
Abstract
Background:
Tyrosine-protein kinase receptor Ret (RET), a proto-oncogene, is considered as an attractive drug target for cancer and neurodegenerative diseases, including Alzheimer’s disease (AD).
Objective:
We aimed to identify potential inhibitors of RET kinase among natural compounds present in the ZINC database.
Methods:
A multistep structure-based virtual screening approach was used to identify potential RET kinase inhibitors based on their binding affinities, docking scores, and interactions with the biologically important residues of RET kinase. To further validate the potential of these compounds as therapeutic leads, molecular dynamics (MD) simulations for 100 ns were carried out and subsequently evaluated the stability, conformational changes, and interaction mechanism of RET in-complex with the elucidated compounds.
Results:
Two natural compounds, ZINC02092851 and ZINC02726682, demonstrated high affinity, specificity for the ATP-binding pocket of RET and drug-likeness properties. The MD simulation outputs indicated that the binding of both compounds stabilizes the RET structure and leads to fewer conformational changes.
Conclusions:
The findings suggest that ZINC02092851 and ZINC02726682 may be potential inhibitors for RET, offering valuable leads for drug development against RET-associated diseases. Our study provides a promising avenue for developing new therapeutic strategies against complex diseases, including AD. Identifying natural compounds with high affinity and specificity for RET provides a valuable starting point for developing novel drugs that could help combat these debilitating diseases.
Keywords
INTRODUCTION
Proto-oncogene tyrosine-protein kinase receptor Ret (RET) is a receptor tyrosine-protein kinase (RTK) from the protein-kinase superfamily and Tyrosine protein-kinase family that plays a crucial role in cellular mechanisms like cell proliferation, cell migration, neural navigation, and cell differentiation [1, 2]. RET is expressed in the thyroid gland, elements of the central nervous system (CNS) and peripheral nervous system (PNS), kidney and adrenal gland [3]. The glial cell-derived neurotrophic factor (GDNF) family of ligands (GFLs) signals through RET. It acts as a signaling receptor for GFLs, including Neurturin, Persephin, and Artemin. It is crucial in promoting differentiation, survival, and chemotaxis of various cell types, including neoplastic epithelial cells [4]. RET is essential for the balance of the molecular mechanism during intestine organogenesis, involved in the enteric essential for molecular mechanisms; it balances during intestine organogenesis. RET is involved in the enteric nervous system and renal organogenesis in embryonic life promotion of payer’s patches-like structure, a major part of gut-associated lymphoid tissue [5]. It regulates cell adhesion via its cleavage by caspase in sympathetic neurons and arbitrates cell migration in an integrin-dependent manner, being involved in neural crest development [6].
RET mediates various diseases like medullary thyroid carcinoma [7], hirschsprung disease 1 [8], multiple neoplasia 2A [7], multiple neoplasia 2B [9], colorectal, neuroendocrine cancers, etc. [10]. RET activates Mitogen-activated protein kinase (MAPK) and Protein kinase B (Akt/PKB) signaling pathways [11]. Tumor regression is observed in medullary thyroid carcinoma when treated with Withaferin A [10]. RET kinase activity can be activated from the extracellular environment by forming a complex between GFLs and their glycosylphosphatidylinositol-anchored co-receptor, the GDNF receptor-α family (GFRα1–4). The GFL-GFRα complex then assembles and activates RET [2]. All these observations indicate that RET plays a significant role in carcinogenesis and is responsible for developing resistance to chemotherapy. RET plays a pivotal role in developing both PNS and CNS [12]. Thus, RET can be a good target for neurodegenerative complexities and several types of cancers, including thyroid, lung, and colon cancers, as well as certain neuroendocrine tumors.
Current RET kinase inhibitors, such as Selpercatinib and Pralsetinib, have limited clinical efficacy due to acquired resistance mechanisms and off-target effects [13, 14]. Therefore, there is a pressing need to identify and develop novel RET kinase inhibitors that may move beyond these obstacles and progress the development of therapeutics. A key strategy for enhancing the clinical outcomes of patients with RET-driven malignancies is the development of novel RET kinase inhibitors. These inhibitors can potentially improve therapeutic applications, enhance efficacy, and improve patient outcomes by overcoming acquired resistance mechanisms, lowering off-target effects, and minimizing side effects [15]. Utilizing advances in structural biology, computational modelling, and translational research, a multifaceted strategy is needed to overcome the challenges involved in developing the RET kinase inhibitors. The integration of rational combination strategies will pave the way for the successful translation of novel RET kinase inhibitors into therapeutic development.
RET kinase has a long polypeptide chain that contains 1,114 amino acid residues [16], with amino acids 730-738 nucleotide-binding regions. The tyrosine kinase domain is present in the intracellular region residues from amino acids 724-1,016. It has Lys758 as ATP binding site and Asp874 as the active site residue (proton acceptor). RET have four cadherins-like domains in its extracellular regions, a calcium-binding site and a cysteine-rich domain [17]. Structure-based drug design is a powerful method that can be used to design small molecule inhibitors that bind to the active site of RET kinase and block its activity. It has become an impactful and essential part of the drug design and discovery computational approach. It reduces the time and human effort in this process, which is vital in finding highly specific and effective small molecules to be used as lead molecules in drug development [18].
The process of virtual screening is a highly efficient technique utilized in the computer-aided drug design process for identifying lead compounds [19]. It is a computational approach to screening compounds to identify the best drug-like compounds from a huge library of chemical compounds to estimate the possibility of high binding affinity of the ligand to the protein [20]. This technique is exceptionally reliable and reduces cost and time, which was very expensive and time-consuming in wet-lab experiments to shortlist the potential leads [21]. For large chemical libraries, virtual screening consists of filters like pharmacophore modelling, structure and ligand-based docking, Lipinski Rule of five, ADMET (absorption, distribution, metabolism, excretion, and toxicity), and carcinogenicity to make the lead-finding process easy and effective.
