
Abstract
Host responses to anti-amyloid-β (Aβ) antibody therapy are evident in neuroimaging changes and clinical symptoms in a subset of clinical trial subjects receiving such therapy. The pathological basis for the imaging changes and clinical symptoms is not known, nor is the precise mechanism of Aβ clearing. We report the autopsy findings in a 65-year-old woman who received three open label infusions of the experimental anti-Aβ drug lecanemab over about one month. Four days after the last infusion, she was treated with tissue plasminogen activator for acute stroke symptoms and died several days later with multifocal hemorrhage. Neuropathological examination demonstrated histiocytic vasculitis involving blood vessels with cerebral amyloid angiopathy. Fragmentation and phagocytosis of vascular Aβ were present throughout the cerebral cortex. Phagocytosis of parenchymal Aβ plaques was noted. Changes suggestive of Aβ and phosphorylated tau “clearing” were also noted. The findings overall suggest that anti-Aβ treatment stimulated a host response to Aβ, i.e., target engagement. The findings also provide evidence that amyloid-related imaging abnormalities might be indicative of an Aβ phagocytic syndrome within cerebral vasculature and parenchymal brain tissue in some cases.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is the most common neurodegenerative disease, with currently available therapies showing limited to no disease modification [1, 2]. The cause of AD is unknown. The most widely explored theory regarding AD pathogenesis is the amyloid cascade hypothesis, initially conceptualized on the basis of Mendelian forms of early-onset AD and Down syndrome [3]. According to this hypothesis, amyloid-β (Aβ) excess in the form of neurotoxic, low-n assembly intermediates, elaborated through secretase cleavage, drive the disease process in AD, with neurofibrillary degeneration, neuronal loss, and neurological deterioration occurring as downstream events [4]. By extension, mitigation of Aβ would be a logical strategy for therapeutic intervention.
Clinical trials targeting Aβ have been on-going for more than 20 years. Among the Aβ species targeted to date are soluble Aβ, oligomeric Aβ, Aβ protofibrils, aggregated Aβ, and Aβ plaques [1]. The various anti-Aβ constructs include anti-Aβ monoclonal antibodies, γ-secretase inhibition, β-site amyloid precursor protein cleaving enzyme (BACE) inhibition, and anti-Aβ vaccines [1]. Of 19 Aβ-targeting phase III clinical trials to date, 15 showed lack of efficacy, two showed toxicity, and two trials were halted for unstated reasons [1]. Recent phase III clinical trials investigating therapeutic benefit of aducanumab (EMERGE and ENGAGE trials) were terminated early due to futility analysis [5]. However, subsequent data analysis showed evidence of mixed results [6]. Aducanumab then received approval by the US Food and Drug Administration (FDA) [7]. A recently concluded phase III study of lecanemab, a humanized monoclonal anti-Aβ antibody therapeutic thought to target soluble Aβ protofibrils, demonstrated a 27% reduction in the rate of cognitive decline at 18 months [8].
The potential for adverse reactions to Aβ-targeting experimental therapies has been recognized since the phase II clinical trial of the anti-Aβ vaccine AN-1792, which was halted due to meningoencephalitis in about 6% of subjects [9]. Subsequent studies showed successful clearing of Aβ plaques, but no obvious clinical benefit or reduction in neurofibrillary pathology [10]. In 2009, a phase II multiple ascending dose trial of bapineuzumab (a humanized monoclonal anti-Aβ antibody) reported vasogenic edema on T2/FLAIR in a subset of subjects [11]. In 2011, a workgroup introduced the acronym ARIA for amyloid-related imaging abnormalities (subtypes include ARIA-E (edema), and ARIA-H (incidental microhemorrhages)) that appeared during the course of the trials in some subjects [12]. In recent phase III clinical trials of aducanumab, ARIA-E was noted in up to 35% of all subjects (10 mg/kg dose group) and up to 43% of APOE ɛ4 carriers receiving aducanumab, compared to 2.7% of placebo subjects [13]. ARIA-H was present in up to 19% of subjects receiving aducanumab, compared to 6.6% of placebo subjects. In a recent phase III clinical trial of lecanemab, ARIA-E was present in 12.6% of subjects receiving lecanemab, compared to 1.7% of subjects receiving placebo [8]. ARIA-H was noted in 17.3% of subjects receiving lecanemab, compared to 9.0% of control subjects. A higher percentage of APOE ɛ4 carriers had ARIA of either type, compared to APOE ɛ4 non-carriers. ARIA-E was noted in 32.6% (46/141) of APOE ɛ4 homozygotes. Among symptomatic ARIA cases, commonly reported symptoms were headache, visual disturbance, confusion, and dizziness.
