
Abstract
Alzheimer’s disease (AD) is the most common neurodegenerative disease, characterized by progressive memory loss and cognitive impairment due to excessive accumulation of extracellular amyloid-β plaques and intracellular neurofibrillary tangles. Although decades of research efforts have been put into developing disease-modifying therapies for AD, no “curative” drug has been identified. As a central player in neuro-inflammation, microglia play a key role inbrain homeostasis by phagocytosing debris and regulating the balance between neurotoxic and neuroprotective events. Typically, the neurotoxic phenotype of activated microglia is predominant in the impaired microenvironment of AD. Accordingly, transitioning the activity state of microglia from pro-inflammatory to anti-inflammatory can restore the disrupted homeostatic microenvironment. Recently, stem cell therapy holds great promise as a treatment for AD; however, the diminished survival of transplanted stem cells has resulted in a disappointing long-term outcome for this treatment. This article reviews the functional changes of microglia through the course of AD-associated homeostatic deterioration. We summarize the possible microglia-associated therapeutic targets including TREM2, IL-3Rα, CD22, C5aR1, CX3CR1, P2X7R, CD33, Nrf2, PPAR-γ, CSF1R, and NLRP3, each of which has been discussed in detail. The goal of this review is to put forth the notion that microglia could be targeted by either small molecules or biologics to make the brain microenvironment more amenable to stem cell implantation and propose a novel treatment strategy for future stem cell interventions in AD.
INTRODUCTION
Alzheimer’s disease (AD) is the most common type of dementia, mainly manifested as progressive memory loss and cognitive impairment. According to the latest World Alzheimer’s Disease Report, there are approximately 55 million people worldwide suffering with AD, a figure projected to increase to 139 million by 2050 [1]. Although the disease imposes a heavy burden on societies and families, there are still no effective treatment to curb or prevent the progression of the disease yet.
Pathologically, AD is characterized by the presence of extracellular amyloid-β (Aβ) plaques and intracellular neurofibrillary tangles (NFTs), as well as synaptic and neuronal loss. Over the past decades, therapeutics research has mainly focused on eliminating or inhibiting either amyloid or tau proteins [2, 3]. Although some amyloid-β targeting biologics such as aducanumab and lecanemab have received FDA approval recently, a truly definitive curative disease modifying therapy for AD is still lack [4]. Accordingly, the search for new therapeutic approaches is becoming an urgent and high priority for AD treatment. Since 2000, an increasing body of research emphasizes the role of neuro-inflammation as an important factor in the underlying mechanisms of AD, suggesting that microglia could be a promising target for AD treatment [5, 6]. As resident cells of the brain, microglia constantly survey the surrounding microenvironment and maintain brain homeostasis [7]. The activation of microglia is triggered by various types of pathologic changes in the brain. With AD, the accumulation of Aβ causes the activation of microglia, leading to the subsequent release of a multitude of pro-inflammatory molecules and the deterioration of homoeostasis [8].
In recent decades, stem cell therapy has been widely studied and has emerged as a promising approach for treating AD [9]. As “master cells” of the body, stem cells have the potential to replace lost neurons, restore the disrupted microenvironment, and regenerate cells [10, 11]. Currently, most studies of stem cell therapy for AD have used externally cultured cells. However, many challenges and limitations like ethical issues, tumorigenicity, and rejection, still exist in the advancement of stem cell therapy [12]. Moreover, stem cell viability which is primarily interfered with by microenvironment is another important challenge faced by researchers in AD [13]. The neuro-inflammatory microenvironment of the host brain was found to be hostile for the survival and differentiation of stem cells [13–15]. For example, the elevation of Aβ levels triggers a microglia-dependent neuro-inflammatory response, which in turn suppresses stem cell proliferation and function [16]. Therefore, microglia-targeted modulation of the brain microenvironment is crucial for the successful implementation of stem cell-based therapies for AD.
This article attempts to provide a review of the underlying pathogenesis of microglia-mediated neuro-inflammation in AD, with the aim of evaluating and identifying possible microglial receptors as putative drug targets. We discuss how the signaling pathways of microglia contribute to the homoeostasis of brain microenvironment. In addition, this review explores the potential of preemptive microenvironment regulation before stem cell transplantation, in the hope of discovering an immune-modulatory treatment strategy for researchers advancing stem cell-based therapies for AD.
