
Abstract
Viable Alzheimer’s disease (AD) hypotheses must account for its age-dependence; commonality; association with amyloid precursor protein, tau, and apolipoprotein E biology; connection with vascular, inflammation, and insulin signaling changes; and systemic features. Mitochondria and parameters influenced by mitochondria could link these diverse characteristics. Mitochondrial biology can initiate changes in pathways tied to AD and mediate the dysfunction that produces the clinical phenotype. For these reasons, conceptualizing a mitochondrial cascade hypothesis is a straightforward process and data accumulating over decades argue the validity of its principles. Alternative AD hypotheses may yet account for its mitochondria-related phenomena, but absent this happening a primary mitochondrial cascade hypothesis will continue to evolve and attract interest.
INTRODUCTION
Attempts to conceptualize Alzheimer’s disease (AD) usually emphasize a single component or temporally order the sequence in which components become evident [1, 2]. Some extrapolate findings from distinct but genetically definable disorders to heterogeneous common syndromes to build a narrative [3]. Narrowing the working definition to one or two phenomena, for example to changes in amyloid-β (Aβ) or tau protein, is also popular [4]. These reductionist approaches leverage focused observations to build theoretical frameworks, which is logical but could inadvertently warp a complicated reality. Collectively considered, these efforts evoke the parable of the blind men feeling an elephant.
A different reductionist approach is to use broader but still well-established empirical evidence to define a simplified outline. Rather than adapting the overall story to fit a specific observation, next steps in this case include linking discrete observations to fit an overall story. One example flows as follows: 1) we age, 2) we compensate to age-related changes to the extent that we can, and 3) upon reaching a point that we can no longer compensate, we decompensate [5]. Critics argue this blurs the line between disease and aging, minimizes rather than emphasizes histologic hallmarks, and in general substitutes chaos for focus. Proponents counter the line between aging and age-related disease is more arbitrary than absolute, we do not fully understand the role of AD’s histologic hallmarks, and extending lessons from aging research to AD provides perspective that reduces chaos.
Mitochondria likely play a role in aging [6 –8], and for related reasons investigators previously proposed mitochondria may also contribute to AD [9]. The “mitochondrial cascade hypothesis,” or at least a version initially summarized in 2004, attempts to tie myriad AD phenomena together within an aging-compatible context [10 –15]. This review discusses the status of the hypothesis while incorporating recent pertinent data.
CORE PRINCIPLES
The hypothesis presumes genes determine an individual’s baseline mitochondrial function and resilience to age-related changes. Behavioral and environmental factors should also affect resilience. Baseline mitochondrial function and resilience determine how far from the proverbial AD cliff one starts life, and how rapidly one approaches that cliff.
Age-related changes in mitochondrial function presumably trigger a broader set of molecular and physiologic responses that preserve homeostasis [16, 17]. Eventually, changes progress to a point that successful compensation is no longer possible, and the individual transitions from a state of maintained homeostasis, or healthy aging, to abandoned homeostasis, or pathological aging.
To say that mitochondrial dysfunction initiates or drives AD is technically accurate but overstates the relevance of frank mitochondrial lesions. Mitochondria function within a physiologic range that varies with age. Thresholds for triggering AD-associated changes including Aβ neuritic plaques, tau neurofibrillary tangles, and other protein aggregations may reside within that range. Also, while it is reasonable to envision compensation giving way to decompensation, probably most biological responses triggered by a primary change are in some sense compensatory. Some happen to preserve integrity, while others dismantle it. Extended to and framed from a clinical perspective, we are either aging in a desired way that preserves overall function, or we are not and our mitochondria influence this.
Figure 1 summarizes mitochondrial cascade hypothesis chronology and core principles. Note that AD is remarkably common, and for a majority how long one lives determines whether they die with or without it. The process begins for all individuals at conception, with genes from both parents determining baseline mitochondrial biology and resilience. As we age our mitochondrial function changes, which begins to affect other cell processes. Initially we compensate for and tolerate age-associated changes in mitochondrial function and biology and compensate for and tolerate downstream changes to cell biology. Given enough time, though, most can reach a point where compensation no longer maintains brain function or integrity at a level that permits an adequately preserved level of cognitive function.

Mitochondrial cascade hypothesis chronology and core principles. Solid lines indicate necessary relationships while dashed lines indicate a component or components within a box may or may not confer a reciprocal impact.
HYPOTHESIS ORIGINS
Reports that brains from AD patients contain structurally and functionally altered mitochondria span decades [18 –20]. Investigators initially could not address questions of cause or consequence, but most assumed these changes represented artifacts of other pathologies or of neurodegeneration itself.
Eventually, though, it was shown that mitochondria from AD and control subject non-brain tissues differed [18 , 22]. This suggested AD-associated mitochondrial features were not a sole consequence of neurodegeneration and probably not exclusively driven by brain histology. It also stressed that at a biochemical level, AD was not a brain-limited disorder.