The following case demonstrates the complexities of anti-Aβ immunotherapy and provides a detailed analysis of changes within the brain associated with such therapy. The findings in aggregate illustrate a pathological process that has not previously been described and raise the issues of anti-Aβ therapy-induced structural changes within the brain parenchyma and brain vasculature, and mechanism of Aβ clearance with anti-Aβ therapy.
CASE REPORT
A brief synopsis of this case was previously published in the form of a correspondence to the New England Journal of Medicine [14], which describes the clinical and imaging presentation. In brief, a 65-year-old woman with known APOE ɛ4/ɛ4 genotype, participated in a phase III clinical trial investigating efficacy and safety of lecanemab. Data on whether the patient received drug or placebo are not available. Following the clinical trial, the patient received three intravenous infusions of lecanemab over the course of one month as part of an open-label extension phase. Brain MRI examination performed three months prior to presentation was unremarkable apart from small T2/FLAIR hyperdensities involving periventricular white matter, interpreted as “nonspecific but consistent with mild small vessel disease.” Four days after her last lecanemab infusion, the patient developed acute ischemic stroke symptoms followed by therapeutic intervention with tissue plasminogen activator, and died several days later. The general autopsy showed no significant underlying medical comorbidity.
Macroscopic neuropathology
The brain was fixed in 10% neutral-buffered formalin. External examination showed patchy bilateral subarachnoid hemorrhage, most prominent over the right frontotemporal, inferior temporal, and parietal lobes. Numerous small hemorrhages involving the bilateral cerebral hemispheres were apparent. There was no atherosclerosis. Coronal sections of the cerebral hemispheres demonstrated multifocal intraparenchymal hemorrhage throughout the right and left cerebral hemispheres, involving anterior, middle, and posterior cerebral artery territories, more pronounced posteriorly than anteriorly. Slight mass effect with midline shift and left to right posterior subfalcine herniation were present (Fig. 1). A small (∼1.0×0.5 cm), acute thalamo-capsular infarct noted on imaging on hospital day 3 (described as “new” on day 3) was confirmed. There was no vascular territory ischemic infarct involving the cerebral cortices.

External brain surfaces (left) demonstrate bilateral patchy acute subarachnoid hemorrhage and numerous cortical hemorrhages, more pronounced in the posterior aspect of the cerebral hemispheres. A coronal section at the level of the thalamus (right) shows multiple bilateral acute intraparenchymal hemorrhages involving the cerebral cortex and superficial subcortical white matter. A slight left to right midline shift is present. The hemorrhages appeared following treatment with tissue plasminogen activator. See the Supplementary Material for the specific manipulations performed on this figure to prepare it for publication.
Thirty-nine regions (thirty-five brain regions and four cerebral and leptomeningeal vessels) were sampled for histopathologic examination including bilateral middle frontal gyri (Brodmann area (BA) 8-9), bilateral inferior frontal gyri (BA 44), bilateral superior temporal gyri (BA 22), bilateral inferior parietal lobule (BA 39), anterior and posterior cingulate (BA 23 and 24), bilateral temporal poles (BA 38), bilateral middle temporal gyri (BA 21), pituitary gland, bilateral hippocampi at the level of the lateral geniculate nucleus, and subcortical structures including basal ganglia with nucleus basalis of Meynert, thalamus with subthalamic nucleus, midbrain, pons with locus coeruleus, and cerebellar hemisphere with dentate nucleus.
Histology and immunohistochemistry
Post-fixed samples were dehydrated in graded ethanol and xylene solutions and embedded in paraffin. 5μm thick sections were prepared. Sections from all paraffin blocks were stained with hematoxylin and eosin (H&E). Immunohistochemical stains were performed on selected blocks according to standard protocols using the following antibodies: Amyloid-beta (4G8) (25 blocks), phosphorylated tau (AT8) (25 blocks), alpha-synuclein, phospho-TDP-43, CD163, CD3, CD20, and glial fibrillary acidic protein (GFAP). Special stains included thioflavin-S and Congo red.
In addition to an abundance of senile plaques, neurofibrillary tangles, and vascular hyalinization indicative of cerebral amyloid angiopathy (CAA) throughout the neocortex, H&E-stained sections were remarkable for widespread histiocytic (i.e., tissue leukocytes of the monocyte-macrophage lineage) infiltrates involving small leptomeningeal and cortical arteries and arterioles, capillaries, and occasional venules. The histiocytic reaction often included multinucleated histiocytes (Figs. 2 and 3). Vasculitis (infiltration of blood vessel walls by histiocytes predominantly) was present by H&E in 25 out of 35 brain samples examined, including all cerebral cortical, medial temporal, and cerebellar samples. The co-localization of vasculitis and amyloid was striking, being present only in samples that were also involved by CAA (Table 1). No vasculitis, CAA, or hemorrhage was detected in the corpus striatum, thalamus, or brainstem. 17 out of the 35 brain samples showed evidence of fibrinoid necrosis of blood vessel walls. These changes were only observed in samples that also showed evidence of CAA by Aβ immunohistochemistry.