ALZHEIMER’S DISEASE AND NEURO-INFLAMMATION
The typical pathological characteristics of AD include the extracellular accumulation of Aβ plaques and the formation of intracellular NFTs. Over the past decade, there has been a growing recognition of the pivotal role of microglia-focused neuro-inflammation in the onset and progression of AD [17]. The accumulation of toxic Aβ peptides, the proteolytic products of AβPP, is believed to be an early initiating factor in AD [18]. Compelling evidence suggests that the toxic accumulation of Aβ and certain inflammatory mediators occurs even prior to the onset of clinical symptoms [19]. Microglia, the resident macrophages of the brain, have traditionally been viewed as the cell population responsible for maintaining the brain’s microenvironment [20]. Exposure to Aβ aggregation can stimulate the activation of resident microglia and the subsequent release of pro-inflammatory cytokines, thereby inducing further neuro-inflammation and facilitating the clearance of Aβ. Microglia are typically classified into different phenotypes, including M1 (classically activated, pro-inflammatory) and M2 (alternatively activated, anti-inflammatory) [21]. However, increasing evidence indicates that microglia exhibit greater diversity than can be captured by the traditional M1/M2 categorization. For example, a new phenotype known as dark microglia has been uncovered through ultrastructural analyses over the past decade [22]. Dark microglia exhibit significantly higher activity levels compared to normal microglia, and express higher levels of triggering receptor expressed on myeloid cells 2(TREM2) in the pathological state of AD. Similarly, another newly identified microglial phenotype, known as disease-associated reactive microglia (DAM) has recently been identified through comprehensive single-cell RNA analysis of brain immune cells [23]. DAM cells exhibit remarkable morphological and functional alterations which are distinct from homeostatic microglia that are dependent on the microglial receptors TREM2 and tyro protein tyrosine kinase binding protein (TYROBP) [24]. Rangaraju et al. utilized AD mouse models and flow cytometry to reveal distinct pro-inflammatory and anti-inflammatory phenotypes of DAM [25]. Interestingly, the study also found that pro-inflammatory DAM emerged earlier in the course of AD progression, characterized by the expression of interleukin (IL)-1β, IL-12β, and the surface marker CD44. Phagocytic genes such as IGF-1 and the surface marker CXCR4 were mainly expressed in anti-inflammatory DAM.
THE DETERIORATIVE NEURO-INFLAMMATORY MICROENVIRONMENT (NIME)
Microglia function as phagocytes and play a crucial role in maintaining the homeostasis of the AD brain microenvironment. This includes phagocytosis of Aβ or cellular debris, blood-brain barrier (BBB) maintenance, and synaptic remodeling [26]. In addition, microglia closely interact with astrocytes and neurons in the regulation of homeostasis. It has been recognized that microglia effectively clear Aβ in the early stages of AD, while late-stage AD is characterized by an accumulation of Aβ in the brain, microglia dysfunction, and subsequent cognitive decline [27]. The accumulation of Aβ leads to neuronal death and significantly accelerates the progression of AD. Under normal physiological conditions, the BBB controls the passage of substances from the bloodstream and facilitates the clearance of various neurotoxic macromolecules [28]. In the progression of AD, elevated levels of pro-inflammatory mediators secreted by the microglia lead to changes in the permeability of the BBB through interaction with receptor proteins on the BBB membrane. This compromises the clearance of neurotoxic molecules from the brain, escalating the accumulation of pathological deposits such as Aβ [18]. Furthermore, certain factors produced by microglia, such as brain-derived neurotrophic factor (BDNF) and transforming growth factor-β (TGF-β), play a role in promoting synaptic remodeling. BDNF plays an important role in facilitating the formation of learning and memory by modulating proteins involved in learning-related synapse formation [29]. TGF-β is a key regulator of neuronal C1q expression and synaptic pruning [30]. Therefore, repeated exposure to neurotoxic molecules confers a higher risk of developing synaptic loss in AD.
DECREASED SURVIVAL OF TRANSPLANTED STEM CELLS
The development of stem cell therapy has promised new hope in the pursuit of disease-modifying treatments for AD. Stem cells have the potential to replace lost or damaged cells through processes such as differentiation, neurogenesis, and the delivery of therapeutic agents to the brain [31]. However, most studies report that stem cell transplantation treatment is only effective in the early period (1–2.5 months after transplantation), with the long-term effects remaining uncertain [9]. Furthermore, reports indicate that the long-term (5 months) transplantation of neural stem cells into AD mice failed to improve their cognitive function, did not result in differentiation into new neural cells, and did not increase the levels of brain-derived neurotrophic factor and synaptic density [32]. This suggests that the therapeutic effect of transplanted stem cells in the brains of AD mice is time dependent. This observation is thought to arise from the progressive deterioration of the microenvironment, the significant reduction in the survival and regenerative capacity of the transplanted stem cells, and their failure to differentiate into new neurons and glial cells [33]. Alternatively, the differentiation potential of transplanted stem cells is affected by the microenvironment. For example, the overexpression of AβPP resulted in transplanted stem cells producing more astrocytes than neurons in AD [34]. Therefore, we postulate that transforming the harmful microenvironment in the AD-affected brain into a favorable one prior to stem cell therapy could enhance the survival and normal differentiation of transplanted stem cells.