Accordingly, some proposed mitochondria in AD could contribute to a metabolism systems failure that represented a cause rather than consequence of the disease [23, 24]. This intersected with speculation that bioenergetic metabolism or related factors might drive aging [25 –27]. Such perspective evolved into a mitochondrial theory of aging, in which an age-dependent accumulation of somatic mitochondrial DNA (mtDNA) mutations possibly drove aging phenotypes [27 –30], and perhaps contributed to AD [31]. Table 1 lists some reported age-related changes to mitochondrial function and integrity, with supporting references in [9].
Reported age-related changes to mitochondrial function and integrity
In 1989, Parker proposed a role for mtDNA inheritance in neurologic disorders with sporadic epidemiology [32, 33]. This included common, late-onset neurodegenerative diseases such as AD. He based this hypothesis on the fact that features inherent to mitochondrial genetics, including maternal inheritance, heteroplasmy, threshold, and mitotic segregation created a situation in which mtDNA could still influence the risk of non-Mendelian diseases.
The Parker group measured respiratory chain function in AD patients. They used platelet mitochondria as these were easier to obtain than brain mitochondria and avoided tissue integrity confounds. They reported complex IV (cytochrome oxidase; COX) activity was reduced in AD patient platelet mitochondria [34], and several groups subsequently extended this observation to brain [35 –38].
The COX holoenzyme contains ten nuclear- and three mtDNA-encoded subunits. To test whether mtDNA contributed to lower AD COX activity, investigators leveraged what was then a recently described variation of the cytoplasmic hybrid (cybrid) technique [39 –41]. To generate cybrid cell lines investigators removed all endogenous mtDNA, also called ρ DNA, to create ρ0 cells. They next isolated platelets from AD or control subject blood samples, as platelets contain mitochondria and mtDNA but lack nuclei and nuclear DNA. Mixing ρ0 cells with platelets in the presence of a membrane-permeabilizing detergent permits the mixing of ρ0 cell and platelet cytosols, leading to the formation of cybrid cells that contain the ρ0 cell nucleus and platelet mtDNA. Over repeated cell division cycles all transferred but non-perpetuating platelet components degrade and dilute, but because the transferred mtDNA replicates the resulting line’s respiratory chain comes to contain subunits encoded by the ρ0 cell nucleus and platelet donor’s mtDNA. As cybrid lines will eventually vary only in the origin of their mtDNA, respiratory chain functional differences should solely reflect differences in mtDNA sequence.
Relative to cybrid cell lines containing mtDNA from control subjects, mean COX activity was lower in lines containing AD subject mtDNA [42]. This indicated mtDNA contributed to the AD COX deficit. Subsequent studies replicated this result [43 –46]. AD-to-control cybrid comparisons demonstrated additional differences including increased oxidative stress, decreased mitochondrial membrane potential, diminished mitochondrial calcium uptake, decreased mitochondrial size, increased cristae disruption, apoptosis pathway activation, decreased ATP, decreased respiratory capacity, and decreased glycolysis flux in the AD lines [9 , 42–65]. In several instances findings recapitulated those already or subsequently shown through direct comparisons of AD and control subject brain mitochondria [66 –69].
Blood cell mtDNA accumulates less somatic mutation than brain mtDNA [70]. So, although the cybrid data did not identify specific variants inherited rather than acquired deviations likely account for the biochemical differences. This does not imply somatic mtDNA mutation does not occur in AD. Multiple studies report AD brain mtDNA accumulates excessive age-related, somatic mtDNA mutations [19 , 71–79]. A negative correlation also exists between the number of mtDNA COX gene heteroplasmic mutations and COX enzyme activity [80].
Two studies reported cybrids expressing AD subject mtDNA accumulated more Aβ than those expressing control subject mtDNA [51, 54]. In addition to recapitulating multiple mitochondrial or mitochondrial-related phenomena observed in AD brain, mtDNA transfer from AD subjects alters amyloid-β protein precursor (AβPP) or Aβ homeostasis and influences the biology of a classic AD hallmark.
By 2004, experimental and observational data existed to support the core assumptions of a primary AD mitochondrial cascade hypothesis [12]. Evidence indicated genes, including mitochondrial genes, determined key aspects of an individual’s mitochondrial function, that an individual’s mitochondrial function would over time predictably decline from its baseline, and that mitochondrial function could influence AD-associated pathologies (Fig. 1).
BURDEN OF PROOF
Genetic studies increasingly inform our view of AD. A mitochondrial cascade hypothesis will need to account for findings from these studies. Unbiased genetic screens should implicate mitochondria or suggest a mitochondrial impact. Relevant DNA variants should reside within nuclear and mtDNA genes.
A mitochondrial cascade hypothesis should inform our understanding of classic AD histology. While it is compatible with the possibility that misfolded, oligomerized, or aggregated proteins interfere with or damage mitochondria [81, 82], mitochondria must necessarily alter the biology or homeostasis of these proteins. It should also help explain AD-associated systemic, vascular, inflammation, and insulin signaling changes.