Natural compounds have become a useful source of leads for drug discovery due to their wide range of biochemical specificity, molecular structure, and multidimensional chemical structures [22, 23]. This study used 90,000 natural compounds of the ZINC database for virtual screening [24]. We used the Protein Data Bank (PDB) to download RET kinase structure in 3D form. Next, we screened 90,000 compounds and applied the Lipinski Rule of five, followed by CarinoPRED-EL and got 32,902 compounds [25]. After that, molecular docking of 32,902 compounds with RET kinase was performed, where we selected the top 50 hits based on their binding affinity. Selected compounds were further evaluated based on their specific interaction and hydrogen bonds with RET. After that, MD simulation was used to assess protein-ligand structural flexibility to understand the dynamics and stability of RET kinase and ligand complexes.
MATERIALS AND METHODS
Receptor preparation
Receptor preparation is crucial in structure-based molecular docking studies [26]. The 3D structure of RET kinase was taken from Protein Data Bank (ID: 7JU5, Resolution 1.90 Å) at RCSB (https://www.rcsb.org) in PDB format and further refined using PyMOL [27]. The crystal structure contains residues from amino acids 705 to 1,013. We took only the kinase domain for this study, and the remaining residues out of kinase were removed. Here, 16 residues were missing from this structure file, so we remodeled these residues 827 to 842 using MODELLER [28] and SwissPDB viewer (https://spdbv.unil.ch/) [29] while utilizing the original structure as a template. The remodeled structure was validated using the SAVES v6.0 server (https://saves.mbi.ucla.edu/). It validated the ERRAT score [30], 3D structure [31], PROVEN [32], WHATCHECK [33], PROCHECK [34], and Ramachandran plot [35] scores; all these suggest that this remodeled structure can be considered for further analysis. The refined structure was prepared in AutoDock tools [26] and InstaDock (https://hassanlab.org/instadock/) [36] for adding polar hydrogens and assigning appropriate atom types.
Databases used for screening
Making a chemical database of small molecules for virtual screening is one of the fundamental requirements [24]. It should be diverse in structures and availability to ease finding suitable libraries. Many small-molecule libraries are available in various public domains like PubChem (https://pubchem.ncbi.nlm.nih.gov/), The Binding Database (https://www.bindingdb.org/rwd/bind/index.jsp), NCI (National Cancer Institute) database, and ZINC database (https://zinc12.docking.org/pdbqt). This study used the ZINC 12 database [37], containing 90,000 natural compounds. The ZINC database, which contains approximately 35 million commercially available compounds, is a carefully curated and freely accessible resource for virtual screening. The 3D format of the ZINC database was downloaded and refined according to Lipinski’s rule of five.
Molecular docking-based virtual screening
Molecular docking-based virtual screening was done through InstaDock against the target receptor protein RET using the method of protein-ligand docking. The refined structure of the protein was used in a molecular docking study. The ZINC database’s filtered library of natural compounds was used as a ligand source. Molecular docking was performed using InstaDock to filter the top-hit compounds with their binding affinity and ligand efficiency. We saved the ligand data receptor protein and InstaDock program in the same directory. We generated a conf.txt file which contains all the essential coordinates and grid box size with X, Y, and Z coordinates in Angstroms, with center X = 15.917, Y = 1.887, Z = 65 and size parameters X = 74, Y = 64, Z = 65. Docking results were analyzed using log and out files, and best-docked conformations were selected for further analysis. PyMOL and Discovery Studio Visualizer were used for the structural analysis of hit compounds.
Pains and ADMET filters
After identifying the top hit compounds, we used DS Visualizer to generate SMILE IDs of the selected hits to analyze further their drug-likeliness and filter compounds with PAINS (Pan assay interference compounds) patterns that show a higher tendency to bind with multiple targets. PAINS properties were evaluated through the SwissADME server (http://www.swissadme.ch) [38]. ADMET filter was applied through the pkCSM server prediction (https://biosig.lab.uq.edu.au/pkcsm/prediction) [39]. CarcinoPRED-EL web server (http://112.126.70.33/toxicity/CarcinoPred-EL/about.html) was used for the prediction of the carcinogenicity of the compounds [40].
MD simulation
MD simulation is used to study the dynamics of molecules at the atomic level in a time-evolution manner. To simulate the complexes of RET, RET-ZINC02092851, and RET-ZINC02726682, we employed GROMACS v5.5.1 program for MD simulations [41]. To generate topologies for the receptor-ligand complexes, we utilized the PRODRG server [42]. We simulated all the systems for 100 nanoseconds (ns) using the SPC216 water model in a cubic box with center distance to edges of 1 nm. We added the appropriate amount of counterions (Na+ and Cl–) to neutralize all three systems. Afterwards, we performed energy minimization with 1,500 steepest descent followed by the conjugate gradient method to eliminate steric hindrance in the solvated systems. The volume was kept constant while gradually increasing the temperature from 0 to 300 K at 1 atm pressure. The final MD run was conducted for 100 ns. The results were analyzed using different packages of GROMACS, and graphs and figures were generated through QtGrace.
RESULTS
Lipinski rule of five filtration
The Lipinski rule of five, also known as the “Rule of Five” or “Ro5,” is a set of guidelines used in drug discovery to evaluate the drug-likeness of a molecule [25]. We used Ro5 to filter compounds from the ZINC library to get compounds with higher drug likeliness. We get 32,902 compounds out of 90,000 compounds in the ZINC database after applying the Lipinski Rule of five (https://zinc12.docking.org/pdbqt/). These 32,902 compounds were used in subsequent docking studies.
Molecular docking-based virtual screening
To perform the docking-based virtual screening, a library of 32,902 compounds was utilized in InstaDock. InstaDock is a software program that predicts protein-ligand binding modes and scores the predicted poses using empirical and knowledge-based methods [36]. After completing the docking simulations, the top 50 hits were selected based on the docking score. Selecting the top 50 hits is a crucial step in virtual screening as it helps filter out compounds that are less likely to bind to the protein. The binding affinity of selected compounds lies between –10.5 kcal/mol and –11.1 kcal/mol, a significant score for further analysis. The docking score and ligand efficiency of the selected hits are shown in Table 1. The results of the docking analysis indicate that all of the chosen hits demonstrate greater affinities in comparison to the reference inhibitors, namely Selpercatinib and Pralsetinib.
