
Abstract
Background:
HMGCS2 (mitochondrial 3-hydroxy-3-methylglutaryl-COA synthase 2) plays a pivotal role as a control enzyme in ketogenesis, and its association with the amyloid-β protein precursor (AβPP) in mitochondria implicates a potential involvement in Alzheimer’s disease (AD) pathophysiology.
Objective:
Our study aimed at identifying repurposed drugs using the DrugBank database capable of inhibiting HMGCS2 activity.
Methods:
Exploiting the power of drug repurposing in conjunction with virtual screening and molecular dynamic (MD) simulations against ‘HMGCS2’, we present new in-silico insight into structure-based drug repurposing.
Results:
The initial molecules were screened for their binding affinity to HMGCS2. Subsequent interaction analyses and extensive 300 ns MD simulations were conducted to explore the conformational dynamics and stability of HMGCS2 in complex with the screened molecules, particularly Penfluridol and Lurasidone.
Conclusions:
The study revealed that HMGCS2 forms stable protein-ligand complexes with Penfluridol and Lurasidone. Our findings indicate that Penfluridol and Lurasidone competitively bind to HMGCS2 and warrant their further exploration as potential repurposed molecules for anti-Alzheimer’s drug development.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is an overwhelming challenge in modern drug discovery and healthcare management [1]. AD is characterized by its complex pathophysiology and the escalating global burden it imposes [2]. It is the most general cause of dementia and develops slowly where the symptoms worsen over time [3]. The accumulation of abnormal protein deposits in the brain is one of the major hallmark features of AD [4]. To date, there is presently no therapy for AD where available medications focus on the management of symptoms and improvement of quality of life [5]. The imperative to innovate therapeutic strategies has led to the convergence of computational methodologies, drug repurposing paradigms, and targeted exploration of specific molecular pathways [6]. Among the intricate molecular networks implicated in AD, mitochondrial 3-hydroxy-3-methylglutaryl-COA synthase 2 (HMGCS2) has emerged as a promising target, governing ketogenesis and demonstrating noteworthy interactions with the amyloid-β protein precursor (AβPP) within the mitochondria [7, 8].
Computational approaches have played a key role in medicinal chemistry becoming increasingly important and pivotal over the years. These offer valuable insights to drug design and discovery process for various diseases ranging from COVID-19 [9] to cancer therapeutics [10]. Virtual screening has been an indispensable facet of lead identification that offers a systematic and efficient way to navigate through large chemical libraries [11]. Virtual screening accelerates the drug discovery pipeline while employing structure-based methodologies [12]. It helps in optimizing the identification of bioactive molecules and curtailing the time and resource investments traditionally associated with drug development [13]. Recent studies underscore the efficacy of structure-based virtual screening in identifying potent inhibitors of different protein targets [14, 15]. In this study, we adopt a thorough virtual screening approach while exploiting the DrugBank database of FDA-approved drugs. The initial phase involves a structure-based molecular docking study where the top initial hits were selected based on their binding affinities towards HMGCS2.
The identified molecules then undergo a detailed interaction analysis and the PASS evaluation [16]. This multifaceted approach concludes with the identification of two molecules demonstrating compelling anti-AD potential. To deepen our understanding of complex molecular dynamics (MD), we engage in MD simulations spanning an extensive 300 nanoseconds (ns). These simulations unravel the dynamic conformational changes within the HMGCS2-ligand complexes that shed light on their stability and interaction dynamics at an atomic level. Taken together, this study identified two repurposed HMGCS2 modulators that can significantly contribute to the ongoing pursuit of novel therapeutic avenues for AD. These molecules stand as promising candidates for further exploration and development.
