
Abstract
Background:
Trisomy 21, an extra copy of human chromosome 21 (HSA21), causes most Down’s syndrome (DS) cases. Individuals with DS inevitably develop Alzheimer’s disease (AD) neuropathological phenotypes after middle age including amyloid plaques and tau neurofibrillary tangles. Ubiquitin Specific Peptidase 25 (USP25), encoding by USP25 gene located on HSA21, is a deubiquitinating enzyme, which plays an important role in both DS and AD pathogenesis. However, the regulation of USP25 remains unclear.
Objective:
We aimed to determine the regulation of USP25 by specificity protein 1 (SP1) in neuronal cells and its potential role in amyloidogenesis.
Methods:
The transcription start site and promoter activity was identified by SMART-RACE and Dual-luciferase assay. Functional SP1-responsive elements were examined by EMSA. USP25 expression was examined by RT-PCR and immunoblotting. Student’s t-test or one-way ANOVA were applied or statistical analysis.
Results:
The transcription start site of human USP25 gene was identified. Three functional SP1 responsive elements in human USP25 gene were revealed. SP1 promotes USP25 transcription and subsequent USP25 protein expression, while SP1 inhibition significantly reduces USP25 expression in both non-neuronal and neuronal cells. Moreover, SP1 inhibition dramatically reduces amyloidogenesis.
Conclusion:
We demonstrates that transcription factor SP1 regulates USP25 gene expression, which associates with amyloidogenesis. It suggests that SP1 signaling may play an important role in USP25 regulation and contribute to USP25-mediated DS and AD pathogenesis.
INTRODUCTION
Down’s syndrome (DS), with an incidence of 1 in 700–1000 live births, is the most common genetic cause of intellectual disabilities [1–3]. Cognitive impairment, muscle hypotonia at birth, and dysmorphic features occur to some extent in all affected individuals. Moreover, individuals with DS inevitably develop Alzheimer’s disease (AD) neuropathological phenotypes after middle age. Amyloid plaques are a unique neuropathological hallmark of AD, consisting of amyloid-β (Aβ). Trisomy 21 is an extra copy of human chromosome 21 (HSA21), the smallest human autosome and represents around 1–1.5% of the human genome, and causes the majority of DS cases [4], while the underlying mechanisms are not well understood. Theoretically, the extra copy of HSA21 would cause a 50% increase in the level of transcripts of all genes mapping to HSA21. However, it has been shown that genomic imbalance, either by deletion or duplication, do not always result in the change in transcript level of genes at the same degree within the aneuploid segment [5, 6]. It suggests that other molecular factors, including transcription factors, play essential roles in regulating RNA transcript of genes within the aneuploid segment.
Abnormal protein accumulation in AD is mainly caused by the dysfunction of protein degradation systems, the ubiquitin-proteasome system (UPS) and the autophagy-lysosome pathway (ALP). These two protein quality control pathways are linked by ubiquitination. Deubiquitination is a necessary condition to regulate the degradation of short-lived ubiquitination proteins. Ubiquitin Specific Peptidase 25 (USP25) is a member of the deubiquitinases (DUBs) family that spans over 150 kb at 21q11.2 [7], one of the lowest gene density regions of the human genome. It is made up of 25 exons and encodes a 1087-aa protein, containing ubiquitin-specific protease (USP) domain, ubiqutin-interacting motif (UIM), SUMO-interacting motif (SIM), and ubiquitin-associated (UBA) domain [8]. USP25 can cleave both lysine 48- and lysine 63-linked polyubiquitin chains and its deubiquitinating enzyme (DUB) activity, were dependent on a cysteine residue (Cys178) and a histidine residue (His607) [9]. There are three USP25 isoforms in human, all containing one UBA domain and two UIMs in its N-terminal region for substrate recognition [10]. Its protease activity can be regulated through its post-translational modification, including ubiquitination and SUMOylation. USP25 itself can be monoubiquitinated at the preferential site lysine 99 (K99), which can be auto-deubiquitinated. Conjugation of ubiquitin (activating) K99 favors DUBs’ catalytic action, and therefore promotes USP25’s activity [10]. SUMOylation within these regions impairs USP25’s binding to and hydrolysis of ubiquitin chains.
USP25 is involved in myogenesis [11], immunity [12] and protein degradation [13] through interacting with the key proteins in those signaling pathways. One of the three USP25 isoforms, USP25m, is restricted to be expressed in muscular tissues. Its expression is increased during myogenesis, suggesting its potential role in regulating muscle development. USP25m interacts with several sarcomeric proteins that are critically involved in muscle differentiation and maintenance [11]. Toll-like receptor (TLR) signaling plays a critical role in innate immunity, and USP25 regulates TLR signaling through affecting its ubiquitination [12]. Mice with USP25 deficiency was associated with enhanced production of proinflammatory cytokines and decreased production of interferon-α [12]. It has also been shown that USP25 is involved in regulating the virus-triggered type I interferon (IFN) induction pathway and its mutants lacking DUB activity affects the ability to block induction of the IFN-β trigged by virus infection [14]. Those results suggest that USP25 plays an essential role maintaining a balanced innate immune response. USP25 is also involved in protein quality control that is essential for maintaining the functioning and homeostasis of many cellular pathways. USP25 also regulates endoplasmic reticulum (ER)-associated degradation (ERAD), which is one of the essential parts of the protein quality control that recognize, deglycosylate, ubiquitinate, and extract misfolded proteins into the cytosol [13]. It is localized at the ER and interacts with ERAD components, deubiquitinates the endogenous proteins and rescues several ERAD substrates from degraded by the proteasome.
Recent studies indicated that USP25 plays an important role in AD pathogenesis. First, increased USP25 promotes microglial activation, synaptic damage, and cognitive dysfunction, while genetic ablation of USP25 reduces neuroinflammation and rescues synaptic damage and cognitive function in AD mice [15]. In addition, USP25 promotes AβPP processing and Aβ generation by increasing BACE1 ubiquitination, while inhibition of USP25 reduces Aβ generation and amyloid plaque formation, and rescues cognitive deficits in AD mice [16]. It highly suggests that the dysregulation of USP25 may play a key role in AD pathogenesis. USP25 is overexpressed in DS fetal brain samples, which may be caused by trisomy of USP25 gene [7]. Moreover, viral infection and lipopolysaccharide (LPS) treatment can upregulate the expression of USP25 through interferon regulatory factor 7 (IRF7) by directly binding to the two conserved IRF binding sites on the USP25 promoter [14]. However, there have been few studies on the mechanism of USP25 cell-specific expression and endogenous transcriptional regulation of the USP25 gene. To explore the mechanism of USP25 transcriptional regulation, we have cloned and functionally characterized the USP25 gene promoter region. We identified the transcription start site of human USP25 gene and found that the promoter of the human USP25 gene contained specificity protein 1 (SP1)-responsive elements. SP1 overexpression significantly promotes USP25 transcription in both non-neuronal and neuronal cells, while SP1 inhibition significantly reduced USP25 transcription and subsequent USP25 protein expression. Moreover, SP1 inhibition dramatically reduced amyloidogenesis. The results demonstrate that transcription factor SP1 plays a significant role in regulating USP25 gene expression, which associates with amyloidogenesis. Our study highly suggests that SP1 signaling plays a role in USP25 regulation and may contribute to USP25-mediated DS and AD pathogenesis.