The impact of therapeutic interventions that alter mitochondria or mitochondria-relevant parameters will further inform hypothesis viability. If the mitochondrial cascade hypothesis is correct, such interventions should arrest or reverse AD patient clinical decline to a greater extent than interventions developed through the consideration of alternative hypotheses.
GENETIC EVIDENCE
Edland et al. found AD subjects with a demented parent more often reported an affected mother [83]. Due to better longevity and likely other reasons women carry a greater lifetime AD risk than men [84], which could confound such studies. Maternal inheritance bias in this report, though, was apparent among relatively young probands, which suggests female longevity was not entirely responsible.
Endophenotypes are biomarker-defined states that exist in the absence of a full clinical syndrome. Multiple studies feature middle-aged adults with an AD mother, father, or unaffected parents. Endpoints include fluorodeoxyglucose positron emission tomography (FDG PET)-defined brain glucose utilization [85], amyloid PET-defined Aβ plaque deposition [86], magnetic resonance imaging (MRI) volumetric measurements [87 –91], MRI perfusion-defined blood flow [92], cerebrospinal fluid (CSF) Aβ and oxidative marker levels [93], and cognitive test performance [94]. In each case, children of AD mothers preferentially show AD-like shifts. Another endophenotype study measured platelet mitochondria COX activity in middle-aged, cognitively normal individuals with an AD mother, father, or cognitively intact parents. COX activity was lowest in those with an AD mother [95], a finding consistent with the possibility that mtDNA inheritance influences COX and by extension AD risk.
Mutations in the AβPP, presenilin 1 (PSEN1), and presenilin 2 (PSEN2) genes cause early onset, autosomal dominant familial AD (FAD) [96]. The AβPP protein localizes to mitochondria and can interfere with mitochondrial function [97 –99]. The extent to which presenilin proteins localize to mitochondria proper is debated, but there is consensus they localize at least to mitochondria-associated endoplasmic reticulum membranes and affect mitochondrial function [100, 101].
A 1991 linkage analysis pinpointed the 19q13.32 locus [102], and subsequently associated APOE gene variation with AD risk [103, 104]. APOE encodes apolipoprotein E, a protein whose classically recognized function includes cholesterol transport outside and within the brain [105, 106]. APOE 2, 3, and 4 isoforms exist and APOE4 increases AD risk. The APOE4 product exhibits a unique folding pattern and undergoes a distinct processing event that activates a mitochondrial targeting sequence [107, 108]. This derivative peptide accesses mitochondria and inhibits COX. Brains from young, cognitively normal APOE4 carriers show lower COX activity than brains from non-APOE4 carriers [109]. AD subjects with an APOE4 allele have lower platelet mitochondria COX activity than those without an APOE4 allele [110].
The TOMM40 gene encodes the translocator of the outer mitochondrial membrane 40 kD (Tomm40) protein and sits adjacent to the APOE gene. TOMM40 and APOE variants show linkage disequilibrium [111, 112]. Due to this confounding, it is difficult to know whether TOMM40 contributes independently to AD risk although multiple studies suggest it may [113 –119].
Genome-wide association studies (GWAS) and whole exome sequencing efforts report over 30 loci associate with altered AD risk [120]. The unbiased nature of locus identification represents a true strength of these approaches. Similar to classic linkage studies, though, selecting the critical gene from within a locus could prove challenging and interject bias [121].
Grouping implicated genes by functional properties informs our understanding of AD biology. Some of these genes, such as CLU and PTCD1, encode proteins that localize to mitochondria [122, 123]. Others, such as TREM2 and BIN1, may indirectly affect mitochondrial function [124, 125]. Pertinent modules identified through GWAS include lipid metabolism [120], which intersects with mitochondrial function.
The extent to which specific mtDNA variants influence AD risk remains unsettled for several reasons. Common mtDNA variants tend to segregate in combination, with certain patterns defining “haplogroups” [126]. Haplogroups could mix variants that increase AD risk with ones that decrease risk and consequently minimize valid associations. Within haplogroups, additional variants can variably present to define sub-haplogroup haplotypes. This could in turn obscure genuine haplogroup-level associations. Many rare mtDNA variants exist that are difficult to impute and whose detection requires fully ascertained mtDNA sequences. As few relevant datasets currently report full mtDNA sequences [127], there is inadequate power to confirm or refute their impact. The risk conferred by specific mtDNA variants may also depend on the nuclear DNA background they play out against [128].
mtDNA haplogroup association studies report positive but non-uniform results [129]. One recent study that combined data from a single academic site’s clinical cohort of AD and cognitively normal participants with participants from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) found haplogroup J conferred an increased risk, with an odds ratio of 1.87 [130]. That study also reported haplogroup K conferred a decreased risk, with an odds ratio of 0.49. The data suggested but did not conclusively establish an APOE-mtDNA haplogroup interaction, as those with an APOE4-haplogroup J genotype trended towards an accentuated AD risk, a relationship observed in a previous study [131]. Haplogroup K trended towards greater risk mitigation in APOE4 carriers, a relationship reported in two prior studies [131, 132].