The top 50 hits selected based on binding affinity and ligand efficiency scores
ADMET analysis
The identified top 50 hit compounds selected from the docking study were further analyzed using the SwissADME server to filter the PAINS pattern. The pkCSM web server was then used to predict the ADMET properties of hit compounds. We found five compounds that show desired ADMET properties with no AMES and hepatotoxicity and good GI absorption, metabolism, excretion, and solubility. Thus, we selected five compounds out of 50 hits for further assessment, as described in Table 2. The permeability of the blood-brain barrier (BBB) plays a crucial role in determining whether a drug can effectively access the brain [43]. According to the study, the chosen compounds demonstrated the capability to traverse the BBB. Moreover, when considering CNS permeability, the threshold should be higher than –2. The compounds examined in the study exhibited values lower than the desired threshold, indicating positive outcomes.
ADMET properties of five selected compounds after pkCSM
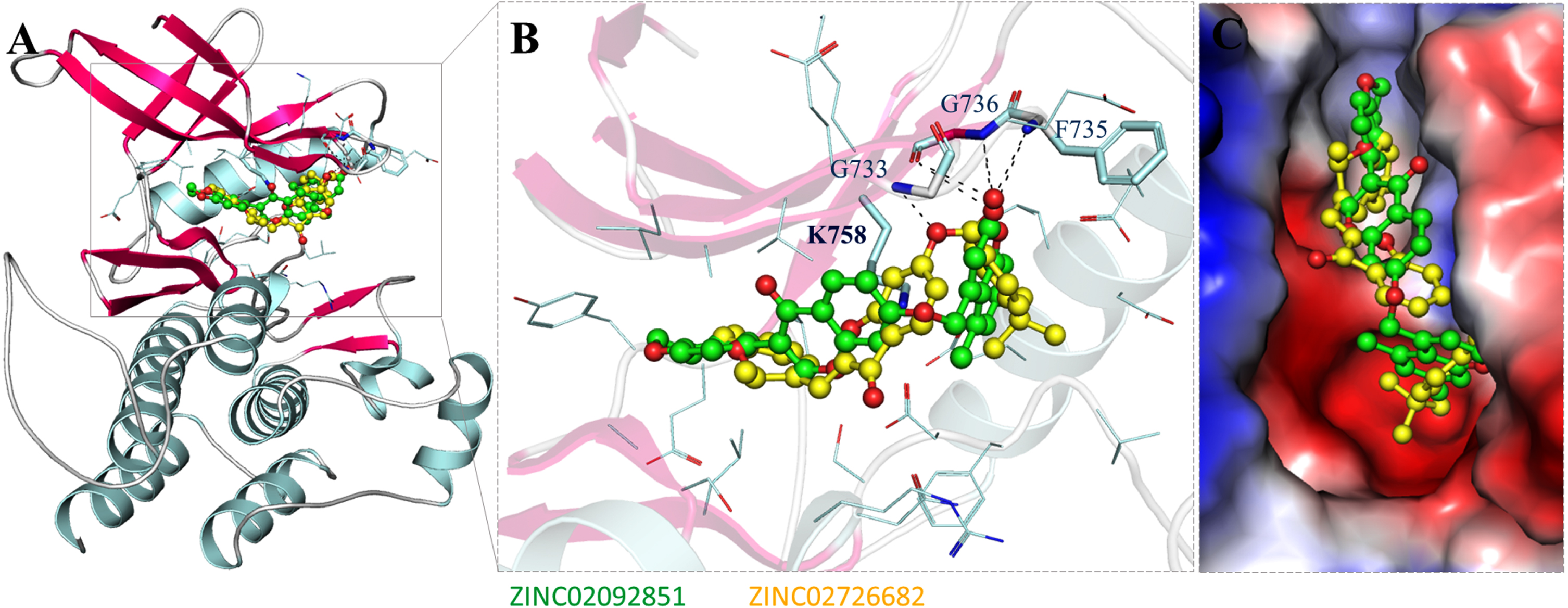
Structure of RET in complex with the selected compounds. A) RET cartoon representation with ZINC02092851 (green) and ZINC02726682 (yellow). B) RET magnifies interaction with ZINC02092851 (green) and ZINC02726682 (yellow). C) Surface potential representation of the RET binding pocket with identified compounds.
Bioactivity and PASS analysis
The biological properties of small molecules play a crucial role in drug discovery and development. By optimizing these properties, we can identify small molecules with improved therapeutic potential and increased likelihood of success in clinical trials. PASS web server was used to assess the biological properties of selected hit compounds [44]. PASS webserver is a Machine Learning based tool which predicts around 4000 different biological properties of small molecules. After analyzing five selected compounds, we found compounds are bioactive compounds which show various desirable biological properties like anti-inflammatory, antineoplastic, chemopreventive, and kinase inhibitory potential with Pa values 0,759 to 0,253. Here Pa is the probability of the compound being active under particular activity. These five compounds were further evaluated in the interaction analysis. When evaluating the reference inhibitors, Selpercatinib and Pralsetinib, the analysis showed that both have kinase inhibitory potential with neurodegenerative diseases properties.
Interaction analysis
After PASS analysis, the interaction analysis was done for all the split conformers of the selected five hits. We used PyMOL and Discovery Studio Visualizer for interaction analysis. After interaction analysis, we found that two compounds, ZINC02092851 and ZINC02726682, out of five compounds, showed the best interactions forming hydrogen bonds and other close interactions. A detailed analysis was done with both compounds for their interaction at the active site of RET kinase, i.e., Lys758. Interaction analysis shows that both ZINC02092851 and ZINC02726682 have several common interactions with important residues of RET binding pocket. Both compounds block the binding site of RET and bind to the deep cavity.