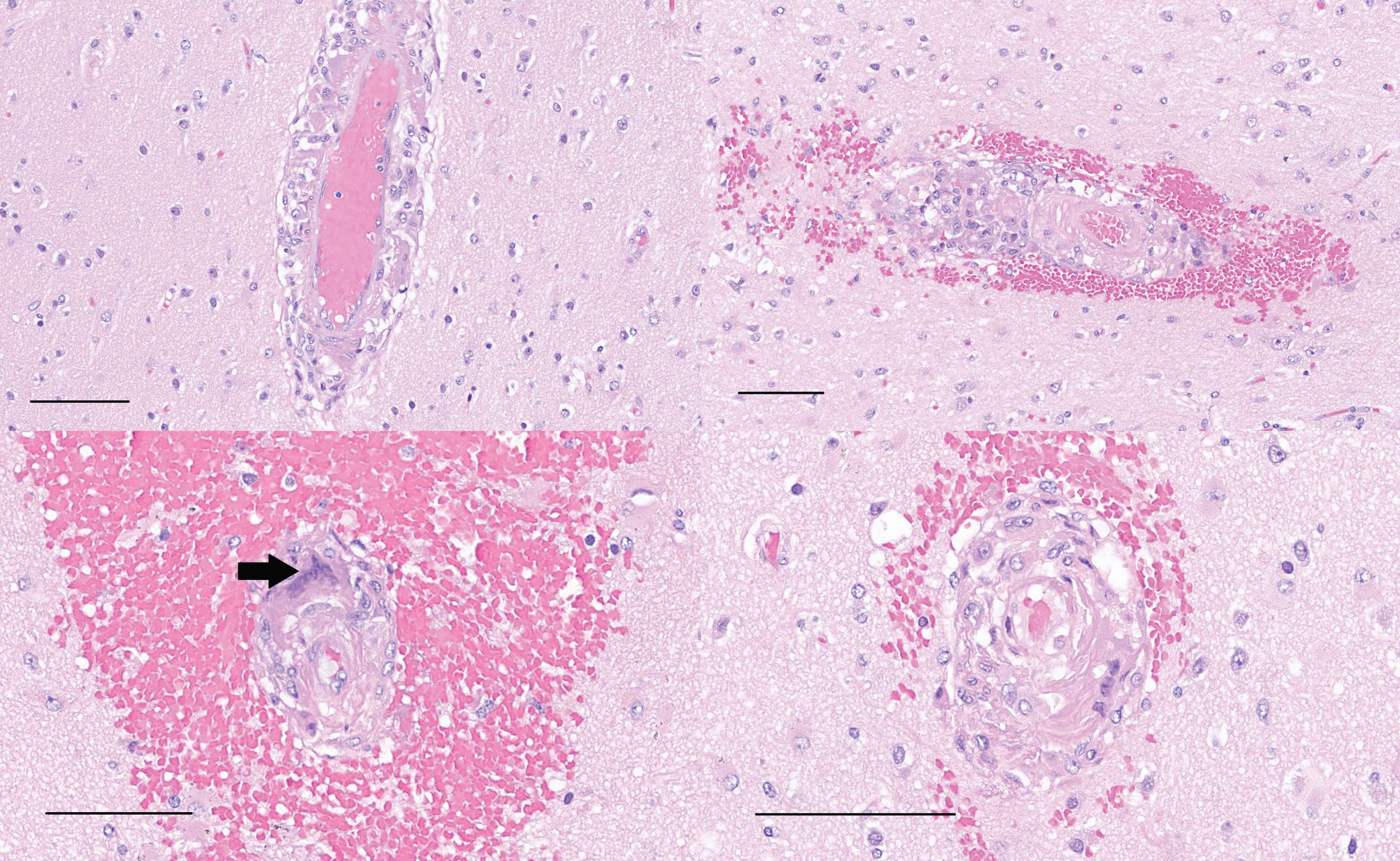
Samples of left and right superior temporal cortex (upper), and left and right inferior temporal cortex (lower) stained with H&E show histiocytic vasculitis with apparent phagocytosis of Aβ within blood vessel walls. A multinucleated histiocyte in the blood vessel wall is present in the left middle temporal cortex sample (arrow). Variable hemorrhage accompanies the changes (scale bars = 100μm).
