiologies in a variety of psychiatric and neurological diseases, such as AD, Huntington’s disease, intellectual disabilities, schizophrenia, major depression [12–14], and infantile neuronal ceroid lipofuscinosis (INCL) [15]. Furthermore, a striking 41% of synaptic proteins are substrates for palmitoylation [16]. Additionally, PSD proteins are frequently palmitoylated, and a high level of PSD-95 palmitoylation is also associated with accumulation of Aβ in AD. In particular, several studies have shown that PSD-95 is a key target of signaling pathways related to post-translational palmitoylation, the latter of which influences the function of PSD-95 its synaptic stability in AD neuropathology [17]. Furthermore, the role of synaptic proteins has recently received increased attention in regard to AD prognosis, in terms of potentially providing a theoretical basis for multitarget AD treatments.
The neuroprotective and neurotrophic agent cattle encephalon glycoside and ignotin (CEGI) is extensively utilized in China for treating central-nervous-system and peripheral-nervous-system damage, including from stroke, neonatal hypoxic ischemic encephalopathy, and diabetic peripheral neuropathy. Since CEGI has been demonstrated to be highly safe and to induce few side effects, it was approved by the Chinese Food and Drug Administration (drug approval no. H22025046, Jilin Sihuan Pharmaceutical Co. Ltd., Jilin, China) in 2010. CEGI is a mixture of gangliosides (especially GM1), polypeptides (mainly carnosine), free amino acids, total nitrogen, and nucleic acids. Although 1 mL of CEGI contains fewer gangliosides than other components, gangliosides are the main active ingredients and also are the precursor enhancers of endogenous neurotrophic factors [18, 19]. Our previous studies have shown that CEGI exhibits multi-target amelioration of memory impairments in an amyloid precursor protein/presenilin 1 (APP/PS1; APPswe/PS1dE9) mouse model of AD via decreasing Aβ levels and inhibiting oxidative stress [18]. However, the role of CEGI on protein palmitoylation and synaptic protein expression in APP/PS1 mice has not yet been reported.
In the present study, we primarily investigated the roles and mechanisms of CEGI in modulating PSD-95 palmitoylation, Aβ pathology, and/or expression of synaptic proteins (i.e., PSD-95, NR2B, and synaptotagmin1 [SYT1]) in APP/PS1 mice. We used behavioral tests, acyl-biotinyl exchange (ABE), immunofluorescent staining, and enzyme-linked immunosorbent assays (ELISAs) to detect changes in the aforementioned proteins in the frontal cortex of APP/PS1 mice. Our findings demonstrate that CEGI treatment improved PSD-95 palmitoylation status, reduced Aβ levels, and increased synaptic protein expression in the frontal cortex of APP/PS1 mice, which may provide a valuable theoretical foundation for further exploring the therapeutic potential of CEGI in AD.
MATERIALS AND METHODS
Mice
A total of 60 male mice (five months old) were used in this study and were acquired from Beijing HFK Bio-Science co., Ltd. (Beijing, China), among which 12 were normal C57BL/6J wild-type littermates and 48 were APP/PS1 AD mice (experimental animal breeding license number, SCXK (Jing) 2014-0004). All of the mice were provided food and water ad libitum and were individually housed in a bio-clean facility with a 12 h light and 12 h dark cycle under a room temperature of 22–25°C and a relative controlled humidity of 55–60%. All of the procedures were implemented under an animal protocol permitted by the Institutional Animal Experiment Committee of the 5th Medical Center of the Chinese People’s Liberation Army General Hospital.
Experimental groups and pharmacological treatments
Mice were randomly separated into the following five groups: 1) nTg group (C57BL/6J wide-type mice, n = 12, saline intraperitoneal [i.p.] injection); 2) Tg group (APP/PS1 mice, n = 12, saline i.p. injection); 3) Tg + CEGI group (APP/PS1 mice, n = 12, CEGI at 6.6 mL/kg/day via i. p. injection, Jilin Sihuan Pharmaceutical Holdings Group Ltd., Jilin, China); 4) Tg + donepezil group (APP/PS1 mice, n = 12, donepezil at 2 mg/kg/day via i.p. injection, Eisai China pharmaceutical Co. Ltd., Suzhou, China); and 5) Tg + N-(tert-Butyl) hydroxylamine (NtBuHA) group (APP/PS1 mice, n = 12, drinking water with 1 mM of NtBuHA, Sigma-Aldrich, St. Louis, MO, USA). The use of CEGI, donepezil (acetylcholinesterase inhibitor) [18] and NtBuHA (a nontoxic de-palmitoylation drug to treat diseases including several infantile neuronal ceroid lipofuscinosis [INLs]) [15] were based on previous work. The mice were weighed every 3 days. Figure 1 shows the schematic representation of the experimental protocol.
Schematic representation of the experimental protocol. After treating mice with CEGI, donepezil, and NtBuHA or equivalent saline for 6 weeks, MWM test, IHC, ELISA, ABE, and IF were performed sequentially.
Morris water maze (MWM) test
After 6 weeks of drug treatment (Fig. 1), the spatial learning and memory function of the mice were assessed in the MWM, as previously described [18, 20]. The apparatus consisted of a black circular pool (120 cm in diameter and 25 cm in height), divided into four equal quadrants filled 2/3 with water (21±2°C) and additional nontoxic titanium dioxide. A hidden platform was submerged 2 cm underwater during the spatial learning trials. The MWM test consisted of two parts, a training session to allow the subject to learn of the hidden platform and a probe trial session. The mouse (n = 12/group) was trained in the MWM to find the hidden platform for 4 consecutive days with four sessions per day (60 s per session). When the mice reached the submerged platform, they were allowed to stay on it for 10 s. If the mouse failed to find the platform within 60 s, it was manually guided to the hidden platform by the experimenter and let it rest on it for 10 s. The time that the mouse took to locate the hidden platform was recorded as the escape latency for its learning ability score. The escape latency and swimming trials were recorded and analyzed using ANY-maze software 5.0 (Stoelting, Co., Wood Dale, IL, USA). In probe trials (day 4), the mice were allowed to swim freely for 60 s, but the platform was not in the tank. The time each mouse spent swimming in the target quadrant and the swimming speed were recorded.