MATERIALS AND METHODS
Cloning and plasmids
The 5’-flanking regions of the human USP25 gene were amplified by PCR from human BAC DNA clone (BACPAC Resources Center, CHORI, Oakland, CA, USA). Primers were designed to including restriction enzyme sites such that PCR products could be cloned into multicloning sites of pGL3-Basic (Promega, Madison, WI, USA). Eleven fragments covering the 5’-flanking region of USP25 gene from –2031 bp upstream to +226 bp downstream of the transcription start site at +1 (adenine) were amplified by PCR and inserted in front site at +1 (adenine) were amplified by PCR and inserted in front of the luciferase reporter gene (Luc) in the pGL3-basic expression vector. The primers, including restriction enzyme sites, were synthesized as follows: forward 1) Xho12075 ccgctcgagaggaggacaacgccattcc; 2) HindIII1976 cccaagcttgtgaccctccgggcggc; 3) Xho11967 ccgctcgagcaatgtagggttagggcgg; 4) Bam1742 tttgggtaagctagggatccgac; 5) Nh1211 ctagctagcgttcccagattagctcctg; 6) Nh57 ctagctagcgtaatgtagcagtttagaagtt; Reverse 1) Hind2313 cacaagcttaaacgccgactgtgagg; 2) Bam1781 ctgcgcccagggcttgtcggatc; 3) BI1292 gaagatctggaattgtaaggaaaatct; 4) BI2313 gaagatctaaacgccgactgtgagg; 5). HindIII2169 cccaagcttgttcgcacgttcctttg; 6) HindIII2095 cccaagctttccctccgcgagtcctc.
Switching mechanism at 5’end of RNA transcription (SMART) RACE cDNA amplification
Total RNA was extracted from HEK293 using TRI reagent (Sigma) following the manufacturer’s protocol. SMART-RACE was performed using the SMARTertrademark RACE cDNA Amplification Kit (Clontech) following the user’s manual. Simply, the first stand cDNA was synthesized from total RNA extracted from HEK293 cells with oligo (dT) primer in the presence of SMARTer IIA oligonucleotide (5’-aagcagtggtatcaacgcagagtacxxxxx (X is undisclosed base in the proprietary SMARTer oligo sequence). The SMARTer IIA oligonucleotide is able to anneal to the 5’-end of the first stand cDNA and serves as template to extend the 5’-end cDNA tail. A USP25 reverse primer (5’-cgtggccctcacagtcg) was specifically designed to recognize the +201 to +221 bp of human USP25 gene downstream of the translation start site (ATG). The PCR products containing USP25 promoter region were amplified using SMARTer IIA oligonucleotide and USP25 reverse primer and the first stand cDNA as template. The resulting PCR products were cloned into pcDNA4/myc-His A vector for sequencing and the first nucleotide linking with the adapter sequence was identified as the transcription start site of the human USP25 gene.
Cell culture, transfection, and luciferase assays
HEK293 ((human embryonic kidney cells), SH-SY5Y (human neuroblastoma cells), N2a (mouse neuroblastoma), C6 (rat glioma cells), and SH-APPs (SH-SY5Y cells with human Swedish APP overexpression) cells were cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum, 1 mmol/L of sodium pyruvate, 2 mmol/L of L-glutamine and 50 units of penicillin and 50μg of streptomycin (Invitrogen Carlsbad, CA, USA) [17]. All cells were maintained at 37°C in an incubator containing 5% CO2. Cells were seeded onto 24-well plates or 35-mm-diameter plate 1 day prior to transfection and grown to approximately 25% confluence by the day of transfection. Cells were transfected with 0.5μg of plasmid DNA per well using 1.5μL or 2μg plasmid DNA per 35-mm-diameter plate for RNA extraction by 6μL PEI reagent (Cat#. 23966, Polysciences Inc.) The Renilla luciferase vector pRluc was co-transfected to normalize the transfection efficiency of various luciferase reporter constructs. Cells were harvested at 48 h after transfection and lysed with 100μL passive lysis buffer (Promega) per well. Firefly luciferase activities and Renilla luciferase activities were measured sequentially using the Dual-luciferase reporter assay system (Promega). The firefly luciferase activity was normalized according to Renilla luciferase activity and expressed as relative luciferase units to reflect the promoter activity.
Electrophoretic mobility shift assay (EMSA)
HEK293 cells were transiently transfected with SP1 plasmid and nuclear extracts were collected. Cells were rinsed and harvested with 1×Phosphate-buffered saline. After centrifugation, cell pellets were resuspended with 5×volume of buffer A [10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM dithiothreitol (DTT), 0.5 mM phenylmethylsulfonyl fluoride (PMSF)]. Cells were pipetted up and down gently and maintained on ice for 15 min. The cell suspension was transferred to a Kontes all glass Dounce tissue grinder and ruptured by 10 strokes. 10% NP40 was added into the cell suspension for a final concentration of 0.5%. The samples were placed on ice for 15 min and stroked 5 more times. Crude nuclei were collected by centrifugation at 2000 g for 10 min and washed three times with buffer A containing 0.5% NP40 and resuspended in buffer C [20 mM HEPES pH 7.9, 0.4 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF), 10% Glycerol] at 4°C for 15 min. The samples were centrifuged at 12000 g for 4 min at 4°C, and the supernatant containing nuclear proteins was collected. Oligonucleotides containing the putative SP1 binding site on human USP25 promoter region were synthesized for detecting the binding ability of SP1 to USP25 promoter. The sequences of the oligonucleotides were USP25 –150 –125, forward, cggccccgccccgccgcccggagggt and reverse, accctccgggcggcggggcggggccg; USP25 –111 –83, forward, tagggcggccggcgggcggggctctgcgg and reverse, tagggcggccggcgggcggggctctgcgg; USP25 –80 –53, forward, gaggagccgccgggcgggggcggggatg and reverse, catccccgcccccgcccggcggctccac; mutant SP1, forward, attcgatcggttgagggagagc and reverse, gctctccctcaaccgatcgaat; The probes corresponding to SP1 binding sites were labeled with IRDye-700 (IDT). Prior to incubation with nuclear extract, oligonucleotide probes were heated at 98°C for 5 min and annealed at 65°C for 10 min. SP1 consensus oligo 0.5 pmol of annealed probes were incubated with 2μl of nuclear extract for 20 min at 22°C and the reaction mixtures were separated on a 4% Tris-glycine-EDTA gel in darkness. The mobility of probes on the gel was visualized using the LI-COR Odyssey (LI-COR Biosciences). For the competition assay, wild-type SP1 consensus oligonucleotides and mutant SP1 oligonucleotides were used as positive and negative controls, respectively.