Haplogroups J and T arise from a common phylogenetic branch and differ through a limited number of variants. In the Swerdlow et al. study haplogroup T did not increase AD risk [130], which suggests one or several haplogroup J-defining variants truly does increase AD risk. The defining haplogroup J variants include a synonymous substitution, two non-synonymous substitutions in complex I-encoding subunits, and three variants that change mtDNA control region nucleotides. The mtDNA control region mediates mtDNA replication and transcription and influences mtDNA copy number [133]. COX activity appears particularly sensitive to mtDNA copy number [65 , 135], and in one cybrid study mtDNA copy number was lower in AD than control cybrid lines [65]. This raises the possibility that inherited mtDNA control region variants may influence AD risk by affecting mtDNA copy number. Interestingly, several studies report AD brains contain less intact mtDNA than control brains [72 , 137]. Prior studies also report elevated levels of heteroplasmic control region variants that affect mtDNA replication in AD patient brains and lymphocytes [72, 138].
HISTOLOGY EVIDENCE
Experimental data show Aβ can disrupt mitochondrial function [81 , 139–141]. Such findings help fuel an emerging interest in the organelle’s AD contribution, because if Aβ initiates AD then mitochondria could potentially mediate Aβ toxicity [142]. This scenario is consistent with the possibility of a secondary mitochondrial cascade [14].
Other data indicate mitochondrial function alters Aβ homeostasis by altering AβPP processing [143 –145]. Figure 2 illustrates additional ways through which mitochondria could influence AβPP and Aβ. Revving mitochondrial turnover by enhancing mitophagy reduces Aβ in AβPP/PSEN1 transgenic mice [146] while interrupting mitophagy enhances Aβ accumulation [147]. Transgenic mouse studies demonstrate mitochondrial integrity, inherited mtDNA variants, and acquired mtDNA somatic mutations influence Aβ accumulation [148 –152]. Mitochondrial-synthesized ATP affects general intracellular amyloid solubility, and mitochondria influence chaperones that impact protein folding and aggregation [153, 154]. These studies emphasize the mitochondria’s contribution to AD could lie upstream of Aβ’s contribution.
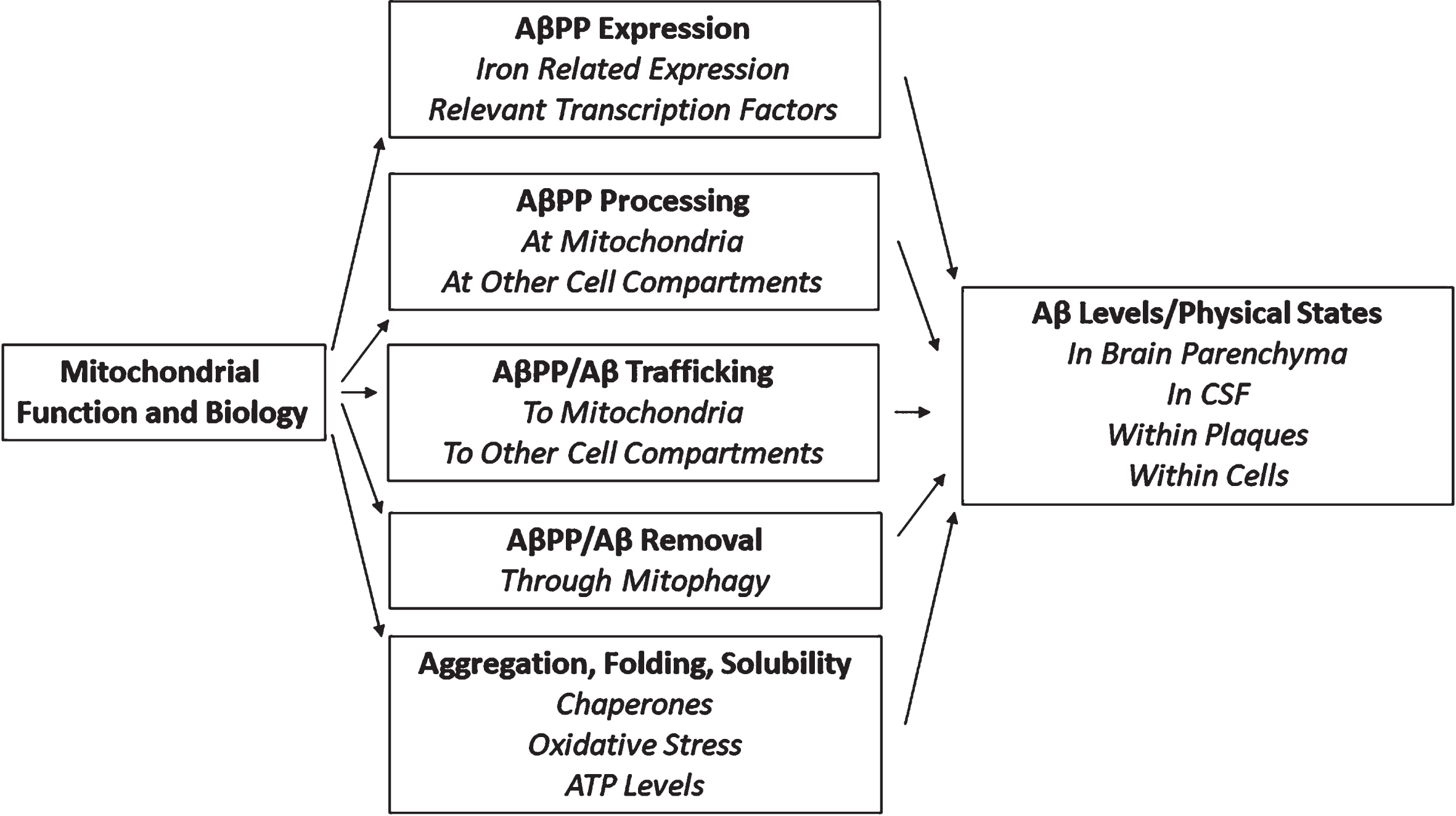
Impact of mitochondria on AβPP and Aβ.