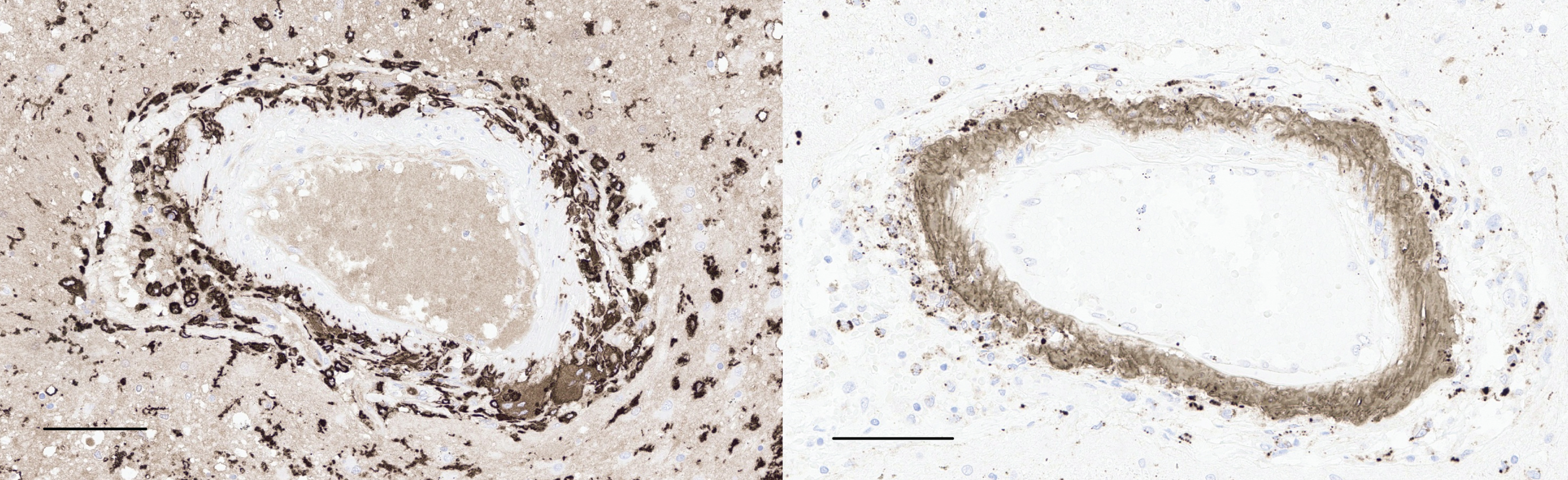
CD163 immunohistochemistry (left) shows robust labeling of histiocytes within a blood vessel wall near the left amygdala (left, scale bar = 100μm). The same vessel is immunoreactive for Aβ (right), with a band of immunolabeling along the vessel wall, as well as fragments of Aβ within histiocytes (scale bars = 100μm).
Neurovascular pathology by brain region
Accompanying the vasculitis was fibrinoid necrosis of cortical blood vessel walls, particularly in samples with accompanying hemorrhage (Fig. 4). Fibrinoid material caused stenosis and occasional occlusion of small blood vessels. Both the vasculitis and the fibrinoid necrosis of blood vessel walls were observed only in areas involved by CAA. The amyloid of CAA was often fragmented and partly phagocytosed (Fig. 5). Vasculitis was present in all cortical and cerebellar samples along with CAA, whereas fibrinoid necrosis of blood vessels was generally observed in areas of hemorrhage. Brain regions without CAA did not demonstrate acute hemorrhage.