PASS analysis table for selected compounds and their biological activity predicted by Way2Drug PASS server
Detailed interaction analyses are required to investigate non-covalent interactions and their types in a protein-ligand complex. The two-dimensional interaction plots were generated using LigPlot v.4.5.3 tool to plot protein-ligand interactions, showing detailed interactions for both compounds with RET kinase residues. The identified interaction patterns of ZINC02092851 and ZINC02726682 towards RET were investigated in 2D plots. Figure 2 shows the 2D plots of all possible interactions for ZINC02092851 and ZINC02726682 with RET, which have almost the same binding pattern as common interactions. The ZINC02092851 interacts with the ATP binding site ‘Lys758’ while ZINC02726682 interacts with the active site ‘Lys758’. Previous research on RET kinase inhibitors has elucidated that they tend to engage in specific interactions with key residues, including Glu805 and Ala807. These interactions are particularly significant as they form crucial polar interactions [16]. Based on the interaction analysis, both compounds exhibited similar interactions with the binding site residue of RET, as illustrated in Fig. 2A and 2B. Furthermore, these compounds demonstrated comparable interactions with several common residues, resembling those observed with the reference inhibitors, Selpercatinib and Pralsetinib (Fig. 2C, D). Interaction analysis suggests that both ZINC02092851 and ZINC02726682 bind to the binding cavity of RET and may inhibit RET kinase’s ATP availability; therefore, they can inhibit its kinase activity. The chemical structures of the selected compounds and reference inhibitors, along with their corresponding names, are presented in Table 4.
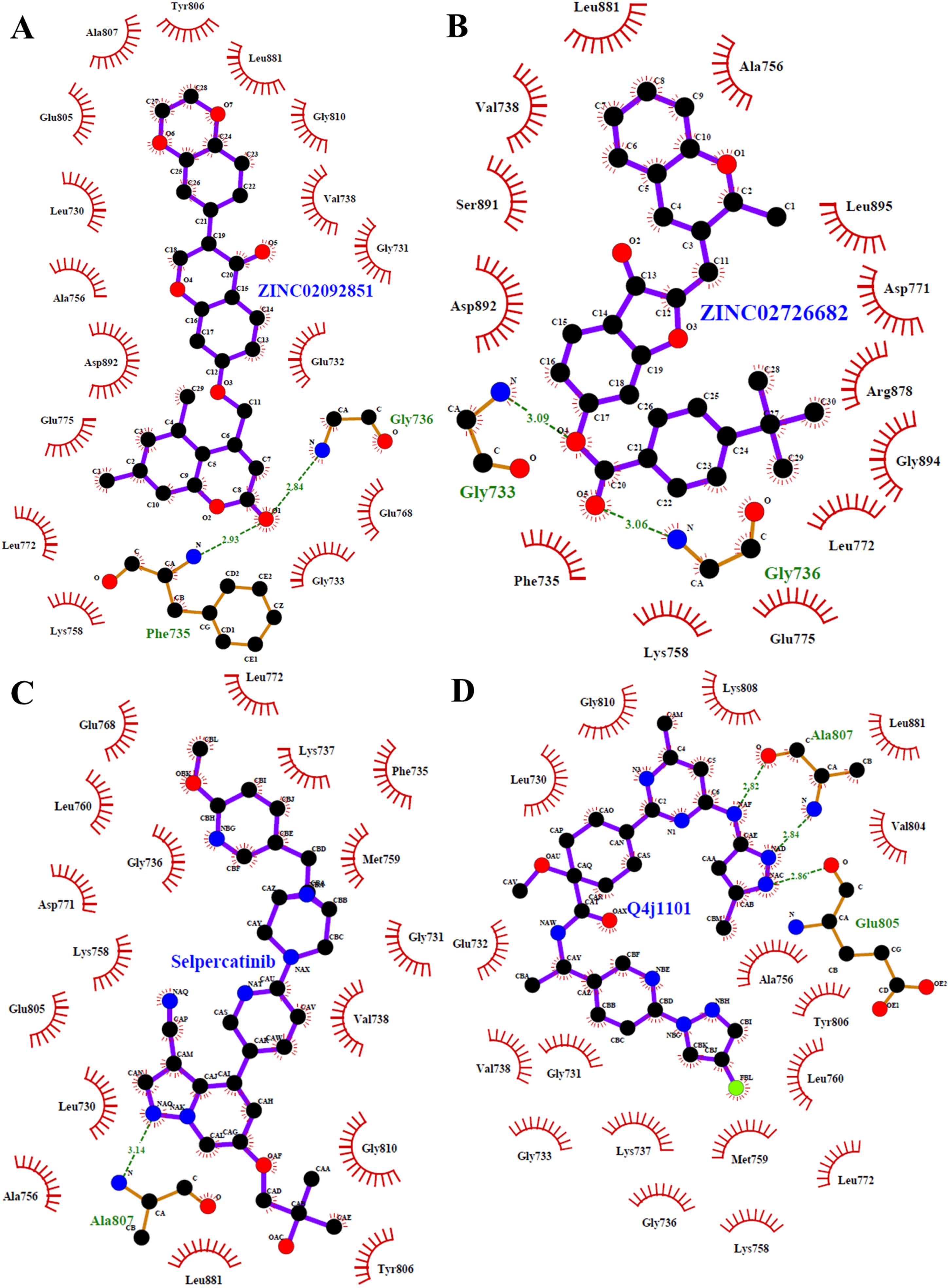
2D plots of RET binding pocket residues and their interactions with (A) ZINC02092851, (B) ZINC02726682, (C) Selpercatinib, and (D) Pralsetinib.
List of selected compounds and the reference inhibitors with their chemical structures
MD simulations
MD simulations are used to understand the dynamics of protein-ligand complexes at the atomic level [45]. To check the stability of RET and its complexes with ZINC02092851 and ZINC02726682, all three systems were simulated for 100 ns in solvent conditions. However, longer simulations are indeed valuable for capturing more detailed dynamics. To get insights into the dynamics and compactness of RET and its docked complex, various parameters were calculated and analyzed, as discussed in the succeeding sections.
Structural deviation and compactness
Root mean square deviation (RMSD) is an important parameter for studying a protein’s dynamics and structural changes. It helps in understanding structural changes in protein under stress over a time period. Figure 3A shows the RMSD values of all three systems plotted in a time evolution manner. The RMSD plots indicate that protein-ligand complexes have negligible fluctuations compared to the free RET during the simulation. The results show that the systems are in equilibrium during the simulation period of 100 ns (Fig. 3A).