Metabolism in general, and energy metabolism specifically, clearly influences AβPP biology and amyloidosis. Transgenic mouse studies in which increasing synaptic activity increases plaque deposition or Aβ demonstrate this [155 –159], and in humans with traumatic brain injury emergence from coma associates with increasing Aβ [160].
Mitochondria contribute to proteostasis and protein quality control [161, 162]. Depressed mitochondrial function increases a cell’s general propensity for protein aggregation, and could potentially explain why multiple proteins, including tau, TDP43, and α-synuclein aggregate in AD.
A recent study revealed mtDNA depletion increased the amount of tau protein a cell contained, increased tau oligomerization, and shifted monomer tau to oligomer tau [65]. AD cybrid cell lines, which showed reduced mtDNA copy numbers, demonstrated a similar increase in tau oligomers and a tau monomer to oligomer shift [65]. Earlier studies also report toxin-induced respiratory chain impairment induces tau phosphorylation and aggregation [163 –168].
PROTEOMIC AND METABOLOMIC DATA
A recent AD proteomic study revealed prominent changes, characterized by an overall decline, in a defined mitochondrial protein network [169]. In the brains and CSF assessed, a sugar metabolism protein network module, summarized as overall protein increases in astrocytes and microglia, associated especially strongly with neuropathology and clinical measures.
A prior metabolomic study from this group reported AD brains exhibit evidence of decreased AD glycolysis flux [170], a finding consistent with an earlier study that directly showed decreased AD brain glycolysis flux [171]. Joint consideration of the proteomic data that showed an increase in sugar metabolism proteins and metabolomic data that showed overall decreased glucose flux, in the setting of decreased mitochondrial proteins, suggest an initiated but failed attempt at bioenergetic compensation.
Relevant to this point, cultured cells will compensate for decreased respiration by upregulating glycolysis [172]. This relationship, though, breaks down in AD cybrid cell lines [44]. A shift in AD cybrids towards a more reduced redox state may account for this. Clearly, links exist between mitochondrial function and sugar metabolism. These links provide context to the proteomic study.
AGING-AD HEURISTIC CONSIDERATIONS
AD prevalence increases so much with advancing age that in some demographics, such as very elderly women, the number of those with AD may exceed the number of those without AD [5, 173]. Many non-demented elderly individuals with low CSF Aβ or elevated Aβ plaque burdens meet criteria for a newly defined AD “pathological change” category [4]. AD pathological change can precede cognitive decline by decades and cognitive decline itself evolves over an extended period [174, 175]. The commonality of this disease and its insidious nature argue substantial mechanistic overlap exists between AD and fundamental aging biology. Given mitochondria’s suspected role in aging [6], an AD mitochondrial cascade hypothesis specifically addresses these points.
LINKS TO OTHER FEATURES
Mitochondrial and inflammation biology intertwine [176, 177]. AD cybrid cell lines show NFκB activation [53]. Mitochondrial components or detritus, when present beyond cell confines, act as damage-associated molecular patterns and activate microgliosis [178, 179]. TREM2, a gene highly expressed by microglial cells, affects bioenergetics [124].
Because mitochondria consume intracellular oxygen, they help determine cell oxygen levels. This affects hypoxia-inducible factor 1α (HIF1α) signaling, and through this vascular structure [180]. Tissue oxygen demand further influences perfusion [181].
One commonly used biomarker of microvasculature status, MRI white matter hyperintensity, additionally reflects myelination. Myelin constitutes a major brain lipid depot [182]. Evidence indicates an energy-starved brain may catabolize myelin lipid as it attempts to reverse bioenergetic insufficiency [183]. Mitochondrial biology, therefore, can add perspective to AD’s vascular component and biomarkers used to assess it.