Semi-quantitative RT-PCR
HEK293, SH-SY5Y, N2a, and C6 cells were harvested 48 h after the treatments and total RNA was extracted using TRI reagent (Sigma). Reverse transcription was sequentially performed using ThermoScripttrademark RT-PCR system kit following the manufacturer’s protocol (Invitrogen). The USP25 gene specific primer F-tgacacccagatactacagc and USP25 R- gccaacattctttagcccaacg were used to amplify a 433 bp fragment of human USP25 gene in HEK293, SH-SY5Y, and N2a cells. The USP25 gene specific primer F-gcagtcggcaataaaattagag and USP25 R-ctctgtatggacccttggaca were used to amplify a 422 bp fragment of rat USP25 gene in C6 cell. The GAPDH gene was also amplified using the forward primer (tgcaccaccaactgcttagc) and reverse primer ggcatggactgtggtcatgag), which produced an 87 bp fragment. The PCR products were analyzed on 1.5% agarose gels.
Immunoblotting
HEK293, SH-SY5Y, N2a, C6, and SH-APPs cells were lysed in triton lysis buffer (150 mM sodium chloride, 1.0% Triton X-100, 50 mM Tris-HCl (pH 8.0) and protease inhibitor cocktail (Roche), followed by brief sonication. Protein concentration was measured by Bradford assay (Bio-rad) and 4×sodium dodecyl sulfate (SDS) sample buffer was added to each sample. Cell lysates were resolved by 7.5% Tris–glycine SDS–polyacrylamide gel electrophoresis (PAGE) for detecting USP25 and SP1-HA, respectively. Rabbit anti-USP25 polyclonal antibody, mouse anti-β-actin monoclonal antibody AC-15 (Sigma) and mouse anti-HA monoclonal antibody 12CA5 were used as primary antibodies. Polyclonal rabbit antibody C20 was used to detect C-terminal fragments (CTFs) of APP [18]. Blots were enhanced with a chemiluminescence detection reagent kit (Fisher, 32106) and visualized with a Bio-Rad imager and Quantity One software. Signal intensities from each band were quantified with Bio-Rad Image Lab software, and the bands were analyzed relative to their controls from the same membrane and experiment.
Aβ ELISA assay
SH-APPs cells overexpressing Swedish APP were treated with 75 nM MTM or control vehicle for 24 h. Conditioned medium was collected. Protease inhibitor cocktail and AEBSF were added to prevent Aβ degradation. Human Aβ42 ELISA kit (Invitrogen, KHB3441, Thermo Fisher Scientific-CN) was used to detect Aβ42 according to the instructions.
Statistical analysis
All results were presented as mean±SEM and analyzed by 2-tailed Student’s t-test or one-way ANOVA using GraphPad Prism software. Statistical significance is accepted when p < 0.05 (*p < 0.05).
RESULTS
Cloning the human USP25 gene promoter and mapping the transcription start site of the USP25 gene
Human USP25 gene spans a large area (>150 000 bp) on the chromosome 21q11.2 region. The gene is located at 17 102 344 -17 252 377 on Chromosome 21. Based on the predicted coding sequence of this gene in the Ensembl genome database (Ensembl ID: ENSG00000155313), the human USP25 gene contains 26 exons and has 3 spliced variants (Fig. 1A). We extracted human genomic DNA from HEK293 cells and cloned a 2, 500- bp 5’ flanking region of the USP25 gene. The DNA fragment was sequenced (Fig. 1B). To identify the transcription initiation site of the human USP25 gene, 5’ RLM-RACE was performed. Products of the nested PCRs (Fig. 1C) were sequenced, and the transcription initiation site was identified (Fig. 1D). The transcription initiation site, an adenine labeled as +1, is located 251-bp upstream of the translation start site (Fig. 1E). Sequence analysis revealed that the human USP25 gene promoter has a very high GC content (>70%) in the region upstream of the transcription initiation site. A transcription factor binding site searching engine MatInspector2.2 (Genomatrix, Oakland, CA, USA) revealed that the 5’ flanking region of USP25 contains several putative regulatory elements, indicating that USP25 gene transcription is under tight regulation. Predicted putative transcription factors that may participate in the transcriptional regulation of USP25 gene promoter including NF-AT1, c-Jun, p53 and SP1 (Fig. 1B).