SIGNALING PATHWAYS FOR MODULATING MICROGLIA ACTIVATION STATE
Although microglia only account for approximately 5% of the cells in the central nervous system (CNS), they play a primary role in immune surveillance, pathogen clearance, and homeostatic maintenance. As previously discussed, dysregulated microglia activation results in a homeostatic imbalance; therefore, the transition of microglia activation from a pro-inflammatory to an anti-inflammatory state holds the potential to restore cognitive function and establish a more balanced neurophysiological environment. These conditions may prove beneficial for the implementation of stem cell therapy (Fig. 1). In the following section, we review in detail the potential signaling pathways involved in microglial modulation in the CNS (see Table 1). We searched the PubMed database by using the keywords “microglia”, “signaling pathway”, and “Alzheimer’s disease”. With these keywords, all articles published before November 20, 2023 were screened. Considering microenvironment regulation as well as the reliability of the study, we mainly included animal experiments which showed increased surveillance and phagocytosis of microglia, clearance of toxic products, reduced production of pro-inflammatory mediators or change in microglial phenotype.
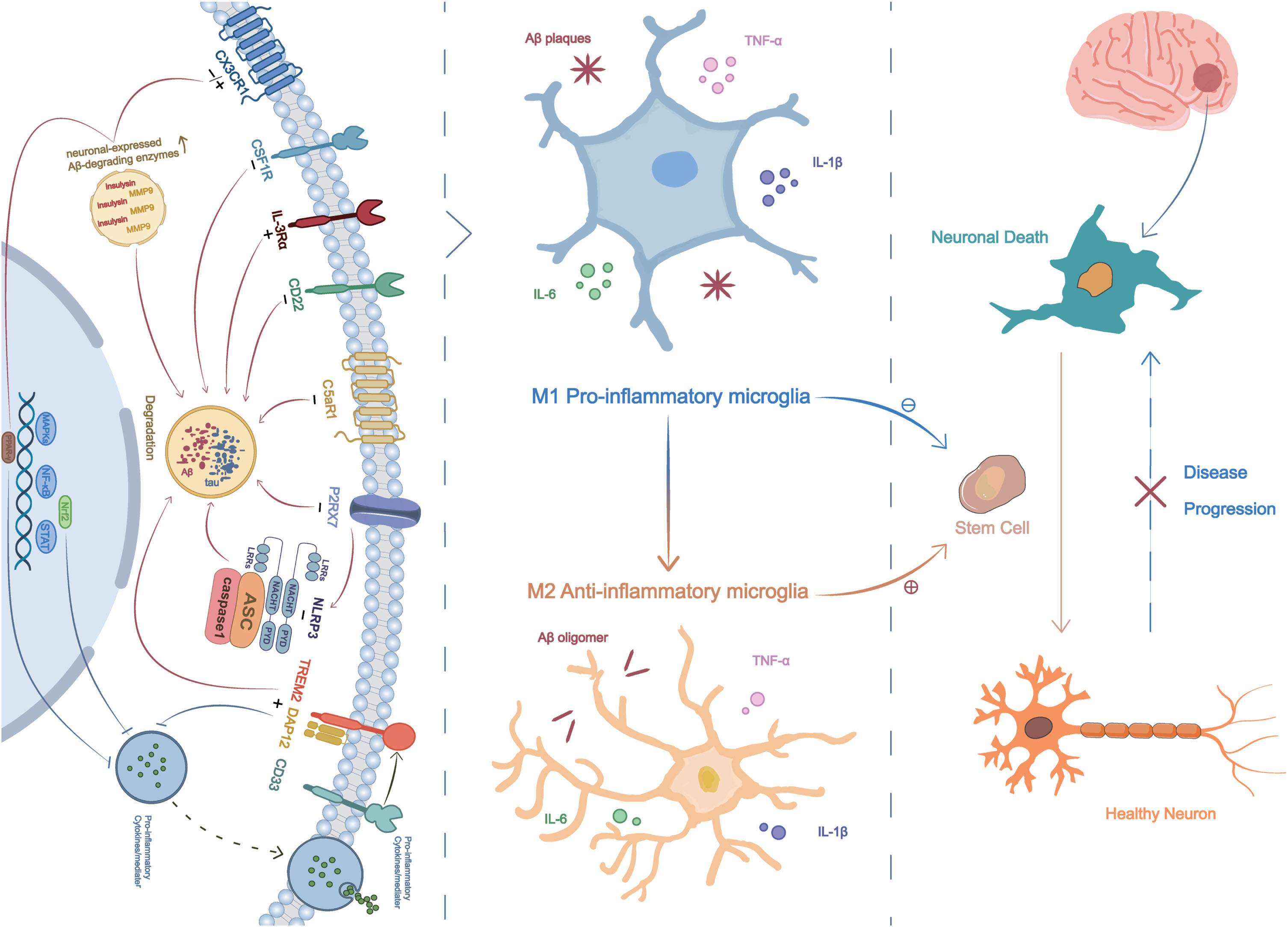
Signaling pathways, neuro-inflammatory microenvironment, and stem cell therapy. Eleven signaling pathways, including TREM2, CSF1R, IL-3Rα, CD22, C5aR1, CX3CR1, P2X7R, CD33, Nrf2, NRLP3 and PPAR-γ, can modulate microglial polarization, shifting them from a pro-inflammatory to an anti-inflammatory state. This leads to a reduction in neuro-inflammatory responses, decreased Aβ deposits, and lower tau levels, ultimately resulting in an improved brain microenvironment that facilitates the efficacy of transplanted stem cell treatment.