AD brains show downregulation of insulin-activated signaling pathways [184, 185]. This led some to postulate a state of insulin resistance exists that could reflect a unique diabetes mellitus (DM), or “type 3” DM state [186]. Relevant to this are studies that report positive correlations between type 2 DM and AD incidence and prevalence [187, 188]. While AD could increase type 2 DM risk or type 2 DM could increase AD risk, the possibility that a common underlying physiology drives insulin resistance within and beyond the brain requires consideration. As mitochondria help determine insulin sensitivity and resistance [189 –191], it is possible mitochondria contribute to clinical phenotypes that present as AD/type 3 DM, type 2 DM, or both.
We classically think of AD as a brain disease but systemic biochemical and molecular manifestations occur [192 –195]. Platelet and fibroblast COX activities are lower in AD subjects [9 , 18]. It is possible this and other systemic changes arise independent of brain disease but given the specific overlapping nature of AD brain and systemic phenotypes they likely reflect a common underlying biology. Regardless, genes that determine mitochondrial function within the brain could also determine their function outside the brain.
THERAPEUTIC PERSPECTIVE
Cholinesterase inhibitors, which benefit AD patients, enhance brain glucose uptake and may impact bioenergetics [196 –200]. Cholinergic activity increases glycolysis and respiratory fluxes through definable mechanisms [201]. Behavioral interventions, especially those that utilize exercise or improve aerobic fitness, may benefit the brain by promoting brain mitochondrial function and bioenergetics [202 –204].
An AD hypothesis that does not lead to effective therapies is an inadequate hypothesis. It is too early to conclude mitochondria-targeted therapies benefit AD patients, but exploratory data from ketotherapeutics-based pilot studies so far suggest a potentially more robust benefit than Aβ–or tau-targeted interventions [205 –209]. A recent study of another bioenergetics-targeted intervention, oxaloacetate, showed it is possible to engage brain bioenergetics in persons with AD [210]. Currently our ability to assess brain mitochondrial engagement by targeted interventions remains limited but attempts to overcome this barrier are underway [211 –213].
The mitochondrial cascade hypothesis suggests effective treatments will need to improve fundamental biological processes and likely alter bioenergetics [214]. This could require overcoming the challenge of restoring an individual’s mitochondria or bioenergetic function to a more youthful state. How well mitochondria-targeted therapeutics ultimately benefit AD patients will nevertheless critically test the mitochondrial cascade hypothesis.
Figure 3 shows how bioenergetic medicine approaches conceptually compare to other AD therapeutic strategies. The figure positions therapeutic strategies specifically tested in or proposed for the treatment of AD and summarizes situations under which a given strategy seems most logical. Different AD hypotheses conceptually align with fundamentally different therapeutic approaches. If in AD an unwanted toxic entity causes a breakdown in function, removing the toxic entity or preventing its genesis could help. If, however, inadequate function arises independent of a toxic factor, an effective intervention could require an actual repair or reversal of the problem. In general blocking compensatory adaptations should make things worse, addressing phenomena that do not contribute to dysfunction should not help, addressing phenomena that contribute a little to dysfunction should help a little, and addressing phenomena that make major contributions to dysfunction should confer the most benefit.