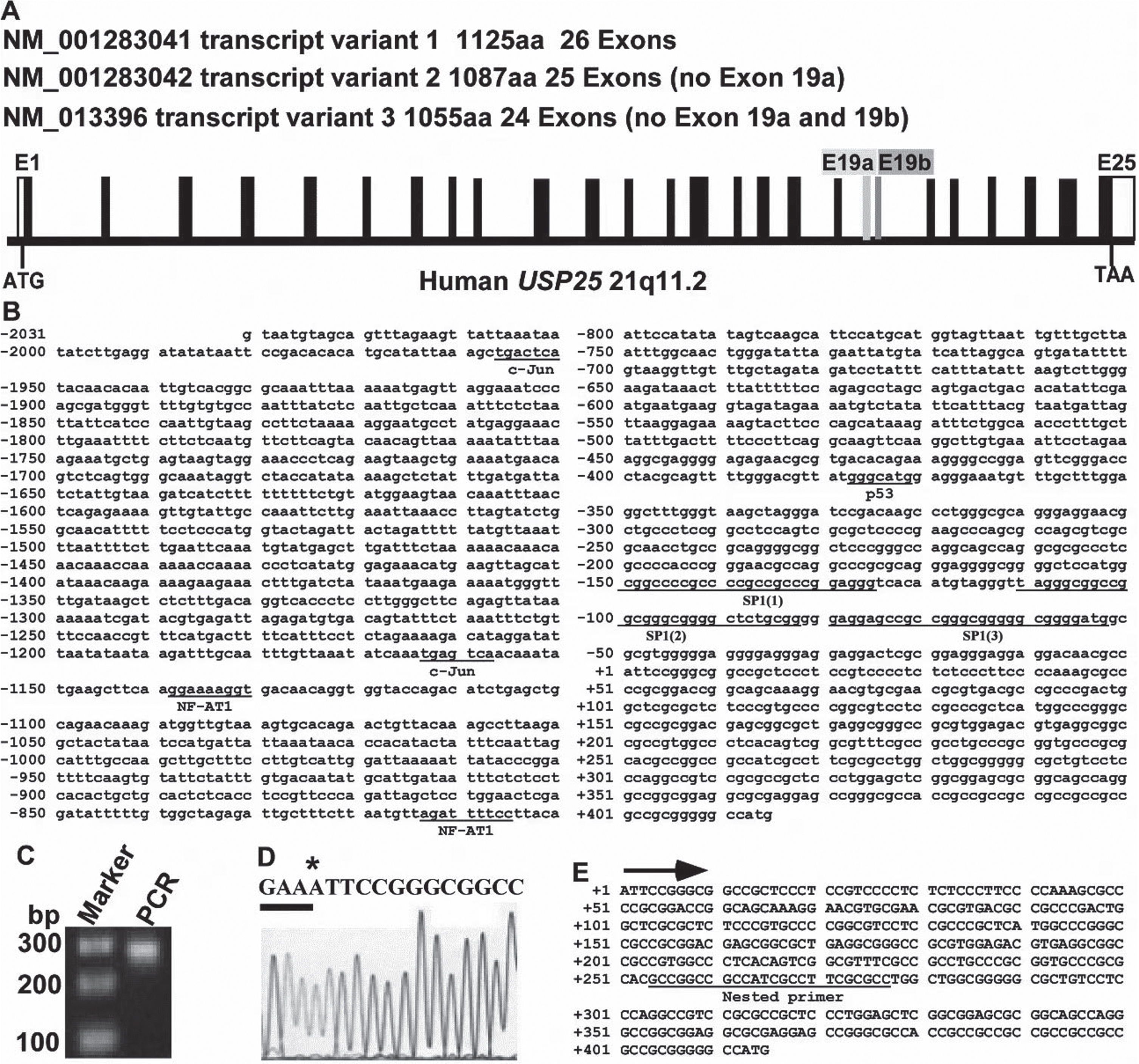
Identify the transcription start site (TSS) of the USP25 gene. A) Based on the predicted coding sequence in the Ensembl genome database (Ensembl ID: ENSG00000155313), the human USP25 gene contains 26 exons and has 3 spliced variants, B) 2.5-kb fragment of the 5’flanking region of the human USP25 gene. The adenine +1 represents the TSS. C) Smarter RACE cDNA amplification kit was used to amplify full-length cDNA from HEK293 cells. Nested PCR was performed, and the product was resolved on 1.5% agarose gel. D) Sequencing results of (B). * represents the TSS. E) Sequence of the 5’UTR region upstream of the UPS25 gene translation start site. Mapped transcription initiation site (+1) is labeled by an arrow.
Functional analysis of the human USP25 gene promoter
A series of nested deletions of the cloned 5’UTR fragment were subcloned to analyze the activity of the USP25 gene promoter. Fragments were cloned into pGL3-Basic for the luciferase reporter assay. The inserts were verified by gel analysis following restriction enzyme digestion and sequencing (Fig. 2A). These fragments with various lengths cover different parts of the cloned 5’UTR region. We transfected these reporter plasmids into HEK293 cells and performed luciferase assays to examine their promoter activities. The pGL3-Basic vector lacks a functional eukaryotic promoter served as a negative control of firefly luciferase activity. However, once a fragment with a functional promoter activity is inserted into its multiple cloning sites, the construct will express firefly luciferase and the level of expression reflects the promoter activity of the inserted fragment. After controlling for transfection efficiency using a co-transfected pRLuc plasmid, the promoter activities of the nine fragments were assayed (Fig. 2B). The construct containing the region from –2031 to +226 (USP25p-A) showed a significant promoter activity, compared with the negative control (pGL3-basic empty vector), indicating that this region contains the functional promoter of human USP25 gene. Further deletions from both 5’ and 3’ (USP25p-C, E and K) did not show significantly reduced promoter activity, indicating that these deletion regions lack important regulatory elements that are required for an optimum promoter function. A region from –122 to +226 has significant promoter activity (3.00±0.18-fold, p < 0.05), while the deletion from –122 to –14 resulted in no promoter activity, suggesting that the fragment from –122 to –14 contains important regulatory elements which are essential for the regulation of promoter activity, and SP1-responsive elements might be the potential elements (Figs. 2 and 3A).

USP25 promoter deletion constructs and functional analysis of USP25 promoter activity. A) Schematic diagram of the USP25 promoter constructs consisting of the 5’ flanking region with serial deletions cloned into the pGL3-basic vector. Arrow shows the direction of transcription. The numbers represent the end points of each construct. USP25 promoter constructs were verified by restriction enzyme digestion and the digested products were resolved on 1.5% agarose gel, which was further confirmed by sequencing. B) The deletion plasmids were cotransfected with pCMV-Luc into HEK293 cells. 24 h after the transfection, the luciferase activity was measured and expressed in relative luciferase units (RLU). The pCMV-Luc was used to normalize for transfection efficiency. The pGL3-Basic plasmid served as the negative control. The values represent means±SEM. n = 3, *p < 0.05, by one-way ANOVA followed by post hoc Tukey’s multiple comparisons test.
The USP25 gene promoter contains SP1 binding sites
By analyzing the DNA sequence, we found three putative SP1-responsive elements with the promoter region. The elements are located at –150 to –125bp (5’cggccccgccccgccgcccggagggt), –111 to –83bp (5’ tagggcggccggcgggcggggctctgcgg), and –80 to –53bp (5’gaggagccgccgggcgggggcggggatg), which are labelled as SP1 (1), SP1 (2), and SP1 (3), respectively (Fig. 3A). These sequences are homologous to the SP1 consensus sequence 5’-(G/T)GGGCGG(G/ A)(G/A)(C/T)-3’ as the GC box element. To examine these putative binding sites, we performed the gel-shift assay (Fig. 3B). The double-stranded oligonucleotides corresponding to the USP25 gene promoter –150 to –125, –111 to –83, and –80 to –53 were synthesized. The probe corresponding to SP1 consensus oligo was labeled with infrared-dye. The shifted bands were shown up (Fig. 3B, lane 2) after the probe incubated with the nuclear extract of HEK293 cells over-expressing SP1. The intensity of shifted band was remarkably reduced to 29.75±11.53% (p < 0.05) when a 10-fold excess competitor (SP1 consensus oligo) was added, and further reduced 7.52±3.92% (p < 0.05) when a 100-fold excess competitor was added (Fig. 3B, lanes 3 and 4). When USP25-SP1 consensus oligos (oligo 1–3) were added, the intensity of shift bands was decreased to 31.51±6.65%,7.57±3.72%, 31.68±3.57%, 7.74±5.26%, 30.02±4.51%, and 8.13±5.60%, respectively, p < 0.05 (Fig. 3, lane 5 to 10). However, mutated SP1 oligo had no significant effect (Fig. 3B, lane 11 and 12). Taken together, our gel-shift assay reveals a physical binding between SP1 and the human USP25 gene promoter in vitro.