Preclinical studies of potential signaling pathways by modulating microglial inflammatory state in AD
TREM2
TREM2, a member of the TREM immunoglobulin superfamily, has been identified as one of the most significant genetic risk factors for AD [52, 53]. A recent study in 5×FAD mice (a model of AD) showed that mice with increased TREM2 expression have a considerable reduction of amyloid burden and improved cognitive functions [35]. Furthermore, an in vitro study investigated the underlying mechanism by which TREM2 regulates microglial activation, using hydroxysafflor yellow A (HSYA), an extract derived from the dried flowers of Carthamus tinctorius L. [54]. This study revealed that upregulating TREM2 can switch microglia from a pro-inflammatory phenotype to an anti-inflammatory phenotype. Additionally, it can regulate the production of inflammatory cytokines via the downstream transcription factor nuclear factor kappa B (NF-κB) [54]. Another study demonstrated that the expression of TREM2 increased in response to elevated Aβ levels during disease progression. The overexpression of TREM2, in conjunction with its adapter protein DAP12, significantly decreased Aβ deposition, neuro-inflammation, and neurological dysfunction [36]. Yoo et al. also reported that replacing TREM2 knockout microglia with TREM2 wild-type microglia-like cells derived from systemically transplanted hematopoietic cells restores microglial dysfunction in a mouse model of amyloidosis [55]. These findings highlight that TREM2 is intimately involved in the development of AD and is a potential target for treatment; however, the implementation of a TREM2-targeted strategy may be contingent on the stage of disease progression. For example, one study proposed that TREM2 activation is beneficial in the early stages of AD, while in later stages it could lead to immune tolerance, the inhibition of microglial functions related to Aβ phagocytosis, and lysosomal degradation [56]. In addition, TREM2 biochemistry is complex and most studies of TREM2 have focused on biologics, such as TREM2-activating antibody [35, 58].
IL-3Rα
Reducing Aβ levels is a crucial aspect of the homeostatic mechanisms that microglia employ to maintain brain health. Crosstalk between astrocytes and microglia has been reported to lead to increased degradation of Aβ aggregates [59], although the precise connection between microglia and astrocytes is poorly understood. Recently, McAlpine et al. revealed that IL-3, a multifunctional cytokine that regulates inflammation, is a key mediator of astrocyte-microglia crosstalk and endows microglia with an enhanced ability to cluster and clear Aβ oligomers [37]. It was also reported that microglial expression of IL-3R was higher in AD transgenic mice compared with wild-type animals. Furthermore, IL-3R signaling was found to increase the expression of TREM2-dependent immune response genes including Spp1, Itgas, Apoe, Lyz2, and Clec7a, which are associated with the clustering and clearance of Aβ aggregates [37]. Thus, astrocytes have the capacity to regulate the neuroprotective phenotypic states of microglia through the secretion of IL-3, implying that it is a key mediator in potential therapeutic interventions for AD. Nevertheless, it should be noted that IL-3 does not readily cross the blood brain barrier and not appear to be of direct therapeutic value [60].
CD22
As established, the functions of phagocytosis and microenvironment surveillance are crucial for microglia to maintain homeostasis in the CNS. Pluvinage et al. have reported that CD22, a B-cell receptor, acts as a negative regulator of microglial phagocytosis and is upregulated in aged microglia [38]. The study also found that inhibiting CD22 led to enhanced clearance of Aβ oligomers in vivo. Additionally, the long-term administration of a CD22 blocking antibody restored microglial homeostasis and improved cognitive function in aged mice [38]. Another study demonstrated a reduced surveillance capacity of microglia in aged mice brains, characterized by a significant reduction of microglial ramification and upregulated CD22 surface levels [39]. Interestingly, this age-related decline in microglial surveillance can be restored through CD22 inhibition [39], whereas CD22 inhibition is mainly antibody-mediated and the task of devising CD22 antagonists remains a high priority. In summary, CD22 emerges as a novel age-related biomarker of microglial phagocytosis and surveillance. Therefore, targeting CD22 presents a promising strategy for restoring homeostasis and improving cognitive function within the CNS.