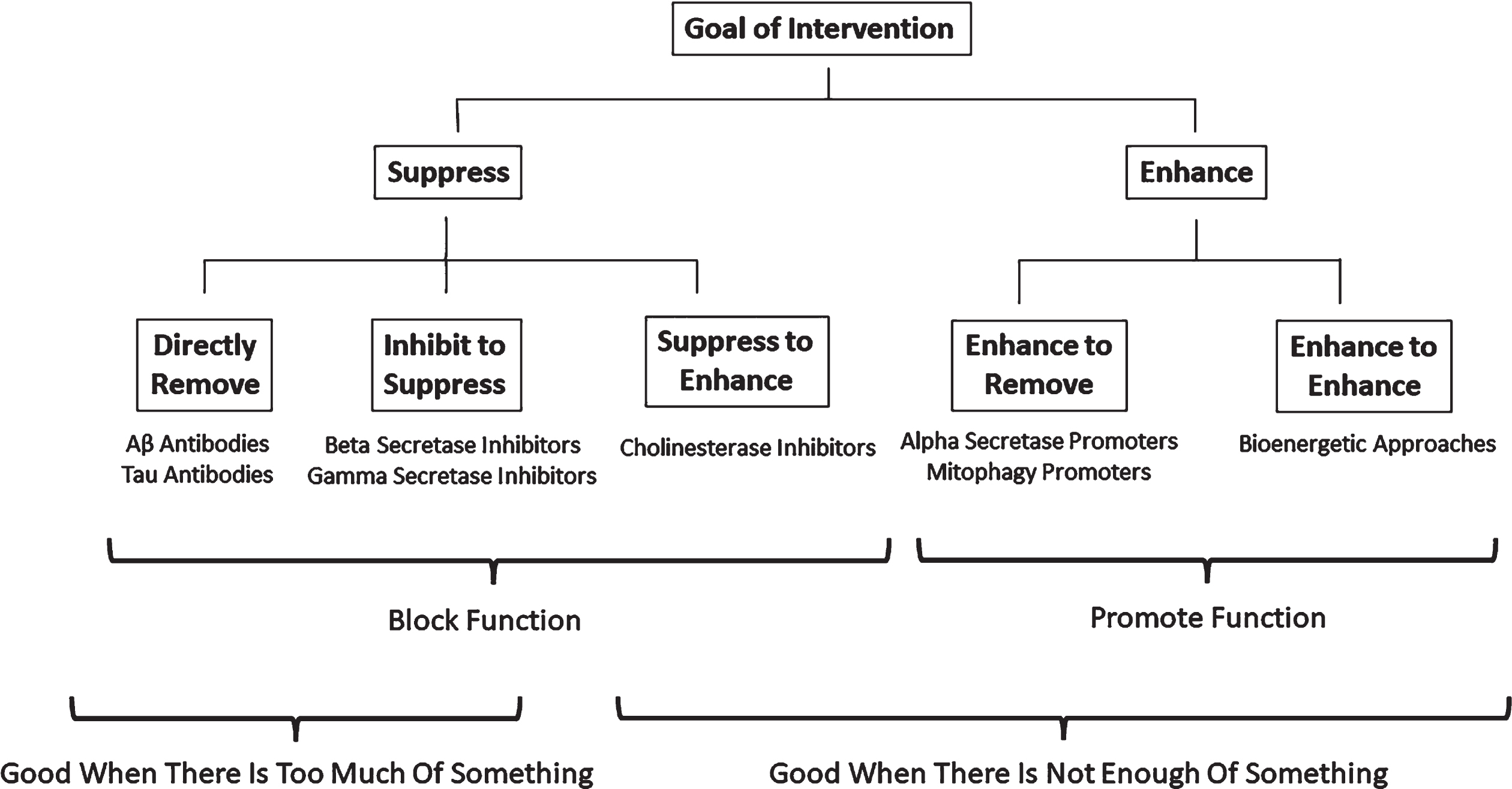
Therapeutic implications.
RELATIONSHIP TO OTHER HYPOTHESES AND BROADER IMPLICATIONS
The AβPP, PSEN1, and PSEN2 mutations that cause FAD increase brain Aβ, and in vitro studies show Aβ can injure or kill cultured cells [215, 216]. A rare AβPP polymorphism that attenuates the processing of AβPP to Aβ reduces sporadic AD risk [217]. Whether Aβ kills or injures cells in the FAD or sporadic AD brain does not impact the integrity of the mitochondrial cascade hypothesis. If Aβ meaningfully contributes to FAD or sporadic AD it likely plays out within the context of mitochondrial function [142].
AβPP and PSEN genetic data more reliably establish the relevance of AβPP biology to AD than they resolve Aβ’s role. Unfortunately, our appreciation of AβPP biology remains incomplete. The mitochondrial cascade hypothesis predicts AβPP’s mitochondrial localization is not coincidental and defining the AβPP-mitochondrial biology nexus will greatly inform our understanding of AD. For example, an AβPP contribution to mitochondrial quality control would suggest mitochondria-AβPP-aging interplay.
Autopsy studies reveal brains from non-demented individuals frequently contain plaques, including neuritic plaques, and neurofibrillary tangles [218, 219]. Our current ability to detect plaques and neurofibrillary tangles within the brains of living individuals confirms this. Recent AD definitions now advocate diagnosing AD solely on the presence or absence of plaques and neurofibrillary tangles [4, 220]. The mitochondrial cascade hypothesis, though, sees plaques and tangles as biomarkers of brain aging and not as a disease. Plaques, especially neuritic plaques, and neurofibrillary tangles may therefore inform (in many but not all cases) how close one is to the AD cliff, and in some cases predict someone is likely over the cliff [221], but by themselves would not establish the presence of frank disease.
The mitochondrial cascade hypothesis questions the order in which AD biomarker shifts presumably occur. One currently influential scheme specifies Aβ changes initially manifest followed in sequence by alterations to tau, brain structural integrity, and cognition [2]. This alignment, though, depends on measurement sensitivities and context. For example, histopathology studies report tau phosphorylation begins in the third decade [222]. Cognitive aptitude evolves throughout life. Retrospective data that show a late-life AD diagnosis predicts early adult linguistic ability and mid-life socioeconomic status suggest cognitive parameters, and possibly changes in cognitive capacity, precede dementia by decades [223, 224]. Middle-aged APOE4 carriers show decreased FDG PET glucose utilization at ages that generally precede plaque deposition [225]. In the absence of detectable Aβ changes, COX activity is lower in brains from young APOE4 carriers than it is in non-carrier brains [109].