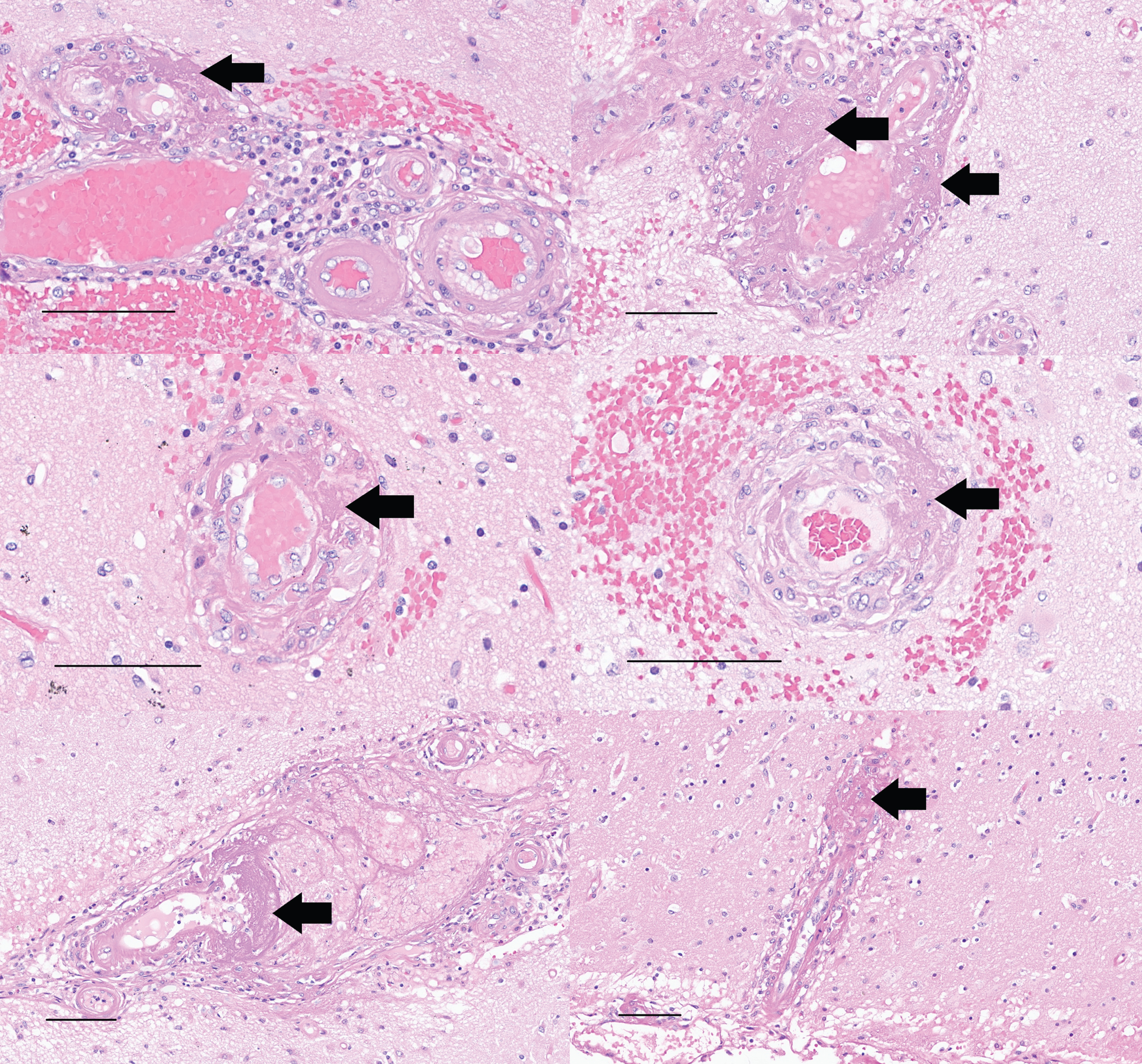
Left and right superior temporal gyrus (upper), left and right middle temporal gyrus (middle), and left and right inferior parietal cortex (lower) samples stained with H&E show fibrinoid necrosis of blood vessel walls (arrows) accompanying the vasculitis, along with variable hemorrhage. These changes were noted only in brain regions that also contained blood vessels involved by CAA (scale bars = 100μm).