MATERIALS AND METHODS
Molecular docking screening
Molecular docking-based virtual screening was used to identify molecules with high binding potential towards HMGCS2. Various computational tools such as MGL AutoDock tools [17], InstaDock [18], PyMOL [19], and Discovery Studio Visualizer [20] were utilized for the docking and interaction evaluation. Three-dimensional structural coordinates of HMGCS2 were obtained from the Protein Data Bank [21] (Accession number: 2WYA), resolution of 1.70 Å and processed for docking study using InstaDock and AutoDock tools. In the preprocessing, the remodeling of missing residues, addition of hydrogens to polar atoms, and assignment of appropriate atom types were performed. A library of repurposed drugs sourced from the DrugBank database was compiled and processed in InstaDock v1.2. The docking simulations were conducted in InstaDock with grid size set to 84, 73, and 70ringA, centrally positioned at –3.378, 40.661, and 10.703 for the X, Y, and Z axes, respectively. After the docking study, all the compounds were screened for their binding affinity and shortlisted for further analysis.
Biological potential and interaction analysis
The biological properties of the chosen molecules were examined using the PASS web server [16]. It evaluates a molecule’s potential by comparing its structure with an integrated training set that encompasses various types of biological properties. It predicts the biological properties of molecules based on their structure-activity relationships. The PASS prediction yields a concise summary of potential biological activities, presented in terms of the ‘probability to be active (Pa)’ and ‘probability to be inactive (Pi).’ A higher Pa value indicates an increased likelihood of a molecule exhibiting the explored biological property. After the PASS analysis, the interactions and the binding prototype of the screened molecules were examined. Polar contacts between the selected molecules and HMGCS2 were visualized in PyMOL [19]. Further, Discovery Studio Visualizer was used for the in-depth exploration of potential interactions of molecules within the HMGCS2 binding pocket. Specifically, molecules demonstrating interactions with critical residues, specifically the substrate binding, Ser258 were selectively chosen for further investigation.
MD simulations
MD simulation is a powerful tool for studying protein-ligand interactions at an atomic level [22]. We performed simulations while utilizing classical mechanics principles at 300K through the GROMACS 2020 beta simulation suite [23]. The CHARMM36-JUL2022 force field [24] was employed to simulate the HMGCS2 systems in both free and ligand-bound forms with the screened molecules. Topology and force field parameters for the screened molecules were generated using the PRODRG web server [25]. Each system was centered in a virtual cubic box while maintaining a distance of 10ringA from the box edges. The solvation of each system in aqueous surroundings was achieved by adding water molecules using the TIP3P solvent model [26]. An appropriate number of counterions were introduced for charge neutrality. For the energy minimization process, we utilized the steepest descent approach for all systems. Subsequently, NVT and NPT ensembles were applied during the position restraint procedure. A 300 ns simulation was conducted for each system, and the resulting trajectories were analyzed through the inbuilt GROMACS utilities.
Principal component and free energy landscape analyses
The exploration of protein folding dynamics was carried out through the application of principal component analysis (PCA), which shed light on the fundamental movements within proteins [27]. PCA, coupled with free energy landscape (FEL) analysis, was conducted on the simulated trajectories. PCA is a mathematical method based on covariance matrices, that facilitates the exploration of the conformational sampling of HMGCS2 and its interactions with the screened molecules. The FELs generated through the PCA matrices are useful for understanding the folding behavior of HMGCS2 in both free and ligand-bound states.
RESULTS
Molecular docking screening
Molecular docking involves predicting the preferred orientation and conformation of a ligand when bound to the binding site of a protein receptor [28]. In this study, a curated set of 3500 FDA-approved drug molecules was retrieved from the DrugBank repository [29]. The docking screening was conducted using InstaDock to identify high-affinity binders of HMGCS2. After the docking of all molecules in the library, the top 10 were fetched out based on their estimated docking scores towards HMGCS2 (Table 1). Notably, the analysis revealed that these selected molecules exhibited substantial affinities with HMGCS2. The docking score for the top 10 hits was ranging from –10.7 to –11.2 kcal/mol. The docking score provides a measure of how well a particular ligand binds to the target protein. Lower docking scores typically indicate stronger binding affinity. All the selected molecules showed appreciable binding affinity towards HMGCS2, which indicated the potential as competitive binders. This underscores the potential of these molecules as promising candidates for further investigation and development as HMGCS2 inhibitors.