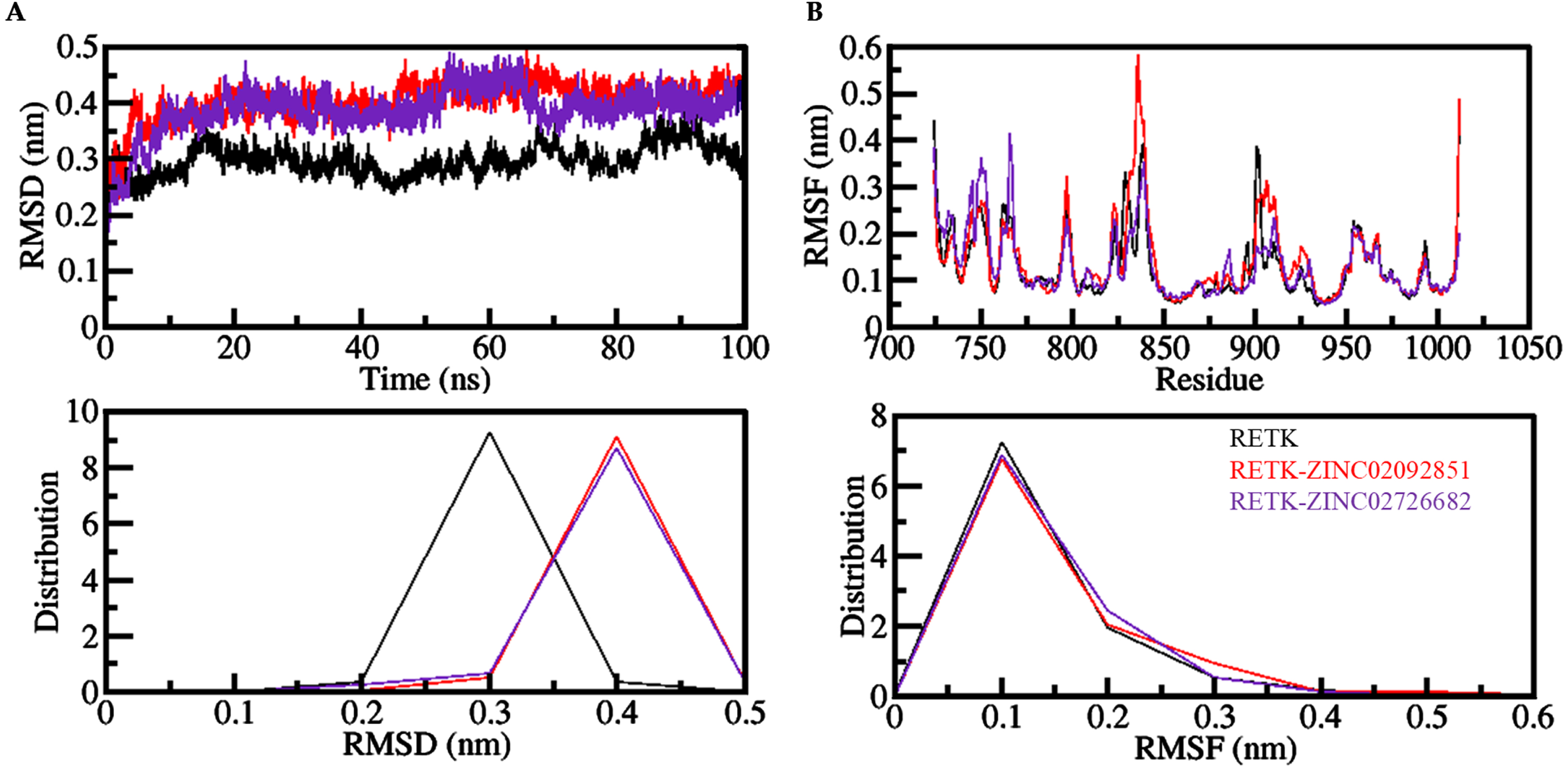
The dynamics of RET before and after it binds to the compounds ZINC02092851 and ZINC02726682 are shown in an (A) RMSD and (B) RMSF plot. The probability distribution function (PDF) is displayed in the lower panels.
Root mean square fluctuation (RMSF) helps us measure residual vibrations in protein structures during the MD simulation process. We checked the flexibility of individual residues of all three systems. The plot shows that all the systems have almost the same RMSF pattern, as shown in Fig. 3B. The figure demonstrates that all systems display a consistent distribution pattern of RMSF. Following the interaction with ZINC02726682, there were some slight changes in the RET structure in the loop and helix areas of the protein. However, the overall RMSFs suggest that the interactions between ZINC02092851, ZINC02726682, and RET are stable during the MD simulations.
The radius of gyration (Rg) is a useful parameter in understanding the protein’s structural compactness and folding dynamics. Here the Rg values were calculated to analyze the structural compactness and folding dynamics of free RET and RET-ligand complexes. Figure 5A shows that the Rg plot remained unchanged throughout the trajectory when ZINC02092851 and ZINC02726682 were present in the binding pocket of RET. The comparison of the results also indicates that RET’s folding and structural dynamics were consistently stable upon binding to ZINC02092851 and ZINC02726682. Furthermore, Fig. 4A lower panel displays the PDF values demonstrating that the distribution of Rg for RET remained consistent before and after binding to ZINC02092851 and ZINC02726682.