Brain-tissue processing
All of the mice in each group were anesthetized with 100 mg/kg of sodium pentobarbital (i. p.) followed by intracardial perfusion with cold 0.9% saline. Mice were then decapitated, and their brains were harvested. Next, each right hemisphere was stored at -80°C for biochemical and molecular-biology analyses. The left cerebral hemisphere was immersed in 4% paraformaldehyde for 72 h and was then dehydrated and embedded with paraffin for histological and immunohistochemical analyses.
Acyl-biotinyl exchange (ABE)
Levels of protein palmitoylation of PSD-95 were quantified by ABE. Brain tissues were triturated on ice via a homogenizer in RIPA lysis buffer (Beyotime Biotechnology, RIPA, Beijing, China) for 1 h at 4°C. Subsequently, these lysed brain tissues were centrifuged (12,000 g, 5 min, 4°C) to gather total supernatant. Then, 10 mg of total proteins were diluted in lysis buffer (LB, pH 7.4) containing 150 mM of NaCl, 50 mM of Tris-HCl, and 5 mM of EDTA. After a chloroform-methanol (CM) precipitation, the dried proteins were re-suspended in 200μl of 4% SDS buffer (SB, pH 7.4; 4% SDS with 50 mM of Tris-HCl and 5 mM of EDTA) and 800μl of lysis buffer (LB) containing 50 mM N-ethylmaleimide (Sigma-Aldrich, USA). Then, the lysate was incubated at 4°C overnight on a shaker. Each sample was transferred into a new centrifuge tube followed by three CM precipitations, after which each protein pellet was dissolved in 400μl of SB and each sample was separated into two equal parts (one for + HA and the other for -HA). Then, each sample was diluted with 800μl of + HA buffer (pH 7.4, 0.7 M of HA (Sigma-Aldrich, Hydroxylamine hydrochloride, St. Louis, MO, USA), 1 mM of Biotin-HPDP (Bio Vision, Milpitas, CA, USA), 0.2% Triton X-100), or equivalent -HA buffer (pH 7.4, 50 mM of Tris-HCl, 1 mM of Biotin-HPDP, 0.2% Triton X-100) followed by an incubation at ambient temperature for 2 h. Each sample was washed via three sequential CM precipitations and the final pellet was suspended in 2% SDS buffer (TB, pH 7.4; 2% SDS with 50 mM of Tris-HCl and 5 mM of EDTA) and was diluted with LB, followed by an incubation with 30μl of streptavidin agarose resin (Thermo Scientific, Rockford, IL, USA) overnight at 4°C on a shaker. After four washes in precooled LB, resin-captured proteins were eluted with LB containing 10% β-mercaptoethanol and were subjected to western blotting.
Western blotting
Equivalent quantities of proteins were fractionated by 12% SDS-PAGE and electro-transferred onto PVDF membranes. After blocking with 5% skimmed milk buffer containing 0.05% Tween-20 for 2 h at ambient temperature, the PVDF membranes were incubated with β-actin (1:1000, Beyotime Biotechnology, Beijing, China) or PSD-95 (1:1000, ab18258, Abcam, UK) antibodies at 4°C overnight. Next, the blots were washed three times with TBST and were then incubated for 1 h with biotin-labeled secondary antibodies at room temperature. Subsequently, membranes were washed three times and were incubated with quantum-dot-labeled streptavidin (1:500, QS-525, Wuhan Jiayuan, Wuhan, China). Blots were exposed via a Gel Doc XR + imaging system (Bio-Rad Universal Hood II, CA, USA) and densitometric quantification was performed and analyzed by an Image-Pro Plus 6.0 image-system.
Enzyme-linked immunosorbent assay (ELISA)
After weighing, brain tissues were triturated on ice via a homogenizer in five volumes of brain weight precooled with guanidine buffer (pH 8.0. 5 M of guanidine-HCI, 50 mM of Tris-HCI), followed by 4 h mixing at ambient temperature. After a dilution in cold standard dilution buffer (Invitrogen, Carlsbad, CA, USA) with 0.05% Tween-20 and 1×PI, the mixtures were centrifuged at 15,000 g for 30 min at 4°C. Next, the supernatants were transferred to new tubes and Aβ42 levels were then quantified by a mouse Aβ42 enzyme-linked immunosorbent assay (ELISA) kit (Invitrogen, Carlsbad, CA, USA).
Immunohistochemical analysis
Four-μm-thick brain coronal sections [21] were deparaffinized in xylene and successively hydrated with ethanol (concentration from 100% to 0%). The slices were subjected to antigen retrieval in formic acid followed by a 5 min incubation with 3% H2O2 to eliminate endogenous peroxidase activity. Next, the slices were washed three times (3 min each) with 1×phosphate-buffered saline (PBS) and blocked with normal goat serum for 30 min, followed by an incubation for 1 h with anti-human β-amyloid antibody (1:100, M0872, Dako, Glostrup, Denmark) at room temperature. After washing, biotin-labeled secondary antibodies against rabbit and mouse and HRP-conjugated streptavidin polymers (ZSGB-BIO, SPlink Detection Kits, SP-9000, Beijing, China) were incubated for 20 min at room temperature. After washing, diaminobenzidine (ZSGB-BIO, SPlink Detection Kits, ZLI-9018, Beijing, China) was used for several seconds to develop the immunoreaction. Finally, sections were dehydrated and coverslips were sealed by neutral resin. We observed all of the sections under a light microscope (Olympus BX60, Tokyo, Japan) and images were acquired with an Image-Pro Plus 6.0 image-system.