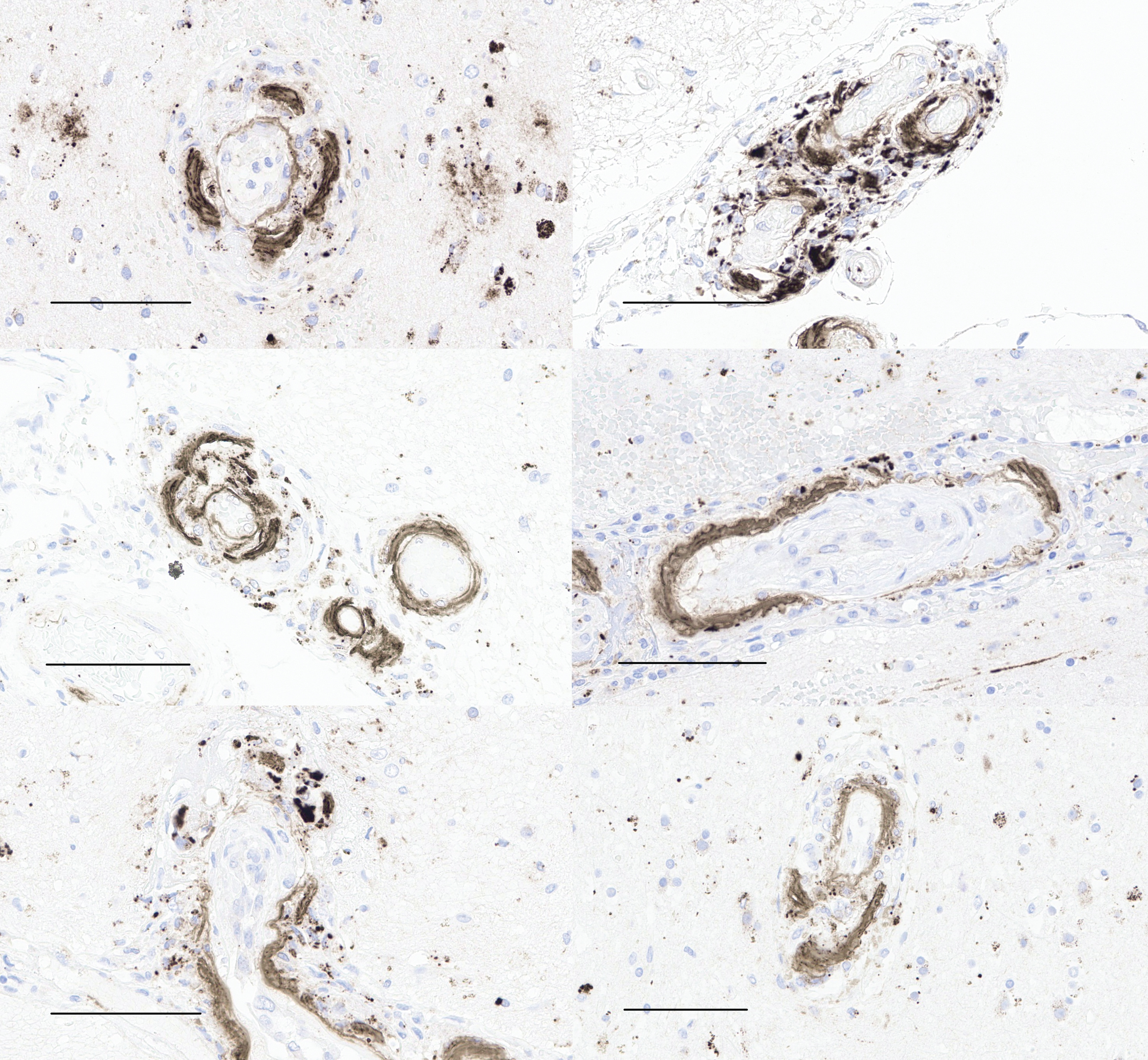
Aβ immunohistochemistry of left and right middle frontal gyrus (upper), left and right superior temporal gyrus (middle), and left and right inferior parietal cortex (lower) shows cerebral amyloid angiopathy involved by vasculitis, with fragmentation and ongoing phagocytosis of Aβ aggregates (scale bars = 100μm).
AD neuropathologic change was assessed as ‘high’ (A3, B3, C3) according to NIA-AA 2012 consensus guidelines [15]. There was no evidence of synucleinopathy, TDP-43 proteinopathy, or aging-related tau astrogliopathy. The extent of Aβ deposition appeared reduced in the superior temporal cortex compared to the frontal cortex (Fig. 6, Table 2). There also appeared to be relatively less phosphorylated tau in the superior temporal cortex relative to the frontal cortex (Fig. 7). Marked microglial activation involving Aβ plaques, or Aβ plaque phagocytosis, was noted in the medial temporal lobe samples and occasionally in the neocortical samples by H&E, as well as by CD163 and Aβ (4G8) immunohistochemistry (Figs. 8 and 9).

Immunohistochemical stains for Aβ (4G8) of the left and right middle frontal gyrus (BA8, upper), and left and right superior temporal gyrus (BA22, lower) show apparent clearing of Aβ plaques in the superior temporal gyrus relative to the middle frontal gyrus (scale bars = 1 mm).
Alzheimer’s disease neuropathologic change and plaque pathology
– = absent; + = sparse; ++ = moderate; +++ = frequent.

Low magnification photomicrographs of left and right middle frontal gyrus (upper), and left and right superior temporal gyrus (lower), show apparent diminution in labeling for phosphorylated tau using AT8 immunohistochemistry (scale bars = 3 mm).

H&E-stained sections of the left subiculum (upper left) and left amygdala (upper right), show Aβ plaque phagocytosis. An Aβ plaque core is apparent in the upper right (arrow). Immunostain for 4G8 (lower left) shows on-going Aβ plaque phagocytosis with partly phagocytosed Aβ (arrow). The macrophage/microglial response with plaque phagocytosis is apparent with CD163 immunohistochemistry (lower right) (scale bars = 100μm).