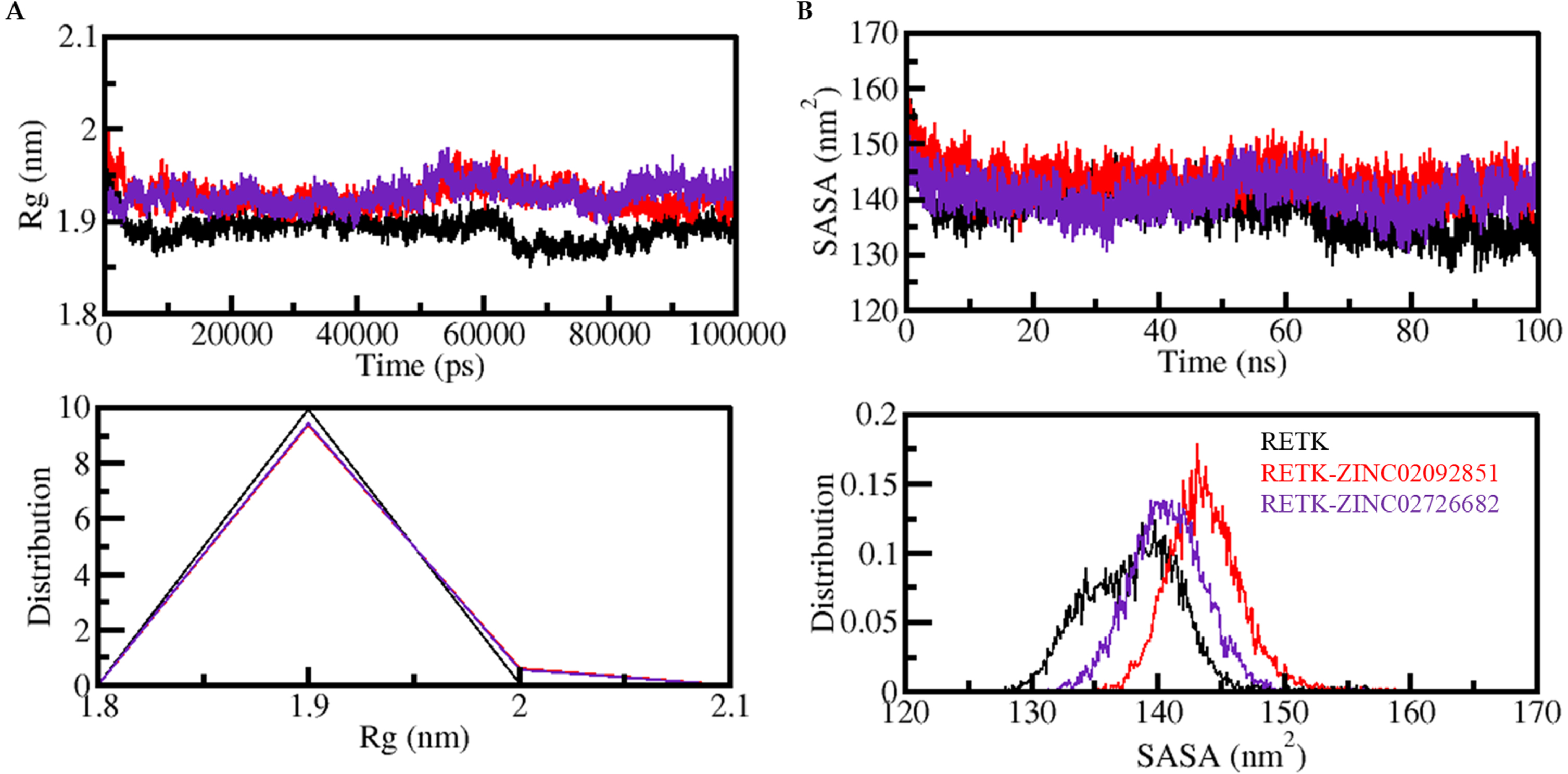
The structural compactness of RET is analyzed over time following its binding to ZINC02092851 and ZINC02726682 by plotting the evolution of (A) radius of gyration (Rg) and (SASA) SASA as a function of time. The probability distribution function values are also shown in the lower panels.

A) Time evolution of hydrogen bonds formed within RET. B) The probability density function (PDF) of intramolecular hydrogen bonds within RET.
The Solvent Accessible Surface Area (SASA) analysis is important for understanding the stability and folding behavior of proteins and protein-ligand complexes. In this study, we performed SASA analysis to evaluate the stability of the RET protein and its complexes with ZINC02092851 and ZINC02726682 during the simulation period. The SASA plots for RET in complex with ZINC02092851 and ZINC02726682 were generated and analyzed. These plots indicate that the systems remained stable throughout the simulation period, with no major changes in their SASA values, as shown in Fig. 4B. The SASA values complimented the Rg trends observed, indicating no significant changes in the structural folding and compactness of the complexes during the simulation. This analysis provides important insights into the stability of the RET protein and its complexes with the selected compounds, suggesting that both ZINC02092851 and ZINC02726682 have the potential to inhibit RET effectively.
Dynamics of hydrogen bonds
Hydrogen bond formation is crucial for the stability of protein-ligand complex formation. The temporal changes of intramolecular hydrogen bonding in RET before and after RET- ZINC02092851 and RET- INC02726682 complex were analyzed to assess their stability. The plot suggests no notable change in hydrogen bonds within the RET when forming a complex with ZINC02092851 and ZINC02726682 (Fig. 5A). The PDF plotting also shows that hydrogen bond formation in RET is constant even after binding with ZINC02092851 and ZINC02726682 (Fig. 5B).
Intermolecular hydrogen bonds were also analyzed to find out the stability of interactions of RET with the elucidated compounds. The average hydrogen bonds were found to be two in RET- ZINC02092851 and RET- ZINC02726682 complex, which rises to five at maximum (Fig. 6). The PDF indicates satisfactory stability for intramolecular hydrogen bonds in both complex systems, with higher PDF values at two hydrogen bonds (Fig. 6, lower).

The formation of hydrogen bonds between (A) RET and ZINC02092851 and (B) RET and ZINC02726682 as a function of time. The lower panels present the probability density function (PDF) of these hydrogen bonds distribution.
PCA and FEL analysis
PCA helps to investigate combined dynamics and conformational sampling in proteins. Using the PCA tool, we analyzed a conformational sampling of RET, RET-ZINC02092851 and RET-ZINC02726682 complexes from the simulated trajectories. Figure 7 shows the conformational sampling of RET, RET-ZINC02092851, and RET-ZINC02726682 through their C α atoms in the essential subspace. The plot indicates that RET docked complexes share similar subspace with the complex in its free state. In both complexes, RET-ZINC02726682 is almost in the same subspace as free-RET, while RET-ZINC02092851 on EV1 has slightly smaller flexibility. The reduced flexibility of both complexes supports their stability during simulations.