The mitochondrial cascade hypothesis assumes an overall chronology in which genes determine mitochondrial function starting from conception, mitochondria change as we age, and this influences Aβ, tau, brain integrity, and cognition. The hypothesis, however, neither depends on nor predicts a specific biomarker chronology. Because we do not fully appreciate the chronology of most biomarker changes and chronologic observation does not critically address underlying relevant mechanisms, it seems premature to conclude Aβ-based biomarkers reside “upstream” of tau, brain integrity, and cognition biomarkers [2].
The mitochondrial cascade hypothesis facilitates links with other age-dependent neurodegenerative diseases. Principles outlined here could play out in other disorders such as Parkinson’s disease [226], explain why protein aggregations typical of one disease frequently occur in the other [227], and address overlap syndromes such as dementia with Lewy bodies.
WHAT THE HYPOTHESIS DOES NOT SAY
The mitochondrial cascade hypothesis does not claim mitochondria solely or uniquely cause AD. Rather it contends age-related mitochondrial changes subtly alter the brain environment in ways that initiate subtle responses. Eventually either the underlying mitochondrial changes or the responses they evoke evolve to a level that impacts the integrity of complex clinical phenotypes such as cognition. Similarly, the hypothesis does not view mtDNA as the primary villain, but simply as one of several unique DNA-level components that mediate aging reserve and resilience, either by contributing to the aging clock or else functioning as an aging clock [7, 8].
The mitochondrial cascade hypothesis does not by itself define what specific phenomena, including Aβ, are toxic in AD or try to predict the extent of a phenomenon’s putative toxicity. It is, though, compatible with a role for AβPP biology and aligns with data that show mitochondria influence AβPP biology and AβPP biology influences mitochondrial function [51 , 143–145]. It does not resolve the relevance of mitochondrial losses or gains of function, although it currently assumes both loss and gain of function contribute. Decreased energy production [43, 44], mitochondrial biogenesis [69, 228], and mitophagy [229] could all play a role, as could an increased production of free radicals [230] or the initiation of inflammation [177].
The hypothesis does not hierarchize the importance of different cell types to the disorder. Various lineages should respond differently to internal mitochondrial stress and to an externally stressed environment. We should not expect neurons, astrocytes, and microglia to respond identically to a given state of mitochondrial dysfunction and given that mitochondrial proteomes are tissue or cell type specific [231, 232], age-related changes should play out in different ways [232].
The hypothesis does not insinuate all AD cases are etiologically identical, which comports with genetic and clinical experience that suggests they are not. It merely lays the groundwork for conceptualizing the common late-onset sporadic cases and their age-dependence. While a primary cascade may extrapolate beyond the sporadic majority, in some cases secondary mitochondrial cascades could still occur [14].
Emphasizing mechanistic links between aging and AD biology does not imply AD is not a disease. Altered function that results in signs or symptoms technically defines the presence of disease. Whether this state develops through exaggerated aging, decompensated aging, or aging gone bad does not abrogate the horror of those experiencing it. On a related point, the mitochondrial cascade hypothesis does not view aging and AD as mechanistically distinct or mutually exclusive entities.
CONCLUSIONS
A mitochondrial cascade hypothesis mechanistically ties AD’s age-dependence, insidious and progressive course, and commonality to aging, the most age-dependent, insidious, progressive, and common condition of all. It mechanistically links the classic AD histologic changes to fundamental biology and explains why aging, and to a greater degree AD brains, accumulate plaques (especially neuritic plaques), neurofibrillary tangles, and other protein aggregations. It tells a story with a beginning, middle, and end and accounts for molecular, systems, and biomarker features that exist within and beyond the brain. The hypothesis accommodates cell-specific contributions without relying solely on the actions of a single cell type. It allows for genetic, lifestyle, and environmental contributions, is open to contributions from intracellular and extracellular phenomena proposed by other pathogenic hypotheses, and in multiple cases explains the late-life appearance of such phenomena.
An influx of supportive data guides the ongoing maturation of a primary mitochondrial cascade hypothesis. While the limitations of other AD hypotheses helped justify its initial formulation, its ability to explain a myriad of AD features now mostly fuels interest. Right or wrong, at this point the mitochondrial cascade hypothesis stands on its own merits. It will continue to evolve as the AD field continues to advance, and hopefully persist in its ability to piece together the many parts of the AD elephant.
Footnotes
ACKNOWLEDGMENTS
This manuscript was submitted as an entry to the Oskar Fischer Prize competition (University of Texas at San Antonio) and received a Bronze Prize Award.
FUNDING
The author is currently the principal investigator for the following grants: P30AG072973 (University of Kansas Alzheimer’s Disease Research Center), R01AG061194, and R01AG060733. He currently receives support from the Thompson Foundation, the Dow Family Foundation, Clune Family Foundation, and the Snyder Family Foundation.
CONFLICT OF INTEREST
The author has no conflict of interest to report. The author is an Editorial Board Member of this journal but was not involved in the peer-review process nor had access to any information regarding its peer-review.