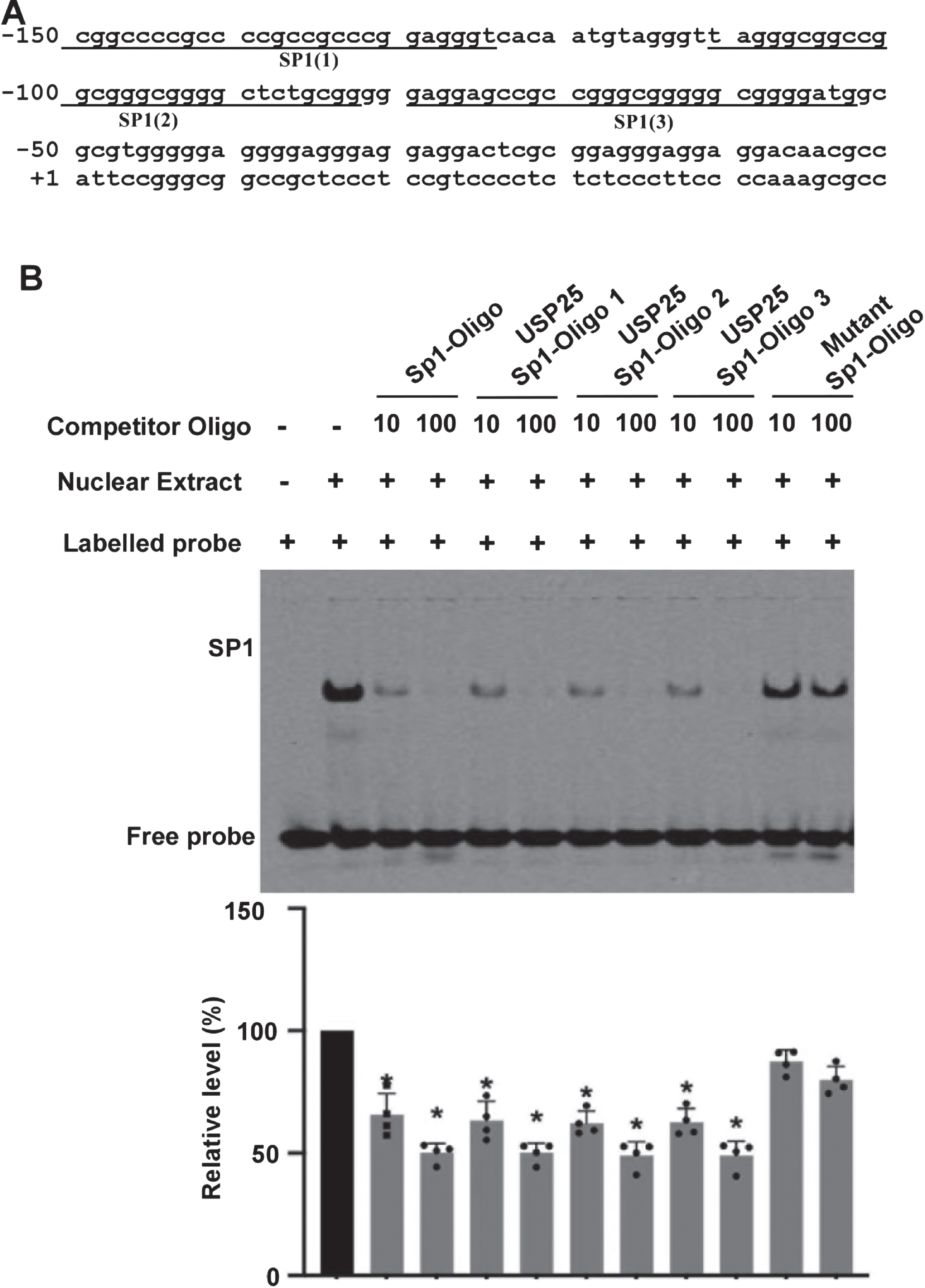
Functional SP1 sites within USP25 promoter was examined. A) –150 to –53 region contain 3 putative SP1-responsive elements. B) Gel shift assays with SP1 consensus probe for examining the interaction between SP1 and the USP25 gene promoter. Lane 1 is labeled probe alone without protein extract. Incubation of the probe with SP1 enriched nuclear extracts forms a shifted DNA-protein complex band (lane 2). Competition assays were performed by further adding different competition oligo-nucleotides. Oligo1, 2 and 3 represent the putative USP25-SP1-responsive elements located within –150 –125bp (SP1 (1)), –111 –83bp (SP1 (2)) and –80 to –53bp (SP1 (3)), respectively. Mutant SP1-oligo represents mutated oligo 2. The values represent means±SEM. n = 4, *p < 0.05, by one-way ANOVA.
SP1 regulates USP25 promoter activity
To examine whether the USP25 gene promoter activity could be regulated by SP1, the USP25-pA, C, E, or I plasmid was co-transfected with either the SP1 expression vector or the corresponding empty vector into HEK293 and SH-SY5Y. Mithramycin A (MTM), a SP1 inhibitor, was added 24 h after transfection. In HEK293 cells, the promoter activity was elevated by overexpression SP1 and inhibited by the treatment of 75 nM MTM. Compared with the control, SP1 significantly increases the promoter activity of USP25-pA, C, E, and I, 103.37 versus 70.03, 159.07 versus 112.02, 214.16 versus 156.51, and 96.52 versus 48.07, respectively, p < 0.05. Compared with the control, MTM reduces the promoter activity, 45.54 versus 70.03, 78.17 versus 112.02, 77.84 versus 156.51, and 37.42 versus 48.07, respectively, p < 0.05 (Fig. 4A). In SH-SY5Y cells, the promoter activity was also elevated by overexpressing SP1 and inhibited by the treatment of 75 nM MTM. Compared with the control, SP1 significantly increases USP25-pA, C, E, or I promoter activity, 102.33 versus 66.30, 160.05 versus 113.69, 231.86 versus 162.23, and 96.61 versus 59.41, respectively, p < 0.05, while MTM reduces the promoter activity, 45.36 versus 66.30, 76.11 versus 113.69, 83.76 versus 162.23, and 39.84 versus 59.41, respectively, p < 0.05 (Fig. 4B).