CX3CR1/CX3CL1
The CX3CL1(fractalkine)-CX3CR1 axis represents a crucial signaling pathway in neuron-glial communication [61]. CX3CL is a unique chemokine produced in neurons and exerts its function by binding with its exclusive receptor, CX3CR1, which is expressed on microglia. The role of CX3CL1 signaling can be either protective or detrimental, depending on the specific disease microenvironment [62]. Multiple studies, conducted on both AD mice and human patients, have indicated a gradual decrease in the levels of CX3CL1 throughout the course of disease progression [63, 64]. Hickman et al. demonstrated that partial CX3CR1 deficiency was associated with reduced Aβ and senile-like plaque burden, leading to improved cognitive function; however, no differences were observed in mice with total CX3CR1 deficiency. This effect could be attributed to elevated levels of Aβ-degrading enzymes such as insulysin and matrix metalloproteinase 9 in APP-PS1-CX3CR1+/– mice [40]. However, in another study conducted in hTau mice, the deletion of CX3CR1 exacerbated tau hyperphosphorylation and increased microglia activation, resulting in spatial memory impairment [65]. These reported differences may be attributed to variations in the inflammatory milieu of the different models, which involve distinct microglial phenotypes. Therefore, the development of CX3CR1 targeted therapy should be carefully considered based on the specific microglial marker of AD patient.
P2RX7
The purinergic receptor P2X7 (P2RX7), predominantly expressed in microglia, plays a critical role in neuro-inflammation, microglial phagocytosis, and AD pathogenesis [66]. Adenosine triphosphate (ATP) acts as the physiological agonist for P2RX7. The interaction between P2RX7 and ATP is believed to mediate various cell functions, such as cell permeability and phagocytosis. Previous studies have not only found that P2RX7 activation tightly correlates with pro-inflammatory effects, such as IL-1β release and reactive oxygen species (ROS) production, but that it also induces anti-inflammatory signals and enhances microglial phagocytosis in non-AD models [66]. The dual effects of P2RX7 on microglia can be summarized as follows: the short-term stimulation of P2X7R leads to neuroprotection, while chronic stimulation leads to neurotoxicity. Recently, an in vitro study using RNA interference demonstrated that silencing P2X7R significantly increased microglial phagocytosis of Aβ1–42. This effect was dependent on the rate of IL-1β release and the inhibition of the COX-2 pathway [67]. Furthermore, a study in AD transgenic mice demonstrated that P2RX7 inhibition significantly improved working memory and memory function, likely via the suppression of P2RX7-induced exosome secretion from microglia [41]. Despite having plenty of P2X7R antagonists, there are no ideal antagonists for AD treatment yet [66]. Interestingly, another study in 6-month-old APP/PS1 mice revealed that 40 Hz transauricular vagal nerve stimulation could attenuate the hippocampal amyloid load and improve spatial learning and memory by inhibiting P2X7R/NLRP3/Caspase-1 signaling [42]. Therefore, modulation of the P2RX7 effect could be a promising therapeutic target for AD treatment. However, further studies are needed to confirm the optimal administration time and detailed mechanisms of the P2RX7 effect due to its diverse roles.
CD33
CD33 is an inhibitory myeloid cell receptor that is exclusively expressed by microglia and infiltrating macrophages in the brain [68]. The activated CD33 receptor is usually associated with the inhibition of cellular phagocytosis, cytokine release, and the induction of apoptosis [68]. A study using CD33 knock-out mice indicated that the absence of CD33 reduced Aβ pathology and significantly improved cognition [43]. Furthermore, this study demonstrated that genes related to phagocytosis and pro-inflammatory factors such as IL-6 and IL-8 were upregulated in CD33 knock-out mice. Conversely, in TREM2 knock-out mice, these genes were downregulated, suggesting that TREM2 acts downstream of CD33 [43]. Emerging evidence has revealed that small molecules and CD33 antibodies, which target the activation sites of CD33, represent two potential avenues for intervention [68], although their efficacy in AD models has yet to be demonstrated. The targeting of CD33 through small molecules requires screening for both competitive inhibitors and allosteric modulators, which is particularly challenging. Notably, CD33 antibodies have undergone extensive human trials for the treatment of acute myeloid leukemia, demonstrating their effectiveness in reducing CD33 expression [69]. In summary, the inhibition of CD33 through gene therapy, small molecules, or antibodies, presents a promising avenue for novel therapeutic approaches in the treatment of AD, achieved through microglial modulation.
Nrf2
The overproduction of ROS creates a state of redox imbalance, potentially resulting in neuronal damage and the progression of AD [70]. The nuclear factor erythroid 2-related factor (NrF2) is considered the primary regulator of redox homeostasis, and its down-regulation typically increases cell susceptibility to ROS damage. A plenty of studies have demonstrated that Nrf2 dysfunction is significantly associated with the pathological exacerbation in AD [71]. In a recent study by An et al., it was reported that rhamnoside PL201 effectively activated the Nrf2 signaling cascade, leading to the upregulation of HO-1 and the downregulation of the NF-κB pathway [44]. After PL201 treatments, a significant reduction in the levels of pro-inflammatory cytokines, including IL-6, tumor necrosis factor-α (TNF-α), and IL-1β, was observed in AD mice. The anti-inflammatory action of PL201 is achieved through the downregulation of the NF-κB pathway and upregulation of HO-1, mediated by the induction ofNrf2 signaling. Moreover, a body of evidence has shown Nrf2 activation not only represses Aβ production but also reduces the level of p-tau [72]. Currently, several Nrf2 activators have been verified in preclinical studies of AD, and some Nrf2 activators have demonstrated a positive impact on AD animal models [72]. However, the Nrf2 pathway is complicated and the development of Nrf2 activators still faces many challenges. Taken together, targeting the Nrf2 signaling pathway holds promise as a therapeutic approach for AD treatment.