Immunofluorescent analysis
After deparaffinating and hydrating, the tissues were heated for antigen retrieval in citrate buffer (pH 6.0) by microwaving (on high) for 20 min and then incubating in 3% H2O2. After washing with PBS, tissues were blocked with 2% BSA for 20 min and were then incubated in PSD-95 (1:300, ab18258, abcam, UK), NR2B (1:80, ab28373, abcam, UK), or SYT1 (1:1000, ab133856, abcam, UK) overnight at 4°C. Then, the sections were washed with PBS and blocked with 2% BSA for 30 min, followed by a 1 h incubation at ambient temperature against biotinylated secondary antibodies to rabbit (1:1000, A0277, Beyotime Biotechnology, Beijing, China), mouse (1:100, A0286, Beyotime Biotechnology, Beijing, China), or chicken (1:100, ab6876, abcam, UK). Then, after another washing and incubation, the slices were incubated with quantum-dot-labeled streptavidin (1:500, QS-525, Wuhan Jiayuan, China). The sections were sealed by 60% glycerol. The images were collected by using a Nikon microscope (Nikon Corporation, Tokyo, Japan) and the captured images were processed using NIS-Elements BR 4.6.
Statistical analysis
All of the data are reported as the mean±standard error of the mean (SEM) and were analyzed with GraphPad Prism (version 7.0, GraphPad Software Inc., CA, USA). A p < 0.05 was considered to be statistically significant. For immunohistochemical/immunofluorescent staining and western blotting, quantitative analyses were implemented via the Image-Pro Plus 6.0 image-analysis system. One-way analysis of variance (ANOVA) and Dunnett’s multiple-comparison post-hoc tests were used for comparing the differences among the five groups.
RESULTS
CEGI improves spatial learning and memory impairments in APP/PS1 mice
Six weeks after treatment, the spatial learning and memory of all mice were assessed by MWM test (Fig. 2A). In order to exclude the influence of individual physical differences, the swimming speed of all mice in the five groups in the probe was determined, and the assessed. Results showed no significant difference in the swimming speed of each group of mice. This indicated that there were no differences in motor ability among mice in the various groups (Fig. 2B).

CEGI treatment alleviates memory deficits of APP/PS1 mice in MWM test. A) Trajectory of each group in place navigation test. B) The swimming speed during the 4 consecutive training days. C) Escape latency of mice in hidden platform test. #p < 0.05 versus nTg group; *p < 0.05 versus Tg group. D) Time spent swimming within the target quadrant in the probe trial. ####p < 0.0001 versus nTg group; *p < 0.05, **p < 0.01 versus Tg group. E) The number of platform location crossing in the probe trial. ###p < 0.0001 versus nTg group; *p < 0.05, **p < 0.01 versus Tg group; n.s., not significant. Data are expressed as the mean±SEM (n = 12 per group).
In the hidden platform test, repeated measures ANOVA showed that the escape latency of the Tg group was significantly longer than that in the nTg group, while CEGI- and donepezil-treated Tg mice reversed this trend (Fig. 2C; p < 0.05). However, NtBuHA-treated Tg mice did not have significantly decreased escape latency relative to the Tg group.
In the probe test (Fig. 2D), the platform was removed from the Morris maze, and the Tg mice spent significantly less time swimming in the target quadrant compared to the nTg mice (Fig. 2D; p < 0.0001), while the CEGI-, donepezil-, and NtBuHA-treated Tg mice spent significantly more time swimming there (Fig. 2D; p < 0.05, p < 0.01, and p < 0.01, respectively). The number of platform crossings was significantly less for Tg mice than for nTg mice (Fig. 2E; p < 0.001). The number of times that CEGI- and donepezil-treated Tg mice crossed the platform was higher compared to the Tg mice (Fig. 2E; p < 0.05, p < 0.01, respectively).
These results, combined with the results of the escape latency, suggest that administration of CEGI for 6 weeks significantly improved the spatial learning and alleviated the memory deficits in APP/PS1 mice.
CEGI ameliorates Aβ42 expression in the frontal cortex of APP/PS1 mice
Treated mice showed no abnormalities during their feeding periods, suggesting that these drugs had no significant toxic effects. Treatment of 5-month-old Tg mice with CEGI for 6 weeks reduced Aβ42 expression and the numbers of Aβ42 plaque deposition in the cortex (Fig. 3A-C). Immunohistochemical analysis showed Aβ42 expression to be 0.00±0.00 (n = 12) in the nTg group, 0.147±0.020 (n = 12) in the Tg group, 0.058±0.016 (n = 12) in the Tg + CEGI group, 0.057±0.013 (n = 12) in the Tg group + donepezil group, and 0.063±0.017 (n = 12) in the Tg + NtBuHA group. There was a significant difference in Aβ42 expression among the nTg group and Tg groups (Fig. 3A-C, p < 0.0001). Treatment with CEGI significantly reduced Aβ42 plaque in the frontal cortex relative to the Tg group (Fig. 3A-C, p < 0.0001). Reduction in Aβ42 plaque also occurred in other treatment groups (Fig. 3A-C, p < 0.001).