List of screened hits against HMGCS2 and their docking parameters
PASS analysis
The PASS server serves as the foundation for predicting the biological activity of a given molecule [16]. In this study, we applied the PASS analysis to predict the biological activity of molecules passed through the docking screening. Notably, four molecules, Penfluridol, Risperidone, Ziprasidone, and Lurasidone, emerged as positive hits in the PASS screening, indicating their potential in desired biological activities (Table 2). The results unveiled that all these molecules exhibit promising anti-AD potential. Importantly, the likelihood of a molecule succeeding the expected biological property is considered high when the Pa is higher than the Pi. Penfluridol, Risperidone, Ziprasidone, and Lurasidone displayed elevated predictions for antineurotic, antipsychotic, neurodegenerative diseases treatment, dementia treatment, nootropic, cognition disorders treatment, and AD treatment potential, with Pa values ranging from 0.126 to 0.954. The results of the PASS analysis highlighted the potential of Penfluridol, Risperidone, Ziprasidone, and Lurasidone as molecules with favorable biological activities, which can further be explored in drug repurposing against HMGCS2.
PASS analysis of the selected molecules with their predicted activity
Interaction analysis
The screened molecules from the PASS analysis underwent a detailed interaction analysis with HMGCS2. All four molecules were splitted into their respective docked conformations and subjected to pose selection in PyMOL based on the substrate binding site of HMGCS2 (Fig. 1). Here, the docked conformers exhibiting direct interactions with the substrate binding site, i.e., Ser258, were selected for further analysis. This site is responsible for the substrate recognition and binding in HMGCS2. Notably, two out of four molecules, Penfluridol and Lurasidone, formed direct hydrogen binds