PPAR-γ/NF-κB
Peroxisome proliferator-activated receptors (PPARs), a subfamily of nuclear receptor transcription factors, and NF-κB are both closely associated with the regulation of inflammatory mediators in the pathogenesis of various inflammatory diseases such as AD and atherosclerosis [73]. EI-Din et al. recently demonstrated that rice bran extract (RBE) acting as a PPAR-γ agonist effectively shifted the microglial phenotype from pro-inflammatory to anti-inflammatory. This was evidenced by a significant decrease in the expression of the pro-inflammatory microglial marker CD45, alongside an increase in the expression of anti-inflammatory microglial and phagocytic markers such as arginase1 and CD36 [46]. Additionally, this study identified the therapeutic role of a PPAR-γ agonist via the enhancement of PPAR-γ expression and the limitation of NF-κB-dependent inflammation. Another study revealed that Bis(ethylmaltolato)oxidovanadium (BEOV), an important vanadium compound, could suppress the Aβ-induced activation of NF-κB signaling and upregulate the expression of PPAR-γ, in a dose-dependent manner [45]. These findings suggest that the upregulation of PPAR-γ and inhibition of NF-κB signaling may hold therapeutic potential in the treatment of AD. Despite good results of PPAR-γ agonists in animal models of AD, the results from human trials were less successful [74]. Thus, there is still a substantial need to understand the underlying molecular mechanisms of PPAR-γ function.
C5aR1
In a healthy brain, the complement system plays an important role in maintaining a robust immune system that effectively clears pathogens and cellular debris. C5a, one product of complement activation, exerts a pro-inflammatory effect through C5aR1, primarily expressed on microglia. Interestingly, a previous study observed a significant upregulation of C5a in microglia in close proximity to amyloid plaques [75]. The genetic ablation of C5aR1 has been reported to confer neuroprotective effects by inhibiting inflammatory genes and enhancing degradation pathways [76]. Gomez-Arboledas et al. recently demonstrated that PMX205, a specific C5aR1 antagonist, significantly decreased dystrophic neurites and amyloid load. Additionally, it shifted microglial gene expression towards a neuroprotective phenotype in the Tg2576 mouse AD model [47]. It is important to note that PMX205 has a high lipophilic nature and thus can easily gain access to the brain [77]. Furthermore, some antagonists of C5aR1 have been tested in human clinical trials for safety [77]. These reports support the proposition that inhibiting C5aR1 signaling could serve as a potential therapeutic strategy for reversing microenvironment deterioration through the polarization of microglial phenotype.
CSF1R
Colony-stimulating factor 1 receptor (CSF1R) is a tyrosine kinase transmembrane receptor that plays an important role in regulating microglial homeostasis within the CNS [78]. The two main ligands for CSF1R, colony stimulating factor 1 (CSF1) and IL-34, initiate downstream signaling pathways, contributing to microglia development and maintenance [79, 80]. However, CSF1R inhibition may result in a widespread depletion of microglia. Recent pharmacological studies have demonstrated that inhibiting CSF1R signaling with the CSF1R kinase inhibitor PLX3397 leads to a significant reduction in both amyloid plaque deposition and soluble amyloid oligomers. This intervention also resulted in a significant improvement in cognitive function in the 5×FAD mouse model [48]. In another study, the application of a CSF1R inhibitor (PLX5622) similarly resulted in a reduced plaque load, ameliorated neuritic dystrophy, and improved cognition in the 5×FAD mouse model [49]. Interestingly, this study found that plaques failed to form following microglial depletion, except in areas containing surviving microglia, whereas a recent study showed that PLX5622 is not microglia specific but also reducing the population of resident macrophages in other organs [81]. Therefore, despite the important role of microglia in initiating plaque pathogenesis, high attention should be paid to the specificity of these inhibitors in the future experimental studies.
NLRP3 inflammasome
The NOD, LRR, and pyrin-domain containing 3 (NLRP3) inflammasome of microglia plays an important role in neuro-inflammation [82]. Normally, it is required for the release of pro-inflammatory cytokines from microglia, which ultimately results in the increase of Aβ deposition [83]. In contrast, the application of NLRP3 inhibitor, such as MCC950, inhibited microglial activation, reduced Aβ accumulation and improved cognitive function in the APP/PS1 mouse model of AD [51]. Moreover, a recent study by Lonnemann et al. analyzed the effect of an oral NLRP3-specific inhibitor OLT1177, which showed inimical microglia were suppressed and cortical plaques reduced in six-month-old APP/PS1 mice [50]. In addition, it has been demonstrated that enhancing Aβ autophagy acts as the downstream effect of NLRP3 inhibition [84]. So far, at least five NLRP3 inflammasome inhibitors have shown high safety in human bodies [85]. Therefore, NLRP3 inflammasome plays a key role in the pathogenesis of AD and targeting NLRP3 inflammasome could be a novel approach for the disease modification.