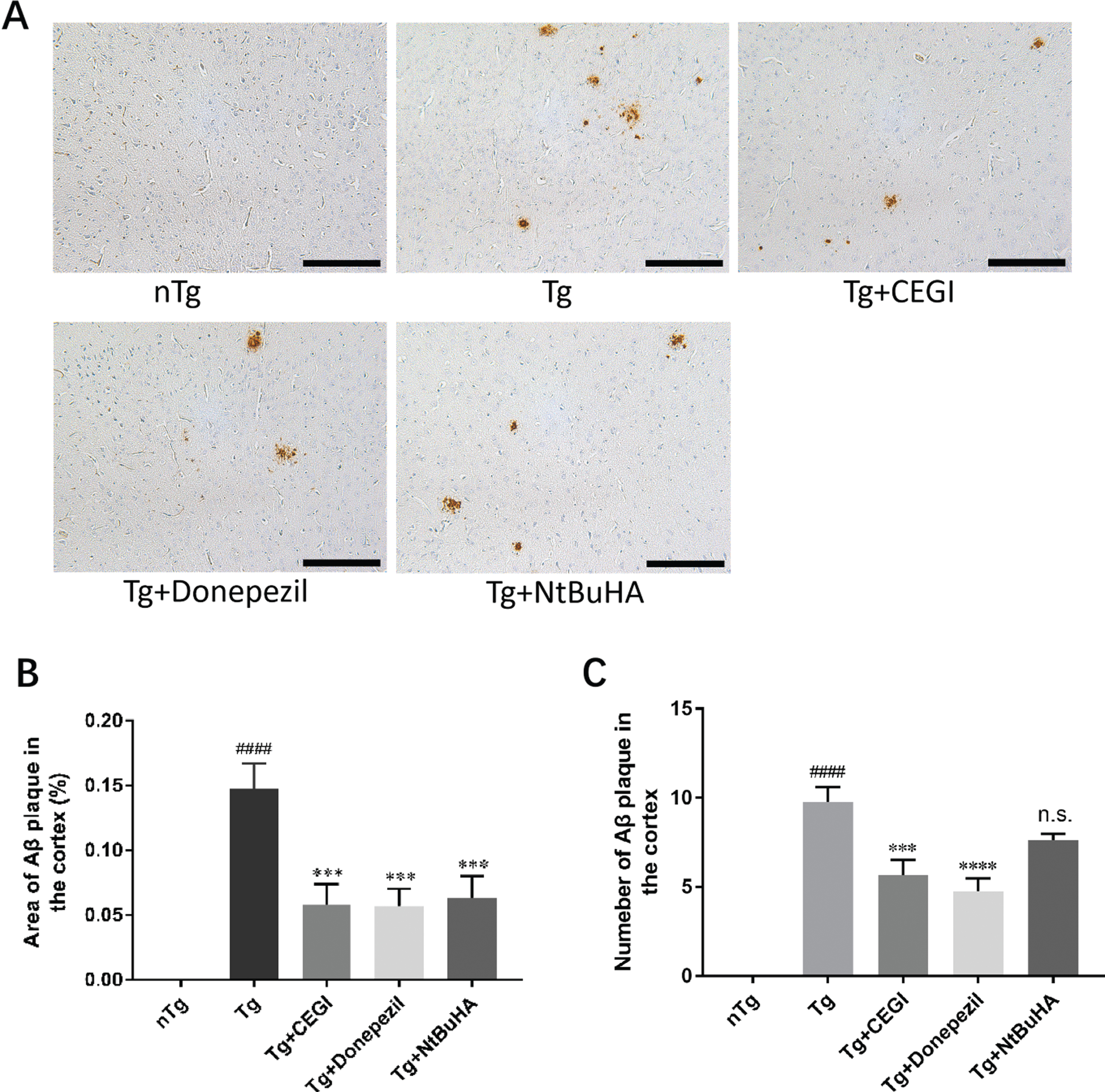
CEGI ameliorates Aβ42 expression in the frontal cortex of APP/PS1 mice. A) Immunohistochemical staining for Aβ42 plaques. B) Quantitative analysis of Aβ42 immunohistochemistry. The y-axis indicates the percentage of area of Aβ42 plaques in the cortex. ####p < 0.0001 versus nTg group; ***p < 0.001 versus Tg group. C) The number of Aβ42 plaque in the cortex of APP/PS1mice. ####p < 0.0001 versus nTg group; ***p < 0.001, ****p < 0.0001 versus Tg group; n.s., not significant. Scale bars = 50μm. Data are expressed as the mean±SEM (n = 12 per group).
CEGI decreases Aβ42 concentration in the frontal cortex of APP/PS1 mice
The concentration of Aβ42 in the frontal cortex of APP/PS1 mice was detected by ELISA (Fig. 4). The results showed that CEGI significantly decreased Aβ42 content in APP/PS1 mice (Fig. 4, p < 0.0001) and further verified the results of Aβ42 immunohistochemistry given above. These data suggested that CEGI is effective in reducing both Aβ42 level and Aβ42 expression.