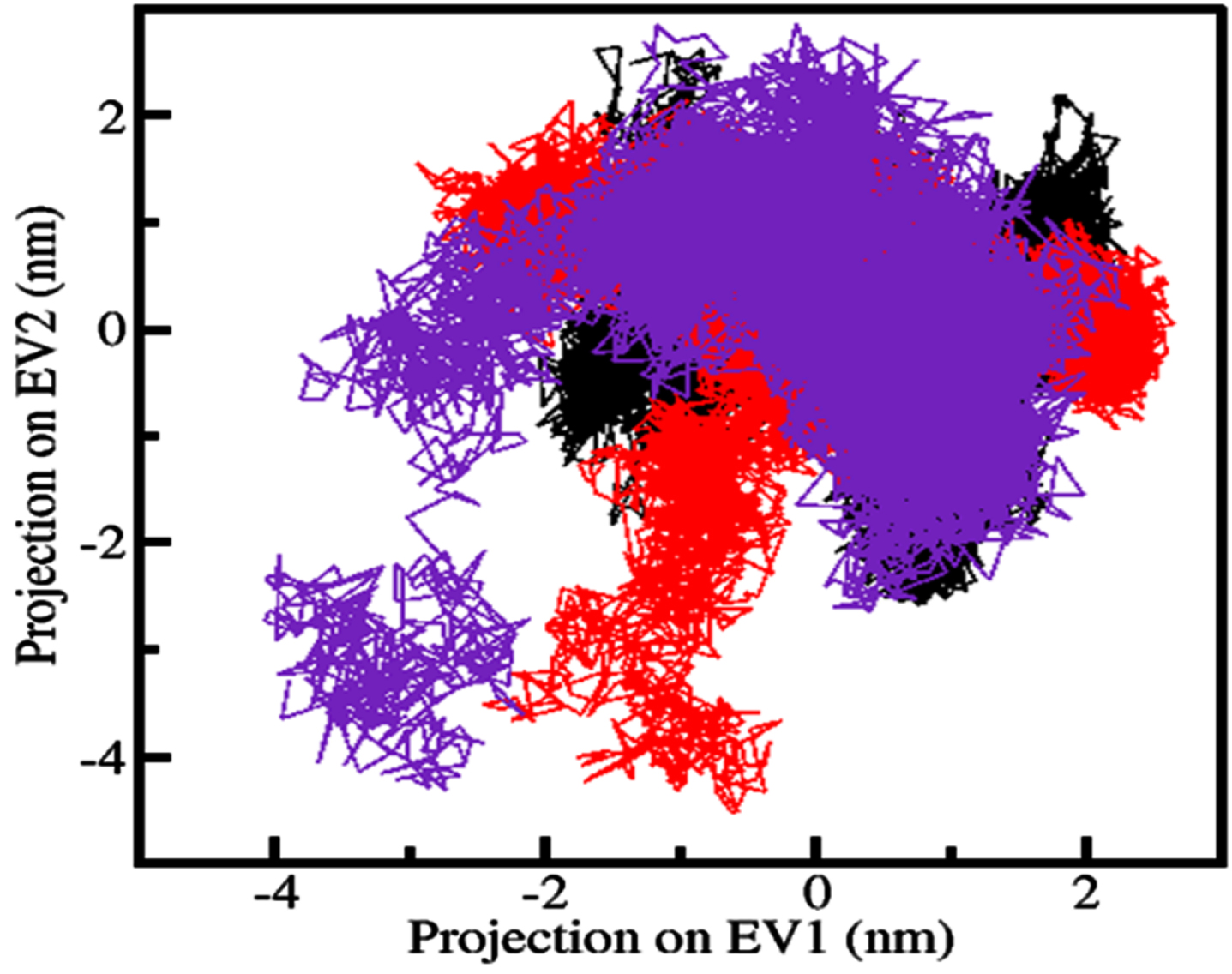
Conformational sampling is displayed using principal component analysis (PCA). The plot includes the conformational states of RET alone (black), as well as in complex with ZINC02092851 (red) and ZINC02726682 (blue). The plot shows a 2D projection of the trajectories, demonstrating the range of conformations sampled by RET.
Free energy landscapes (FELs) depict a protein’s folding and unfolding journey to reach its native form and global minima. FEL determines if the protein and protein-ligand complexes are stable during MD simulations. The lower the energy near-native state of the protein, the darker the blue in the FELs. We used two PCs to generate the energy minima and conformational landscape of RET, RET-ZINC02092851 and RET-ZINC02726682 complexes, shown in Fig. 8. The FELs plots indicate that ZINC02092851 and ZINC02726682 binding with RET has a small impact on the size and location of the phases limited within 1-2 stable global minima, as indicated by the darker blue.
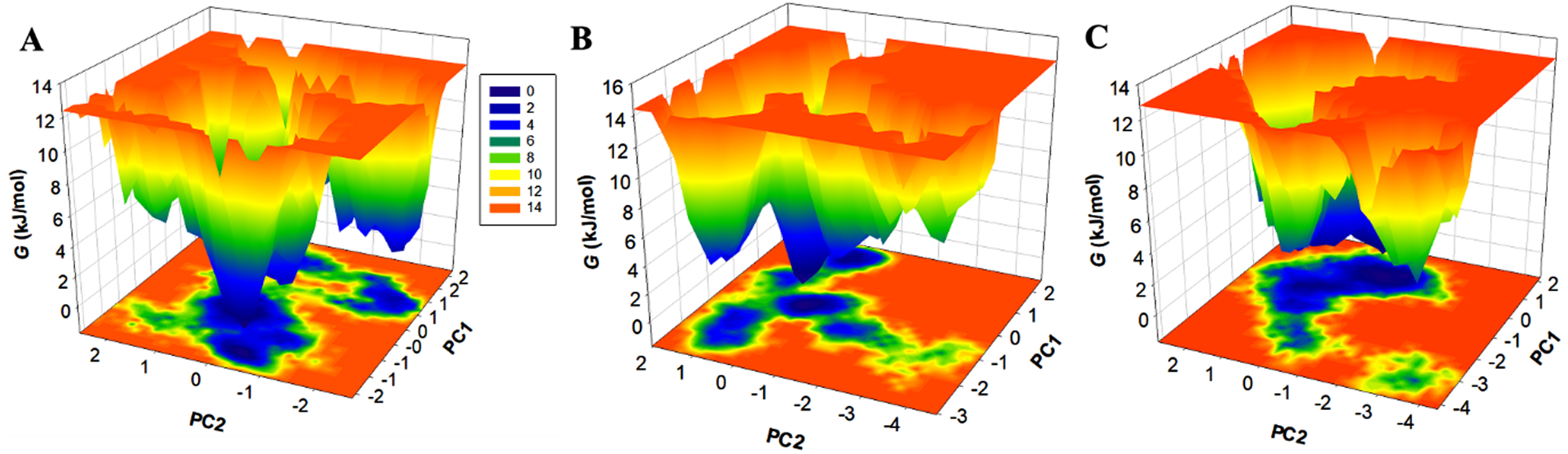
The free energy landscapes of (A) free RET, (B) RET in complex with ZINC02092851, and (C) RET in complex with ZINC02726682.