SURVEILLANCE AND PHAGOCYTOSIS
In a healthy CNS, microglia in their resting state are highly dynamic, actively surveying the microenvironment through their extended or retracted processes, maintaining close contact with astrocytes, neurons, and blood vessels [9]. Furthermore, it has been demonstrated that microglial processes undergo cycles of de novo formation and withdrawal, while the somata of microglial cells remain in the same position [86]. The interactions between CD200 and its receptor (CD200R), as well as CD22-CD45, CD172A-CD47, and CX3CL1-CX3CR1, are reported to be associated with the microglial surveillance state [86]. Typically, CD22-CD45 and CD200-CD200R signals require direct cell-cell contact, while the CX3CL1-CX3CR1 signal can be transmitted through either cell-cell contact or the release of soluble CX3CL1 [87]. The inhibitory CD22-CD45 signaling in aged mice has been demonstrated to greatly enhance microglia surveillance capacity [39].
In addition to their role in microenvironment surveillance, microglia serve as the brain’s professional phagocytes. Toll-like receptors (TLRs) and TREM2 are the primary receptor types involved in microglial phagocytosis [86]. TLR scan recognize damage-associated molecular patterns (DAMPs), such as α-synuclein and Aβ fibrils [88]. TREM2, primarily located in microglia, mediates the protective phagocytosis of cell debris and suppresses the secretion of pro-inflammatory factors [89]. The upregulation of TREM2 facilitates the clearance of Aβ1–42 by binding to the DNAX activation protein 12 (DAP12) [36]. Furthermore, CX3CL1, also known as fractalkine, plays a role in suppressing microglial inflammatory responses, attracting microglia, and stimulating microglial phagocytosis by activating the CX3CR1 receptor [40, 90].
CLEARANCE OF TOXIC PRODUCTS (LYSOSOMAL DEGRADATION)
The main pathological features of AD include extracellular senile plaques, intracellular NFTs, and neural degeneration, resulting in microglia dysfunction, synaptic damage, and neuronal loss. These effects escalate during disease progression, concurrent with cognitive decline. The clearance of toxic products, such as Aβ and tau, is crucial for maintaining brain homeostasis. Enhancing microglial phagocytosis of Aβ oligomers plays a key role in Aβ clearance [91]. Moreover, p38 MAPK inhibition augments microglia-mediated tau phagocytosis [92]. Conversely, activated microglia possess the capability to degrade Aβ by producing proteolytic enzymes [9]. Recently, a study by Pluvinage et al. found that inhibiting CD22 leads to the reprogramming of microglia towards a homeostatic state. This intervention improves cognitive function by promoting the clearance of myelin debris, Aβ oligomers, and α-synuclein fibrils in aged mice [38]. Hickman et al. revealed that partial CX3CR1 deficiency leads to a significant reduction in Aβ levels and senile-like plaque load. This was accompanied by significantly increased levels of Aβ-degrading enzymes such as insulysin and matrix metalloproteinase 9 [40]. Furthermore, another study found that a PPAR-γ agonist reduced both Aβ42 deposition and p-tau protein levels [46]. These findings indicate that microglial reprogramming regulates the expression of Aβ degrading enzymes and enhances microglial phagocytosis. This contributes to the maintenance of a homeostatic microenvironment, leading to delayed disease progression and improved cognition in AD.
REDUCED PRODUCTION OF PRO-INFLAMMATORY MEDIATORS
It is believed that the self-perpetuating cycle between Aβ and pro-inflammatory mediators contributes to the progression of AD pathogenesis. The chronic deposition of Aβ leads to an increase of pro-inflammatory mediators, ultimately resulting in a disrupted microenvironment, tau hyperphosphorylation, and neuronal loss [93]. In return, the release of pro-inflammatory mediators exacerbates the production and accumulation of Aβ oligomers [9]. Thus, targeting the regulation of microglial pro-inflammatory cytokines might be a viable strategy for interrupting or reversing the disease course. He et al. recently demonstrated that a PPAR-γ inhibitor could effectively counteract Aβ-induced NF-κB activation, subsequently inhibiting the production of pro-inflammatory mediators, including TNF-α, IL-6, and IL-1β, in both in vivo and in vitro models [45]. An et al. found that rhamnoside PL201 significantly reduced the accumulation of activated microglia and suppressed the production of pro-inflammatory cytokines, including IL-6, TNF-α, and IL-1β, in APP/PS1 mice [44]. This anti-neuroinflammation effect was achieved through the enhancement of the Nrf2 signaling cascade, resulting in the upregulation of HO-1 and downregulation of the NF-κB pathway [44]. Moreover, Niklas et al. and Dempsey et al. demonstrated that inhibition of NLRP3 inflammasome leads to decreased release of pro-inflammatory factors such as IL-1β and reduced Aβ accumulation in APP/PS1 mice [50, 51]. Therefore, inhibiting the release of pro-inflammatory cytokines from microglia may offer a viable strategy for maintaining the microenvironment homeostasis.