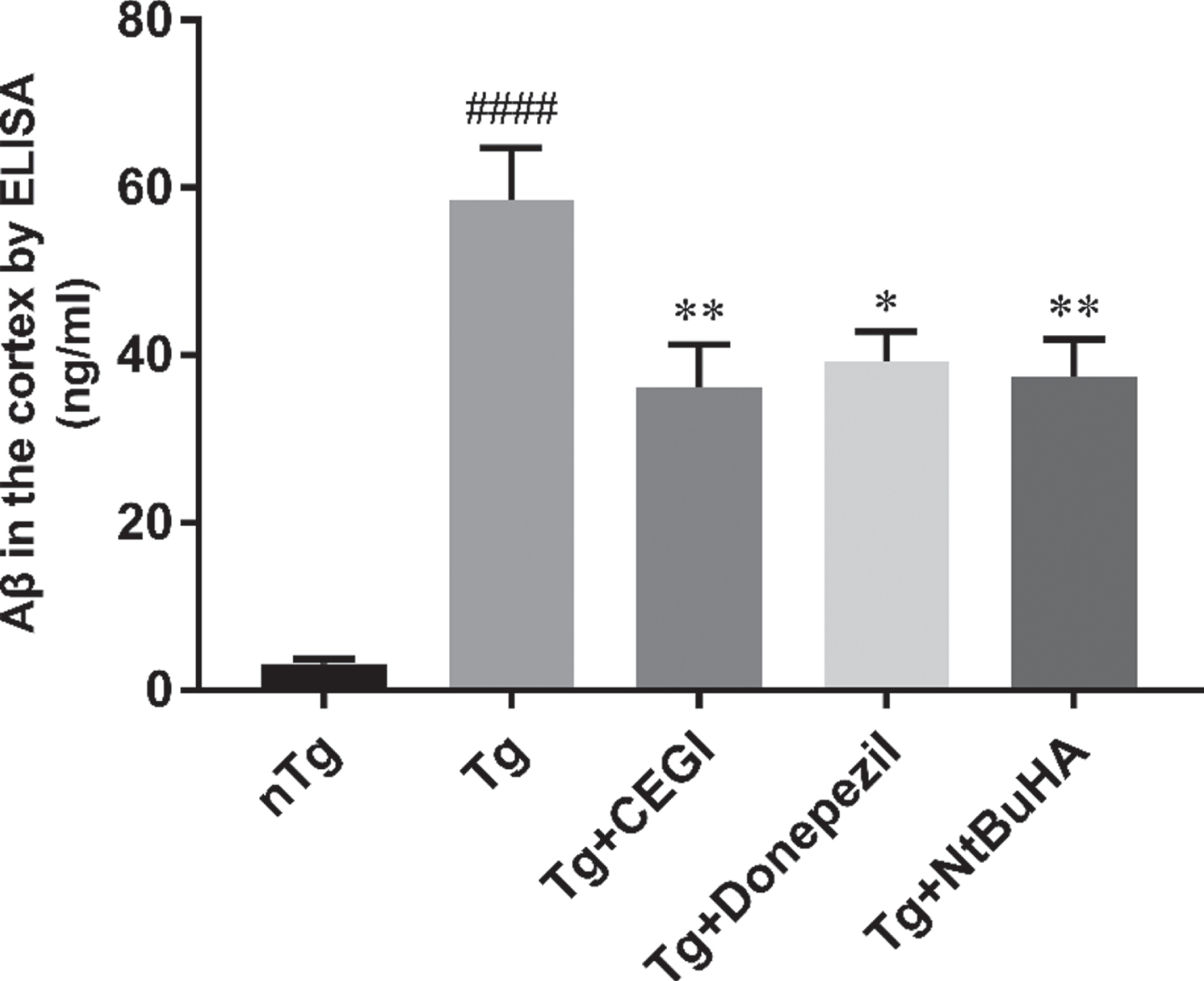
CEGI decrease Aβ42 concentration in the frontal cortex of APP/PS1 mice. ####p < 0.0001 versus nTg group; **p < 0.01, *p < 0.05 versus Tg group. Data are expressed as the mean±SEM (n = 12 per group).
CEGI reduces PSD-95 palmitoylation status in the frontal cortex of APP/PS1 mice
The PSD-95 palmitoylation status in the frontal cortex was analyzed by western blots after ABE test. The results showed that the palmitoylation level of PSD-95 were 41.19±6.12 (n = 12) in nTg group, 92.57±11.34 (n = 12) in Tg group, 45.67±8.36 (n = 12) in Tg + CEGI group, 50.39±9.22 (n = 12) in Tg + donepezil group, and 51.63.±7.50 (n = 12) in Tg + NtBuHA group, respectively. Quantification of western blots showed that compared with that in the nTg group, the palmitoylation level of PSD-95 was significantly higher in the Tg group (Fig. 5A, B, p < 0.01). Additionally, compared with that in the untreated Tg group, the other three Tg groups (CEGI, Donepezil, and NtBuHA) all showed lower levels of PSD-95 palmitoylation (Fig. 5A, B, p < 0.05 and p < 0.01). However, there was no significant difference in the PSD-95 palmitoylation levels among these three treated Tg groups, suggesting that all three drugs were equally effective in reducing palmitoylation levels. These results suggest that CEGI decreased PSD-95 palmitoylation in the frontal cortex of APP/PS1 mice.