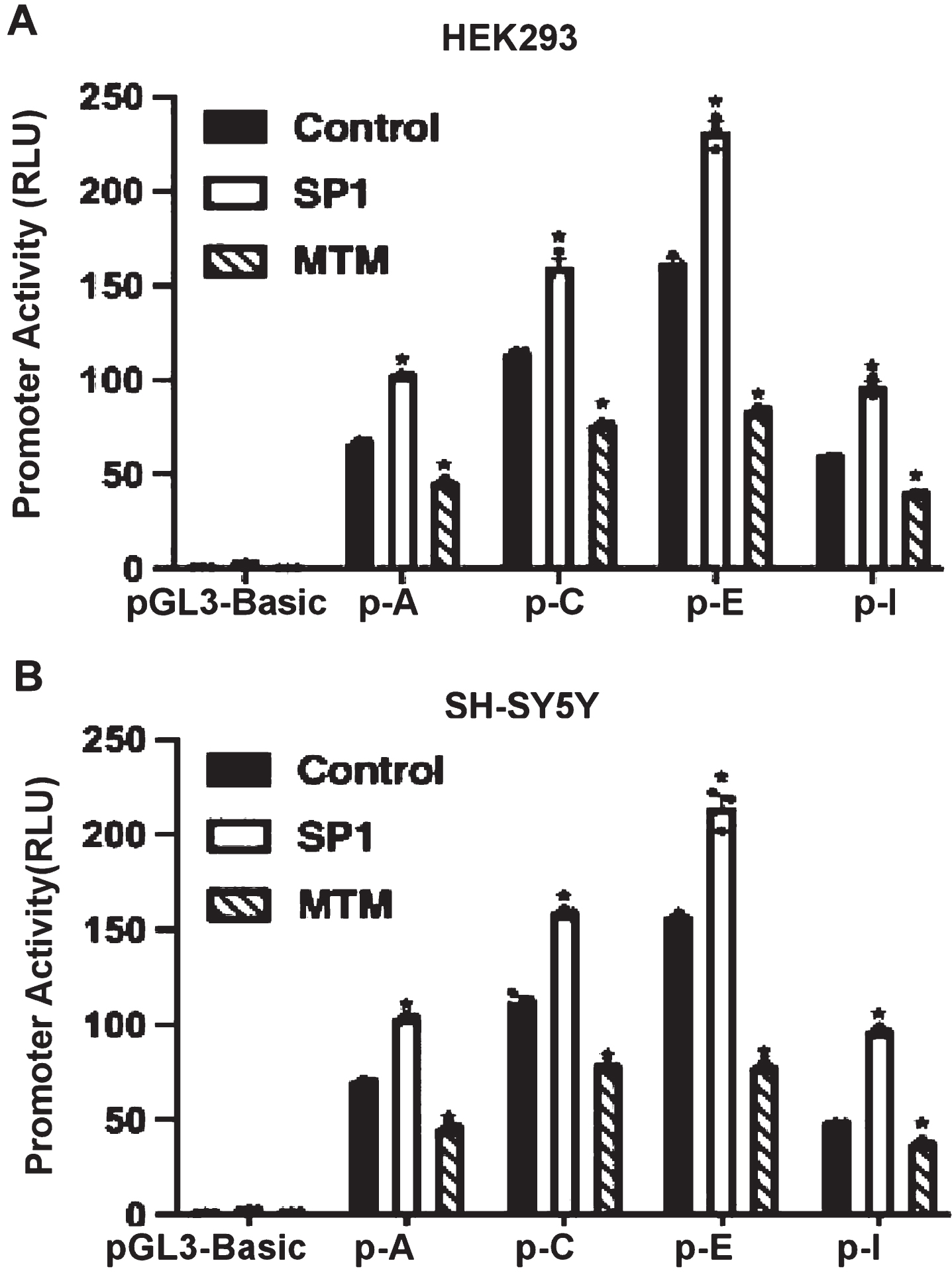
SP1 regulates USP25 promoter activity. To examine whether the USP25 gene promoter activity is regulated by SP1, the USP25p-A, C, E, or I plasmid was co-transfected with either the SP1 expression vector or the corresponding empty vector into (A) HEK293 and (B) SH-SY5Y. MTM (SP1 inhibitor) was added 24 h after transfection. Luciferase assay was performed. The pGL3-Basic plasmid served as the negative control. The values represent means±SEM. n = 3, *p < 0.05, by one-way ANOVA.
SP1 regulates USP25 gene expression in non-neuronal cells
It is known that the physiological functions of neuronal cells and non-neuronal cells are different, and the gene expression regulation pattern might be also different. To evaluate that the regulation of USP25 by SP1 is a neuronal specific effect or a universal effect in cells, we examined the regulation effect in various cell types. To examine the effect of SP1 on USP25 expression in non-neuronal cells, HEK293 and C6 cells were used. The semi-quantitative RT-PCR and western blot were performed to examine whether endogenous USP25 mRNA and protein was affected by SP1 overexpression or SP1 inhibitor. SP1 overexpression increased endogenous human USP25 mRNA to 1.19±0.20 folds, p < 0.05 (Fig. 5A), and increased USP25 protein level to 1.61±0.14 folds in HEK293 cells, p < 0.05 (Fig. 5B). MTM significantly decreased endogenous human USP25 mRNA to 80.45±3.98%, p < 0.05 (Fig. 5C), and decreased USP25 protein level to 49.91±1.76% in HEK293 cell, p < 0.05 (Fig. 5D). In C6 cells, SP1 increased USP25 mRNA and protein to 1.44±0.27 folds and 1.31±0.54 folds, respectively, p < 0.05 (Fig. 5E, F). MTM significantly decreased endogenous USP25 mRNA and protein to 69.11±7.41% and 49.59±4.16%, respectively, p < 0.05 (Fig. 5G, H). It indicated that SP1 regulates USP25 expression in both human and rat non-neuronal cells.