DISCUSSION
We present a comprehensive study on identifying potential inhibitors for RET kinase for neurodegenerative diseases, such as AD. We employed a structure-based virtual screening approach to identify natural compounds from the ZINC database that exhibit high affinity and specificity for the ATP-binding pocket of RET kinase. We conducted a multitier virtual screening of 90,000 natural compounds from the ZINC database against RET kinase. Initially, the Lipinski rule of five was applied to filter the compounds, resulting in a selection of 32,902 compounds for docking study. All these compounds were passed in Lipinski rule of five and showed favorable physicochemical properties. The docking screening selected the top 50 hits based on their binding affinity scores, ranging from –10.5 kcal/mol to –11.1 kcal/mol. The docking results showed that selected compounds have a high potential to bind with RETK, which can be explored further for their drug-likeness. The docking results revealed that all the chosen hits exhibited higher affinities when compared to the reference inhibitors, Selpercatinib and Pralsetinib. This finding emphasizes the potential of these compounds as promising candidates for further exploration as therapeutic agents targeting the RET kinase.
The identified top 50 hit compounds selected from the docking study were further analyzed for ADMET properties and PAINS patterns. This analysis found five compounds that show desired ADMET properties with no AMES and hepatotoxicity. By carefully evaluating the predicted ADMET parameters, we discovered that the elucidated compounds possess superior ADMET properties compared to the reference inhibitors. Unlike the reference inhibitors, the elucidated compounds do not exhibit any toxic patterns and demonstrate the absence of BBB and CNS permeability. In PASS analysis, the selected compounds showed various biological properties like anti-inflammatory, antineoplastic, chemopreventive, and kinase inhibitory potential, which suggested their use in therapeutic interventions.
Following the PASS analysis, interaction analysis was performed on all split conformers of the five selected hits. The interaction analysis results revealed that two compounds, namely ZINC02092851 and ZINC02726682, exhibited the most favorable interactions, including the formation of hydrogen bonds and other closely associated interactions. Based on the interaction analysis, it was inferred that both ZINC02092851 and ZINC02726682 bind to the binding cavity of RET, potentially impeding the availability of ATP for RET kinase. Consequently, these compounds could inhibit the kinase activity of RET. Furthermore, both compounds displayed a high affinity and specificity for the ATP-binding pocket of RET kinase and possessing drug-like properties. This analysis suggests that these compounds possess the potential to act as RET inhibitors, thereby presenting promising prospects for the development of innovative therapeutic agents.
To further validate their potential as therapeutic leads, MD simulations were performed for 100 ns. The MD simulations revealed that the binding of both compounds stabilizes the RET structure and leads to fewer conformational changes, indicating a favorable interaction between the compounds and RET kinase. The RMSD and RMSF analyses suggested that the protein and ligand complexes were stable, as the residual fluctuations were stable and decreased, especially after the binding of ZINC02092851. Rg and SASA also suggested that RET’s structure remained stable after binding to the elucidated compounds. Further, hydrogen bonds analysis suggested that ZINC02092851 and ZINC02726682 does not change their initial docking position on RET because of stable intermolecular hydrogen bonding stabilizing the complex structures. PCA indicates that RET-docked complexes share similar subspace with the complex in its free state. The FELs plots indicate that ZINC02092851 and ZINC02726682 binding with RET has a small impact on the size and location of the phases limited within 1-2 stable global minima.
Overall, the findings of this study suggest that ZINC02092851 and ZINC02726682 can potentially be developed as RET kinase inhibitors for therapeutic targeting of neurodegenerative diseases, including AD. Identifying natural compounds with high affinity and specificity for RET kinase provides a valuable starting point for developing novel drugs to combat these debilitating diseases. One potential limitation of the study is the lack of experimental validation of the identified compounds. While the computational approaches used in this study are valuable for initial screening and lead identification, further experimental studies, such as in vitro and in vivo assays, are necessary to confirm the inhibitory activity of ZINC02092851 and ZINC02726682 against RET kinase.
Conclusions
The complexity of cancer cell metabolism can be better understood by identifying potential inhibitors for critical proteins. In this study, we aimed to identify inhibitors for RET kinase using integrated computational methods. Our approach of utilizing natural compounds as inhibitors for RET provides a model for metabolic reprogramming in cancer and neurological complexities. Combining drug-likeness screening, molecular docking, and all-atom MD simulations, we identified ZINC02092851 and ZINC02726682 as promising candidates for RET inhibition. These compounds showed high binding affinity and specificity towards RET and formed stable complexes during the simulations. This study provides valuable insights into the structure-activity relationship of RET and its potential ligands and emphasizes the importance of bioinformatics in drug discovery. Our results could inform future drug discovery efforts for RET and related targets, thereby speeding up the drug discovery pipeline for cancer and neurodegeneration therapeutics. Further experimental validation is needed to confirm their in vivo activity and efficacy.
Footnotes
ACKNOWLEDGMENTS
G.M.H. thanks the Prince Sattam Bin Abdulaziz University (Grant No. 2023/RV/012).
FUNDING
This work is sponsored by Prince Sattam Bin Abdulaziz University (Grant No. 2023/RV/012).
CONFLICT OF INTEREST
The authors declare no conflict of interest.
DATA AVAILABILITY
The information supporting this study’s findings is available in this article.