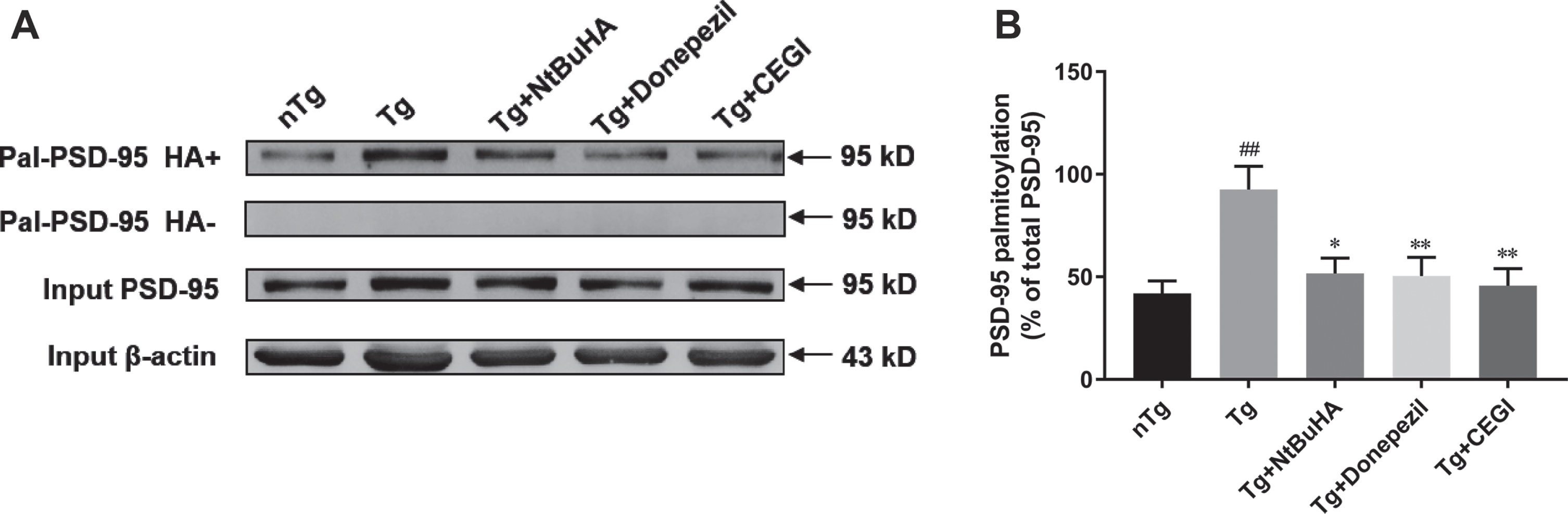
CEGI reduces PSD-95 palmitoylation status in the frontal cortex of APP/PS1 mice. A) PSD-95 palmitoylation was detected in the western blot followed by ABE. B) Quantitative of western blots for PSD-95 ABE. The y-axis represents the PSD-95 palmitoylation percentage of total PSD-95 in the cortex. ##p < 0.01 versus nTg group; *p < 0.05, **p < 0.01 versus Tg group. Data are expressed as the mean±SEM (n = 12 per group).
Aβ42 expression is positively correlated with PSD-95 palmitoylation in the cortex of APP/PS1 mice
The correlation between Aβ42 expression and PSD-95 palmitoylation was further examined by Pearson linear (Table 1). In the Tg group, Aβ42 expression was positively correlated with PSD-95 palmitoylation (r = 0.9858, p < 0.05). Similarly, Aβ42 expression was also positively correlated with PSD-95 palmitoylation in the Tg + CEGI group (r = 0.9982, p < 0.05). This result provides direction and a basis for further research on the clinical application of CEGI.
Correlation between Aβ42 expression and PSD-95 palmitoylation in the cortex of APP/PS1mice
*p < 0.05 indicates a statistically significant difference.
CEGI ameliorates PSD-95 reductions in the frontal cortex of APP/PS1 mice
PSD-95 immunostaining was primarily observed in the neuronal perikarya and in dendrites in the frontal cortex, hippocampus, and entorhinal cortex of wild-type mice [1]. Representative graphs of PSD-95 immunofluorescent study are shown in Fig. 6. PSD-95 expressions in the Tg group were significantly suppressed compared with those in nTg mice (Fig. 6; 49.14±2.64 versus 100.00±4.61, p < 0.0001), which suggests that PSD-95 may play a crucial role in the progression of AD. Treatment with donepezil ameliorated this PSD-95 loss in Tg mice (Fig. 6; p < 0.01). Similarly, CEGI also ameliorated this Tg-induced PSD-95 loss (Fig. 6; 49.14±2.64 in Tg group versus 64.33±2.82 in Tg + CEGI group, p < 0.05), but this effect was slightly less pronounced than that of donepezil. The expression of PSD-95 did not differ significantly between group Tg and group Tg + NtBuHA.

CEGI ameliorates PSD-95 reductions in the frontal cortex of APP/PS1 mice. A) Immunofluorescence staining for PSD-95. B) Quantitative analysis of PSD-95 expressions. The y-axis shows the PSD-95 mean fluorescence intensity in each group relative to the nTg group. ####p < 0.0001 versus nTg group; *p < 0.05, **p < 0.01 versus Tg group; n.s., not significant. Scale bars = 100μm; insets = 25μm. Data are expressed as the mean±SEM (n = 12 per group).
CEGI ameliorates NR2B reductions in the frontal cortexes of APP/PS1 mice
The immunoreactivity study revealed that NR2B was expressed in the frontal cortex, hippocampus, and amygdala [10]. The distribution of NR2B-stained neuronal bodies in frontal cortex is shown in Fig. 7. The expression of NR2B in the frontal cortex of AD mice was determined by immunofluorescence staining. As shown in Fig.7, NR2B expression was 99.99±1.17(n = 12) in the nTg group, 66.67±3.32 (n = 12) in the Tg group, 94.07±4.26 (n = 12) in the Tg + CEGI group, 81.44±3.46 (n = 12) in the Tg + donepezil group, and 70.05±3.43 (n = 12) in the Tg + NtBuHA group, respectively. The expression of NR2B in the untreated Tg group was significantly lower than that in each of the other three groups (nTg, CEGI, and Donepezil) (Fig. 7, p < 0.0001 and p < 0.01). Thus, CEGI ameliorated both NR2B and PSD-95 reductions in the Tg group, suggesting that CEGI may be neuroprotective in an APP/PSI mouse model of AD.