with Ser258 of HMGCS2. Interaction analysis revealed that Penfluridol and Lurasidone shared interactions with key residues within the HMGCS2 binding site (Fig. 1A). Conformation analysis demonstrated that Penfluridol and Lurasidone exhibited consistent interactions with important amino acids of the HMGCS2 binding site (Fig. 1B). Both molecules effectively obstruct the binding site of HMGCS2 and fit snugly into the deep cavity with commendable complementarity (Fig. 1C). Overall, this analysis indicated that Penfluridol and Lurasidone’s stability could impede HMGCS2 accessibility, suggesting potential functional inhibition.
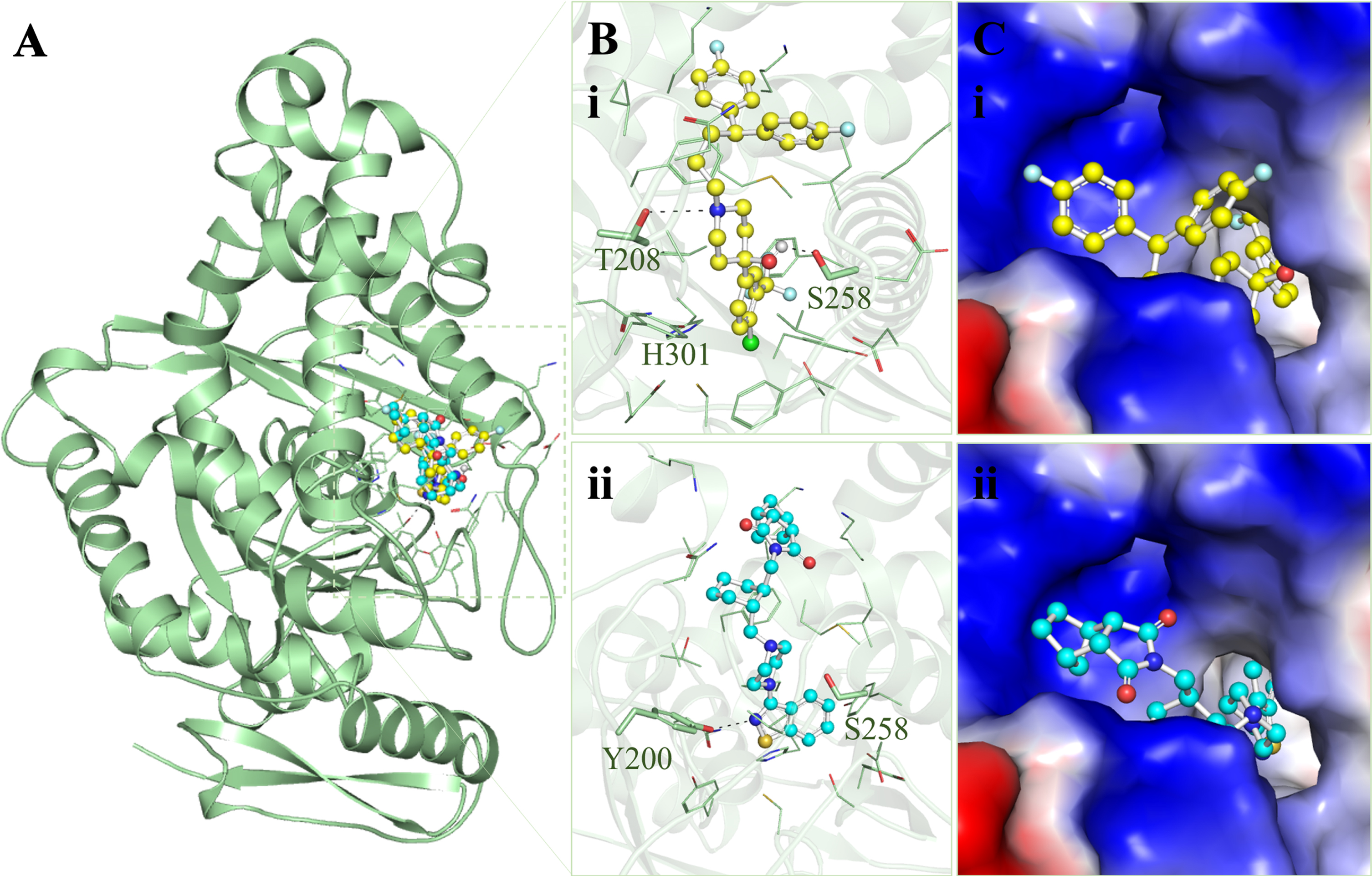
Protein-ligand interactions between HMGCS2 and the screened molecules. A) Ribbon diagram of HMGCS2 with Penfluridol (yellow) and Lurasidone (cyan). B) Magnified cartoon representation of HMGCS2 with the elucidated molecules, (i) Penfluridol and (ii) Lurasidone. C) Charged view of HMGCS2 binding pocket occupied by the elucidated molecules, (i) Penfluridol and (ii) Lurasidone.
The detailed exploration of the selected binding modes of Penfluridol and Lurasidone delved into detailed interactions with HMGCS2. These analyses provided insights into the specific types of interactions and their binding residues. The two-dimensional plots depict all interactions for Penfluridol and Lurasidone are illustrated in Fig. 2. The plots showed that Penfluridol and Lurasidone interacted with the substrate binding site Ser258 and shared common interactions (Fig. 2A-B). The analysis affirms that Penfluridol and Lurasidone exhibit a comparable binding pattern with several common interactions. Both molecules demonstrated multiple interactions with important amino acids in the binding site of HMGCS2. This consistent interaction profile supports the potential of Penfluridol and Lurasidone as promising candidates for further exploration and development as HMGCS2 inhibitors.