CHANGE IN MICROGLIAL PHENOTYPE
The phenotypes adopted by activated microglia are highly contingent on the surrounding microenvironment and the progression of AD pathogenesis. Typically, ineffective clearance of Aβ results in a neurotoxic microglial phenotype, while pro-inflammatory microglia exacerbate the deterioration of the microenvironment. Conversely, anti-inflammatory microglia contribute to a reduced accumulation of Aβ plaques and an overall improvement in the microenvironment. A growing body of evidence has demonstrated that shifting microglial polarization from a pro-inflammatory to an anti-inflammatory phenotype reduces neuro-inflammatory reactions, Aβ deposits, and tau hyperphosphorylation in AD [94]. In recent years, a new disease-associated reactive microglia phenotype known as DAM has been discovered [23]. Microglia undergo a transition from a homeostatic phenotype to a DAM population, often accompanied by specific alterations in their gene expression [23]. In diseased conditions, microglial activation is characterized by two distinct states: the pro-inflammatory Stage I and the anti-inflammatory Stage II DAM [95]. TREM2 is known to play a critical role in inducing the anti-inflammatory activation of microglia, transitioning them form a pro-inflammatory state [96]. Furthermore, studies have shown that anti-inflammatory cytokines can upregulate TREM2 expression [97], whereas pro-inflammatory cytokines downregulate TREM2 expression [98]. Recent work by EI-Din et al. demonstrated that the decreased expression of the pro-inflammatory signaling pathway NF-κB and the pro-inflammatory microglial marker CD45 could shift the microglial phenotype from a pro-inflammatory to an anti-inflammatory state [46].
THE NIME AS A TARGET FOR STEM CELL THERAPY
Based on the compiled body of research, it is plausible that targeted remodeling of the microenvironment, specifically through microglial intervention, could establish a favorable milieu for stem cell treatment. However, the efficacy of this approach may depend on the stage of the disease and the associated signaling pathways. Typically, the pro-inflammatory phenotype of activated microglia dominates in the middle and later stages of AD progression, while the anti-inflammatory phenotype is more prominent in the early stages [99]. Similarly, the microenvironment is primarily compromised when the pro-inflammatory phenotype predominates. Therefore, the innovative combination of microenvironment remodeling and stem cell transplantation may find greater applicability in the middle and later stages of AD. Another aspect to consider is that different signaling pathways engender diverse microglial functions, subsequently shaping the characteristics of the microenvironment in AD. For example, P2X7R, CX3CR1, IL-3Rα, CD22, PPAR-γ, and TREM2 have been shown to enhance Aβ clearance [37, 54], while Nrf2, NLRP3, and PPAR-γ downregulate the expression of pro-inflammatory cytokines[44, 50], leading to improved cognitive outcomes in AD mouse models. Thus, the strategy of microenvironment remodeling encompasses enhanced cell viability, increased Aβ phagocytosis, suppressed pro-inflammatory responses, and restored microglia surveillance capacity. Collectively, these establish a foundation for future stem cell therapy.
CONCLUSION AND FUTURE PERSPECTIVES
AD is a complicated disease without effective disease-modifying treatment. Although stem cell therapy has exhibited encouraging results in animal models of AD, the dampened survival and integration of stem cells due to the hostile microenvironment has prevented the application of this technique to the treatment of AD patients. Since the discovery of microglial activation and its role in microenvironment alteration of AD, the exploration of treatment strategies centered on microglia has emerged as a hottopic. In this review, we teased out some critical signaling pathways of microglia which are involved in the regulation of senescent microenvironment of AD, including TREM2, IL-3Rα, CD22, C5aR1, CX3CR1, P2X7R, CD33, Nrf2, PPAR-γ, CSF1R, and NLRP3. In light of the effectiveness and feasibility in AD patients, TREM2, CD22, CD33, C5aR1, and NLRP3 should be given priority over others. The therapeutic effects of these signaling pathways of microglia are mainly manifested as the following: a reduction in neuro-inflammatory reactions, decreased Aβ deposits and tau levels, and an enhancement in microglial surveillance and phagocytosis. In conclusion, the combined treatment of microglial modulation and stem cell therapy in AD might be a critical consideration to achieve promising results in the future.
AUTHOR CONTRIBUTIONS
Zhiwei Shen (Conceptualization; Data curation; Formal analysis; Methodology; Project administration; Writing – original draft); Xinyi Yang (Investigation; Methodology; Software); Yulong Lan (Supervision; Validation; Writing – review & editing); Gao Chen (Funding acquisition; Supervision; Validation; Writing – review & editing).
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
This work was supported by the Key Research and Development Project of China (No. 2018YFA0108603).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.