CEGI ameliorates NR2B reductions in the frontal cortex of APP/PS1 mice. A) Immunofluorescence staining for NR2B. B) Quantitative analysis of NR2B expressions. The y-axis represents the NR2B mean fluorescence intensity in each group relative to nTg group. ####p < 0.0001 versus nTg group; ****p < 0.0001, **p < 0.01 versus Tg group; n.s., not significant. Scale bars = 100μm; insets = 25μm. Data are expressed as the mean±SEM (n = 12 per group).
CEGI ameliorates SYT1 reductions in the frontal cortexes of APP/PS1 mice
SYT1 immunofluorescence distribution was detected in neuronal bodies and cytomembrane in the frontal cortex and hippocampus, as previously reported [3]. To evaluate SYT1 changes, we compared Tg mice and wide type controls. The levels of SYT1 were 99.38±2.56 (n = 12) in the nTg group, 84.38±4.42 (n = 12) in the Tg group, 96.40±2.23 (n = 12) in the Tg + CEGI group, 94.98±2.22 (n = 12) in the Tg group + donepezil, and 89.58.±1.48 (n = 12) in the Tg + NtBuHA group. Statistical analysis showed that SYT1 levels were significantly lower in the untreated Tg group compared to those in the nTg mice (Fig. 8, p < 0.0001), whereas both CEGI and Donepezil Tg groups exhibited significantly higher SYT1 levels compared to those in the untreated Tg group (Fig. 8, p < 0.0001, and p < 0.01). Collectively, our results suggest that increased Aβ deposition in APP/PS1 mice correlates with a concomitant decline in SYT1 and that CEGI helps to ameliorate both of these phenotypes.

CEGI ameliorates SYT1 reductions in the frontal cortex of APP/PS1 mice. A) Immunofluorescence staining for SYT1. B) Quantitative analysis of SYT1 expression. The y-axis here shows the SYT1 mean fluorescence intensity in each group relative to the nTg group. #p < 0.05 versus nTg group; *p < 0.05 versus Tg group; n.s., not significant. Scale bars = 100μm; insets = 25μm. Data are expressed as the mean±SEM (n = 12 per group).
DISCUSSION
In the present study, five-month-old APP/PS1 mice were intraperitoneally administered CEGI for six weeks. CEGI significantly increased the expression levels of PSD-95, NR2B, and SYT1 in the frontal cortex of Tg (i.e., APP/PS1) mice compared to these levels in untreated Tg mice. Importantly, we found that compared with those in untreated Tg mice, PSD-95 palmitoylation levels were significantly decreased in the frontal cortices of CEGI-, donepezil-, or NtBuHA-treated Tg mice, which correlated with reductions in Aβ deposition. Thus, CEGI treatment may reduce PSD-95 palmitoylation levels to improve synaptic pathology in APP/PS1 mice, although this correlative result requires further investigation.
As a multi-target neuroprotective agent, CEGI consists of a variety of active protein factors, including monosialoganglioside (GM1), carnosine, free amino acids, and total nitrogen, which may collectively play an effective role in the prevention and repairment of diseased nerve cells and tissues. GM1, the predominant component of CEGI, is a sialic acid-containing glycosphingolipids that exist in membranes of all vertebrate cells and is most abundant in the central nervous system. It has been reported that GM1 has neuroprotective and neuroregenerative properties against Aβ-induced toxicity [22]. In this study, we found that treatment with CEGI significantly reduced Aβ42 expression and the level of Aβ42 plaque deposition in the frontal cortex, relative to the Tg group. These data suggest that GM1, as the main component of CEGI, may be the major player in treatment of AD with CEGI. The possible mechanism of GM1 in reducing Aβ load are as follows: 1) being well-accessible and dynamic, GM1 in nanomolar can serve as an inhibitor of membrane-mediated Aβ oligomerization [23]; 2) GM1 is involved in the process of inhibiting Aβ fibril formation and disrupting Aβ fibril into nontoxic structures in the lipid membrane [24]; and 3) GM1 has the ability to cross the blood-brain barrier and is neurotrophic, which allow it to play an anti-neurotoxic and neuroprotective role [25]. Furthermore, another major active component of CEGI, carnosine, knowing to inhibit cell death, apoptosis, and oxidative stress induced by Aβ1–42 oligomers in RAW 264.7 macrophages [26], could also contribute to the effect of CEGI on prevention and treatment of AD.
The results of our present study demonstrated that CEGI treatment for 6 weeks significantly reduced AD pathological damage, palmitoylation of PSD-95 and facilitated learning and spatial memory in AD mice. However, the other two drugs, donepezil and NtBuHA, did not improve these three indicators simultaneously. These findings indicate that CEGI plays a role in ameliorates cognitive impairment, Aβ42 formation, and protein palmitoylation.
Recent studies have indicated that palmitoylation contributes significantly to multiple dynamic adjustments for synaptic plasticity, as well as to regulating synaptic intensity in response to neuronal activities. For example, palmitoylation of PSD-95, a postsynaptic scaffolding protein, is a pivotal element for synaptic plasticity [8]. Furthermore, recent studies have suggested that aberrant palmitoylated proteins are closely related to pathophysiologies in psychiatric and neurological disorders, such as AD, Huntington’s diseases, intellectual disability, schizophrenia, and INCL [12, 28].