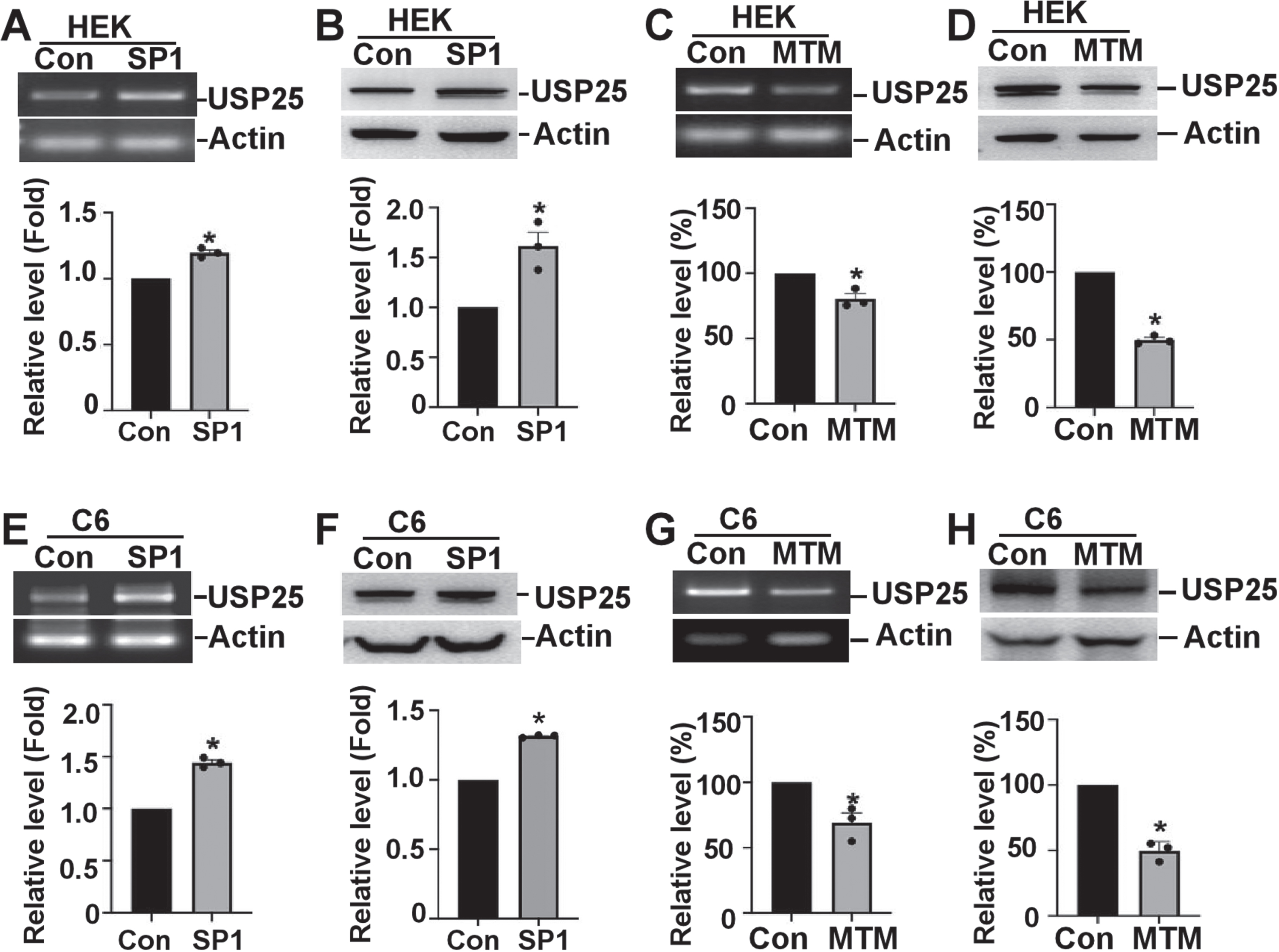
SP1 regulates USP25 mRNA and protein expression in non-neuronal cells. SP1 overexpression increases the endogenous human USP25 (A) mRNA and (B) protein in HEK293 cells. MTM (SP1 inhibitor) decreases the endogenous human USP25 (C) mRNA and (D) protein in HEK293 cells. SP1 overexpression increases the endogenous human USP25 (E) mRNA and (F) protein in C6 cells. MTM (SP1 inhibitor) decreases the endogenous human USP25 (G) mRNA and (H) protein in C6 cells. Quantification was completed by ImageJ software. The values represent means±SEM. n = 3, *p < 0.05, by Student’s t-test.
SP1 regulates USP25 gene expression in neuronal cells
To investigate the effect of SP1 on USP25 expression in neuronal cells. SH-SY5Y and N2a cells was used. The semi-quantitative RT-PCR and western blot were performed to examine whether endogenous USP25 mRNA and protein was affected by SP1 overexpression or SP1 inhibitor. SP1 overexpression increased endogenous human USP25 mRNA by 1.44±0.27 folds, p < 0.05 (Fig. 6A), and increased USP25 protein level by 1.55±0.86 folds in SH-SY5Y cells, p < 0.05 (Fig. 6B). MTM significantly decreased endogenous human USP25 mRNA and protein to 56.17±1.91% and 29.63±7.31% in SH-SY5Y cells, respectively, p < 0.05 (Fig. 6C, D). In N2a cells, SP1 increased USP25 mRNA and protein to 1.25±0.65 folds and 1.37±0.67 folds, respectively, p < 0.05 (Fig. 6E, F). MTM significantly decreased endogenous USP25 mRNA and protein to 76.72±0.82% and 71.06±3.18%, respectively, p < 0.05 (Fig. 6G, H). It indicated that SP1 regulates USP25 expression in both human and mouse neuronal cells.

SP1 upregulates USP25 endogenous mRNA and protein expression in neuronal cells. SP1 overexpression increases the endogenous human USP25 (A) mRNA and (B) protein in SH-SY5Y cells. MTM (SP1 inhibitor) decreases the endogenous human USP25 (C) mRNA and (D) protein in SH-SY5Y cells. SP1 overexpression increases the endogenous human USP25 (E) mRNA and (F) protein in N2a cells. MTM (SP1 inhibitor) decreases the endogenous human USP25 (G) mRNA and (H) protein in N2a cells. The values represent means±SEM. n = 3, *p < 0.05, by Student’s t-test.