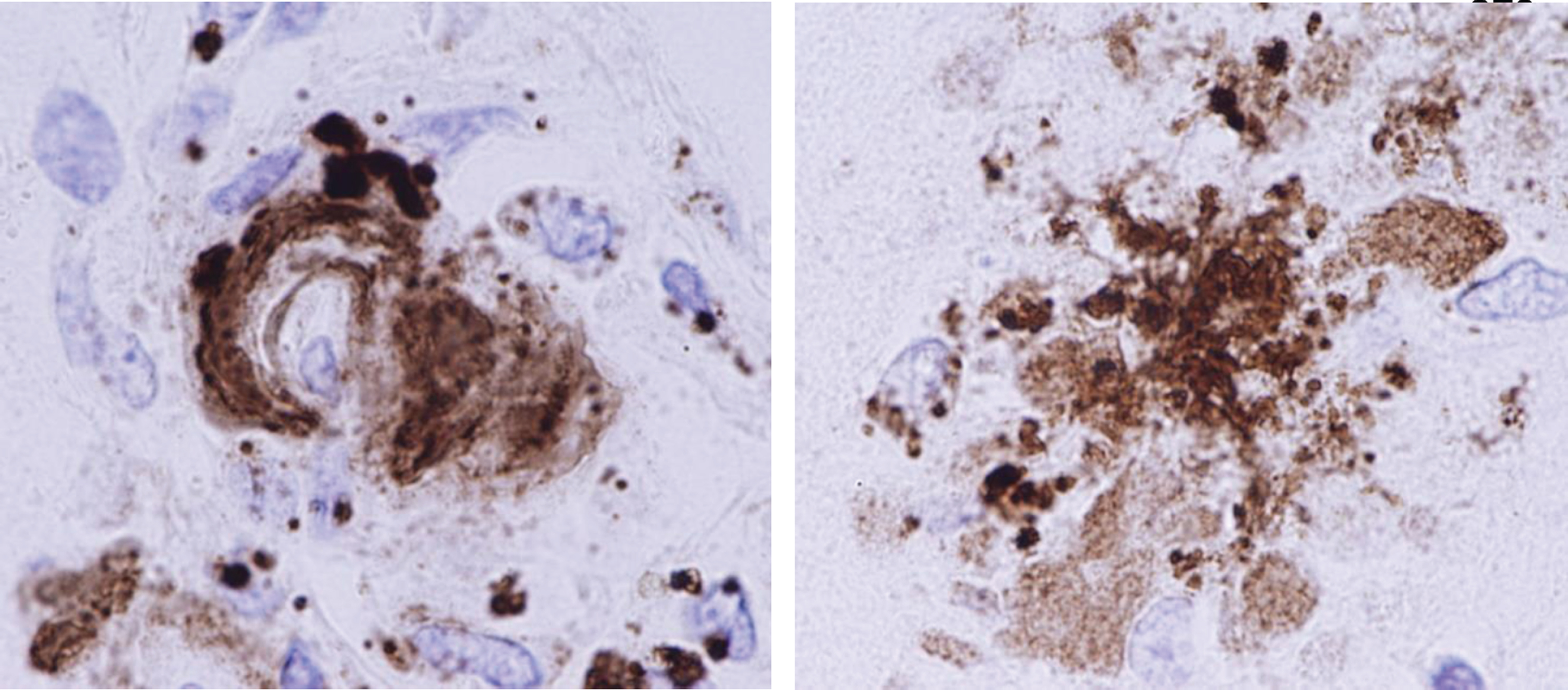
Aβ immunohistochemistry shows cerebral amyloid angiopathy with fragmentation and phagocytosis of Aβ by macrophages (left), and Aβ plaque phagocytosis by macrophages (right) (magnification = 300X before enlargement).
A small, acute white matter infarct (acute coagulative necrosis with no macrophages or other cellular reactions) involving the right posterior limb of the internal capsule was noted and corresponded to the new right thalamo-capsular infarct on imaging that appeared late in the hospitalization. This was interpreted as a small focus of agonal ischemia and was unassociated with vasculitis or CAA.
DISCUSSION
The neuropathological examination in this case indicates the presence of a robust phagocytic response to anti-Aβ therapy that extends beyond protofibrils and includes phagocytosis of insoluble Aβ aggregates of CAA [16]. The findings also suggest that Aβ phagocytosis by macrophages may be an important mechanism of Aβ clearance with anti-Aβ immunotherapy [17]. One recent report that examined post-mortem changes four months following treatment with aducanumab demonstrated plaque engagement by “amoeboid” microglia similar to this case, although with less involvement of cerebral vasculature involved by CAA [18]. More research is needed to better understand the spectrum of clinical symptoms, imaging findings, mechanisms of clearance, and the extent of neurovascular pathology associated with anti-Aβ therapy.