Protein-protein interactions have been reported as one of the mechanisms to coordinate PSD-95 postsynaptic localization [29]. PSD-95 palmitoylation at Cys3 and Cys5 is predicted to represent a central mechanism for such postsynaptic localization because of predicted to represent a central mechanism for such postsynaptic localization because of the role of these residues in PSD-95 anchoring at synapses, which can modulate synaptic strength via AMPARs [30]. As palmitoylation modifications are a conclusive factor in the clustering of PSD-95 at synapses, it has been considered that PSD-95 palmitoylation correlates closely with alterations in synaptic strength. For instance, without influencing NMDARs, de-palmitoylated PSD-95 synaptic clusters were dispersed by glutamate-receptor activation, thus leading to AMPAR selective loss at postsynaptic sites [31]. Moreover, 2-bromopalmitate (2-BP), the inhibitor of protein palmitoylation, weakens the frequency and amplitude of AMPAR-mediated miniature excitatory postsynaptic currents in hippocampal neurons [32]. In the present study, we found significantly elevated palmitoylation levels of PSD-95 in the frontal cortices of Tg mice compared with those in nTg mice, which correlated with increased Aβ accumulation. Furthermore, CEGI-, donepezil-, and NtBuHA-treated Tg mice all had significantly reduced PSD-95 palmitoylation in the frontal cortex compared with that in untreated Tg mice. Hence, we demonstrated for the first time that CEGI regulates PSD-95 palmitoylation and may target brain-specific gangliosides for the alleviation of synaptic damage in APP/PS1 mice.
It has been well demonstrated that as the most abundant postsynaptic-scaffolding protein, PSD-95 has a significant influence on AD pathogenesis. PSD-95 immunoreactivity has been shown to be reduced in APP/PS1 mice and to be associated with the development of memory impairment; a prior study [33] found that this reduced PSD-95 correlated with a loss of neurons in the deep layer of the cortex, which suggests that a decrease of PSD-95 may contribute to neuronal death. It has been suggested that curcumin treatment increases the expression of PSD-95 in hippocampal CA1 in a mouse model of AD [30]. In the present study, we observed that PSD-95 expression was significantly reduced in six-month-old APP/PS1 mice compared to that in wild-type mice, which is consistent with the findings of a previous report [34]. This suggests that Aβ accumulation in APP/PS1 mice is deleterious to neurons and induces a low expression of PSD-95. In our present study, CEGI or donepezil administration in APP/PS1 mice significantly increased PSD-95. It is well-known that there is a link between PSD-95 and the NMDAR subunit, NR2B, particularly in terms of receptor stabilization, excitation-control balance, and synaptic plasticity [3]. Previous studies [35] have shown that NR2B can uniquely heighten memory and cognition. Postmortem analysis in various brain areas of AD patients has revealed a significant decline of NR2B, expression by 40% in the hippocampus and 31% in the entorhinal cortex, compared with that in control samples [36]. Our present data showed that NR2B expression in APP/PS1 mice was reduced and that CEGI significantly ameliorated this reduction, which was consistent with our results on PSD-95 levels.
SYT1 is a synaptic-binding protein in the presynaptic membrane and is mainly distributed in the cerebral cortex and hippocampus. As an essential protein for vesicular exocytosis, SYT1 mediates the release of neurotransmitters via its role as a presynaptic calcium sensor [37]. Some studies have confirmed that SYT1 is significantly reduced and co-localizes with neuronal plaques in the cerebral cortices of post-mortem AD brains [38]. In AD, synaptic loss is correlated with cognitive impairment, and the potential involvement of SYT1 in this phenomenon makes it a candidate molecular marker for the development of clinical manifestations in AD [38, 39]. In our present study, CEGI or donepezil treatments yielded significantly higher SYT1 levels in Tg mice compared with those in untreated Tg mice, suggesting that CEGI and donepezil can elevate SYT1 expression and may contribute to inhibition of Aβ deposition and neurotoxicity in the brains of APP/PS1 mice, although this correlative result requires further investigation.
Overall, in the present study, we examined the possibility that the multi-target neuroprotective agent CEGI has a synaptic protective effect on APP/PS1 transgenic AD mice, the dominant effects of which are as follow: 1) we demonstrated that CEGI treatment in AD mice significantly increased the expression of synaptic-associated proteins PSD-95, NR2B, and SYT1, and remarkably reduced the level of Aβ42 and Aβ42 immunoreactive in the frontal cortex. PSD-95 modulation is a critical step in the neuropathological progression induced by Aβ oligomers [17]. We show here that upregulated of PSD-95 and other synaptic proteins can protect synapses from Aβ42, and its molecular mechanism may be that increasing PSD-95 synaptic content can protect synapses from Aβ-induced synaptic loss and cognitive impairment by reducing NMDA receptor metabotropic function [40]. 2) CEGI treatment significantly decreased PSD-95 palmitoylation in the frontal cortex. PSD-95 palmitoylation was positively correlated with the level of Aβ42 expression (Table 1). This is in good agreement with previous publication, in which high level of palmitoylated PSD-95 in AD patients is associated with Aβ accumulation [41], and the percentage of palmitoylated PSD-95 was greater in the frontal cortices of old mice [42]. Thus, although underlying mechanisms of how CEGI affects PSD-95 level and palmitoylation to protect synapses from Aβ42 remains to be investigated, current study provide multiple lines of evidence showing that CEGI as a multi-target agent has great potential for treatment of AD.
Conclusion
Our current findings provide the first demonstration of neuroprotective effects of CEGI on synapses in APP/PS1 transgenic mice, possibly by reducing PSD-95 palmitoylation and Aβ deposition, as well as enhancing PSD-95, NR2B, and SYT1 levels. These de-palmitoylation and neuro-protective effects suggest that CEGI may represent a candidate agent in the prevention and treatment of AD. However, further investigations on how CEGI regulates both palmitoylation of PSD-95 and expression levels of synaptic proteins in different brain regions of APP/PS1 mice are required before this agent may be considered in AD clinical trials.