SP1 inhibition reduces USP25 expression and Aβ levels
To investigate the effect of SP1 inhibition on USP25 expression and AβPP processing in DS or AD, MTM was applied on SH-APPs cells, SH-SY5Y cells overexpressing human Swedish APP. The C-terminal fragment with 83 amino acids (C83) was the major products of AβPP processing in SH-APPs cells. USP25 protein was increased to 1.59±0.12 folds in SH-APPs cells compared with that in SH-SY5Y cells, p < 0.05 (Fig. 7A). MTM significantly decreased endogenous human USP25 protein to 43.00±4.99% in SH-APPs cells, p < 0.05 (Fig. 7B). Moreover, MTM significantly decreased Aβ42 to 39.80±1.14%, p < 0.05 (Fig. 7C). It indicated that SP1 inhibition reduces USP25 expression and Aβ levels.

SP1 inhibition reduces USP25 expression and Aβ levels. A) USP25 is increased in SH-APPs cells. C83 is the major C-terminal product of AβPP. B) MTM decreases the endogenous human USP25 protein expression in SH-APPs cells. C) SH-APPs cells were treated with 75 nM MTM or control for 24 h. The conditioned medium was collected. ELISA was performed to detect Aβ. Aβ levels are expressed as a percentage of the control group. The value represents the mean±SEM. n = 3, by Student’s t test, *p < 0.05.
DISCUSSION
Our study clearly indicates that the promoter region of the USP25 gene contains a functional SP1 responsive element that regulates USP25 gene expression control at the transcriptional level. SP1-medicated USP25 transcriptional regulation was determined in both neuronal cells and non-neuronal cells of human and rodents.Understanding the interaction between the USP25 gene promoter and SP1 is informative for the study of the regulation of HSA21 genes and the pathogenesis of DS and AD. SP1 is a ubiquitously expressed zinc finger-containing DNA binding protein that binds GC-rich motifs with high affinity and enhances transcription with one of the two glutamine-rich domains [19]. Mithramycin A was discovered to bind to GC-rich sequence with high affinity and it competitively binds to SP1 consensus binding site working as a site-specific inhibitor for SP1 [20]. We identified three SP1 binding sites within USP25 promoter region. Application of MTM resulted in significantly reduced USP25 promoter activity, gene transcription and protein expression.
Increased Aβ generation plays a key role in the pathogenesis of AD. Our data showed that the level of USP25 in SH-APPs cells was higher than that in SH-SY5Y cells. MTM consistently reduced USP25 protein expression in SH-APPs cells, which was associated with Aβ42 reduction. It suggests that SP1 inhibition-induced Aβ42 reduction may mediated by decreased USP25 expression although multiple SP1-associated pathway may also be involved in. The proteolysis of AβPP at the β site is essential to produce Aβ, while BACE1 is a major β-secretory enzyme involved in the cleavage of AβPP at β site [21]. Previous studies showed that SP1 is implicated in transcriptional regulation of both AβPP and BACE1 [21–23]. The current study showed that SP1 is also involved in transcriptional regulation of USP25, while USP25 plays an important role in AβPP and BACE1 degradation [16]. It indicated that SP1 may directly regulate AβPP and BACE1 transcription, and indirectly regulate AβPP and BACE1 degradation via USP25, acting in concert to the regulation of AβPP and BACE1 expression. Thus, SP1 regulates Aβ generation via multiple pathways. However, it remains unclear which pathway is the predominant one for regulating Aβ generation. Further investigation is necessary to elucidate this issue.
SP1 is also implicated in the pathogenesis of other neurodegenerative diseases. SP1 also enhanced the gene transcription of huntingtin and MTM reduced huntingtin gene expression, suggesting that the dysregulation of SP1-mediated huntingtin transcription may partially contribute to the pathogenesis of Huntington’s disease [24]. Mutations in Leucine-rich repeat kinase 2 (LRRK2) gene are associated with familial Parkinson’s disease that contributes to the dopaminergic neurodegeneration. SP1 signaling regulates human LRRK2 gene expression and controlling LRRK2 level by manipulating SP1 signaling may be beneficial to attenuate Parkinson’s disease-related neuropathology [25].
In addition, SP1 is increased in many cancers, including breast, stomach, pancreatic, lung, brain (glioma), and thyroid cancers, which correlates with stage, invasion potential, and metastasis. SP1 levels are associated with survival in almost all cancer patients [26, 27]. Mithramycin A (MTM), an inhibitor of SP1, inhibits the growth of various cancers by reducing Sp1 protein expression. MTM is mainly used in the treatment of testicular embryonic carcinoma and hypercalcemia caused by various malignant tumors, and can also be used in glioma, lymphoma, and myelogenous leukemia [28, 29]. Disruption of the proteasome system by altering USP25 activity is associated with various human cancers including breast cancer, hepatocellular carcinoma, gastric adenocarcinoma, and non-small cell lung cancer [30–32]. Knockdown or knockout USP25 expression significantly reduces characteristics of cancers (proliferation, migration, invasion etc.) shown by in vivo or in vitro studies [33–36]. Our study highly indicates that targeting USP25 may be a mechanism of SP1 inhibitor for cancer treatment although further investigation is needed. The main side effects of MTM are gastrointestinal reaction, bleeding, liver and kidney damage, including nausea and vomiting, diarrhea, stomatitis, rash, and fever. In some patients, central nervous system symptoms, including restlessness, fatigue, lethargy, and headache, may be produced. USP25 plays an important role in ubiquitin-proteasome degradation system, while homeostasis of ubiquitin-proteasome degradation is essential in maintaining physiological function of human body [30, 37]. Thus, its inhibition may cause a serious toxic effect. Our study highly suggests that USP25 reduction may contribute to SP1 inhibitor-associated side effects. However, other mechanisms may also be implicated in SP1 inhibitor-associated side effects as SP1 is involved in the regulation of a variety of genes such as insulin-like growth factor 1 receptor and damage response factor ataxic telangiectasia mutation [38].
In this study, we demonstrated that SP1 signaling is involved in regulating USP25 gene expression, while both SP1 and USP25 regulation is implicated in the pathogenesis and treatment of neurodegenerative diseases and cancer [29, 40]. More studies will be needed to investigate the effect of manipulating SP1 and USP25 in the treatment of neurodegenerative diseases and cancer, and underlying mechanisms. Moreover, the side effects of SP1 and USP25 inhibition should be investigated. Furthermore, the balance between therapeutic effects and side effects of regulating SP1 and USP25 should be precisely assessed, which will be beneficial for the clinical application of SP1 inhibitors or specific USP25 inhibitors.
Footnotes
ACKNOWLEDGMENTS
A portion of this work was done as part of a thesis/dissertation of Beibei Song, the author of this work.
FUNDING
W.S. was the Canada Research Chair in Alzheimer’s Disease. This work was supported by National Natural Science Foundation of China (NSFC) Grant 82150710557 and 82293642 to W. S, and Grant 81971019 to Y.W.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding authors.