The findings in this case further raise the possibility that an Aβ phagocytic syndrome represents a pathological substrate for clinical and imaging complications of anti-Aβ therapy (i.e., symptomatic ARIA), and by extension might vary based on APOE genotype. Patients with APOE ɛ4 alleles have slightly higher percentages of ARIA, and APOE ɛ4 (as well as ɛ2 alleles) are over-represented in patients with CAA [19, 20]. On the other hand, ɛ3 alleles and ɛ3 homozygosity are common in patients with CAA [19, 21–24]. Complications associated with phagocytosis of CAA therefore cannot be ruled out on the basis of APOE genotype.
Although it is difficult to infer “clearance” of AD pathology on the basis of autopsy material, the marked difference in Aβ labeling between frontal and temporal cortices raises the possibility that target-engagement by lecanemab was on-going. Noteworthy as well was the relatively less phosphorylated tau immunolabeling in the superior temporal gyrus compared to the middle frontal cortex, which is generally not a feature of AD [25, 26]. This raises the intriguing possibility that Aβ-targeting by lecanemab suppresses phosphorylated tau pathology in addition to Aβ, possibly through phagocytosis of dystrophic neurites within Aβ plaques, although given the extensive hemorrhage following tissue plasminogen activator therapy in this case, it is difficult to completely exclude the possibility that immunohistochemical antigenicity was hampered by the acute pathology.
This case has features that resemble CAA-related inflammation (CAA-RI) [27, 28]. Indeed, conceptual and pathogenic similarities between CAA-RI and complications of anti-Aβ therapy have been commented upon in the literature [28–31]. Coincidental disease has been suggested as a potential confounder in this case [8], but this seems unlikely. The prevalence of symptomatic CAA-RI is low (one person per 769,000 according to one study from Japan [32]). The temporal relationship between drug administration and the onset of clinical signs, and an MRI scan negative for features of CAA-RI three months prior to presentation argue further against coincidental CAA-RI. Necrotizing vasculitis is described in CAA-RI only in anecdotal reports [31, 34], with transmural inflammation reportedly less common than perivascular inflammation [35]. In this case there was transmural inflammation with widespread necrotizing vasculitis, suggesting a particularly robust immune response. Perhaps most compelling in favor of a drug response is the evidence of therapeutic target engagement noted above, not just in cerebral blood vessels but in parenchymal brain tissue. Aβ plaque phagocytosis was plainly apparent, along with apparent immunohistochemical evidence suggestive of Aβ clearing, neither of which exist in nature outside of experimental anti-Aβ therapy. The more likely explanation is therefore a generalized drug-induced “amyloiditis” or Aβ phagocytic syndrome, which appears to be consistent with the drug design.
This report has clear limitations. Individual cases may generate hypotheses, but cannot speak to incidence or relative risk in any given patient population. Information on total dosing is also lacking, so extrapolation to a broader treatment group is not possible. Therefore, whether this case represents an idiosyncratic or excessive reaction, or is otherwise rare in the patient population under study, cannot be determined from this individual case. Information on whether the patient received drug or placebo during the clinical trial is also lacking, so it cannot be determined with certainty whether the changes reflect a subacute reaction to the anti-Aβ drug as the findings tend to suggest, or instead represent a more chronic response. More research is needed to understand the likelihood of similar complications in the broader treatment group, especially APOE ɛ4 homozygotes.
In conclusion, we present a patient with recent anti-Aβ antibody infusions and pathological findings that reflect therapy-induced Aβ phagocytosis involving fibrillar Aβ both in the parenchymal brain tissue and in the cerebral vasculature. The findings underscore the importance of close follow up with clinical trial subjects who are diagnosed with ARIA and/or who withdraw from clinical trials. Notwithstanding the anecdotal nature of the observations in this case, the striking and unique findings merit more research and a better understanding of potential neurovascular complications of anti-Aβ therapeutics.
Footnotes
ACKNOWLEDGMENTS
The authors are indebted to the decedent and family for their invaluable contribution to medical science.
CONFLICT OF INTEREST
Rudolph J. Castellani and Margaret E. Flanagan are Editorial Board Members of this journal but were not involved in the peer-review process nor had access to any information regarding its peer-review.
All other authors have no conflict of interest to report.
FUNDING
The authors have no funding to report.
DATA AVAILABILITY
The data supporting the findings of this study are available within the article.