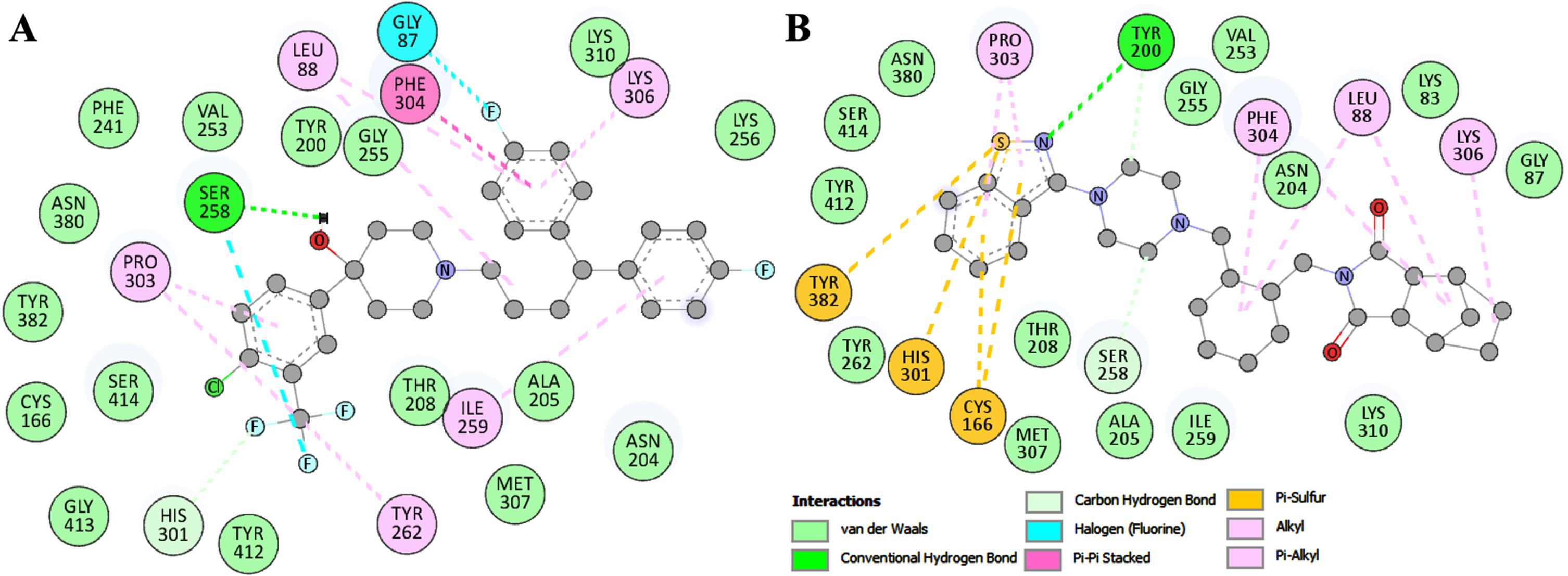
Binding residues of HMGCS2 and their interactions with (A) Penfluridol and (B) Lurasidone.
MD simulations
MD simulations are useful to refine the protein-ligand docking models and gain insights into the structural dynamics and stability [30]. We performed MD simulations of HMGCS2 and its complexes with Penfluridol and Lurasidone. The simulations were performed under explicit solvent conditions for 300 ns. The root mean square deviation (RMSD) analysis is a crucial parameter for studying structural deviations in proteins over time [31]. The RMSD analysis revealed consistent and small fluctuations in the backbone atoms of HMGCS2, HMGCS2-Penfluridol, and HMGCS2-Lurasidone complexes throughout the 300 ns trajectory (Fig. 3A). The analysis showed minimized deviation, particularly in the ligand-bound states with equilibrium reached and sustained stability observed across all three systems (Fig. 3A, upper). The probability density function (PDF) plot further illustrated lower RMSD and increased stabilization in HMGCS2 dynamics when in a ligand-bound state that affirms the stability enhancement conferred by Penfluridol and Lurasidone (Fig. 3A, lower).
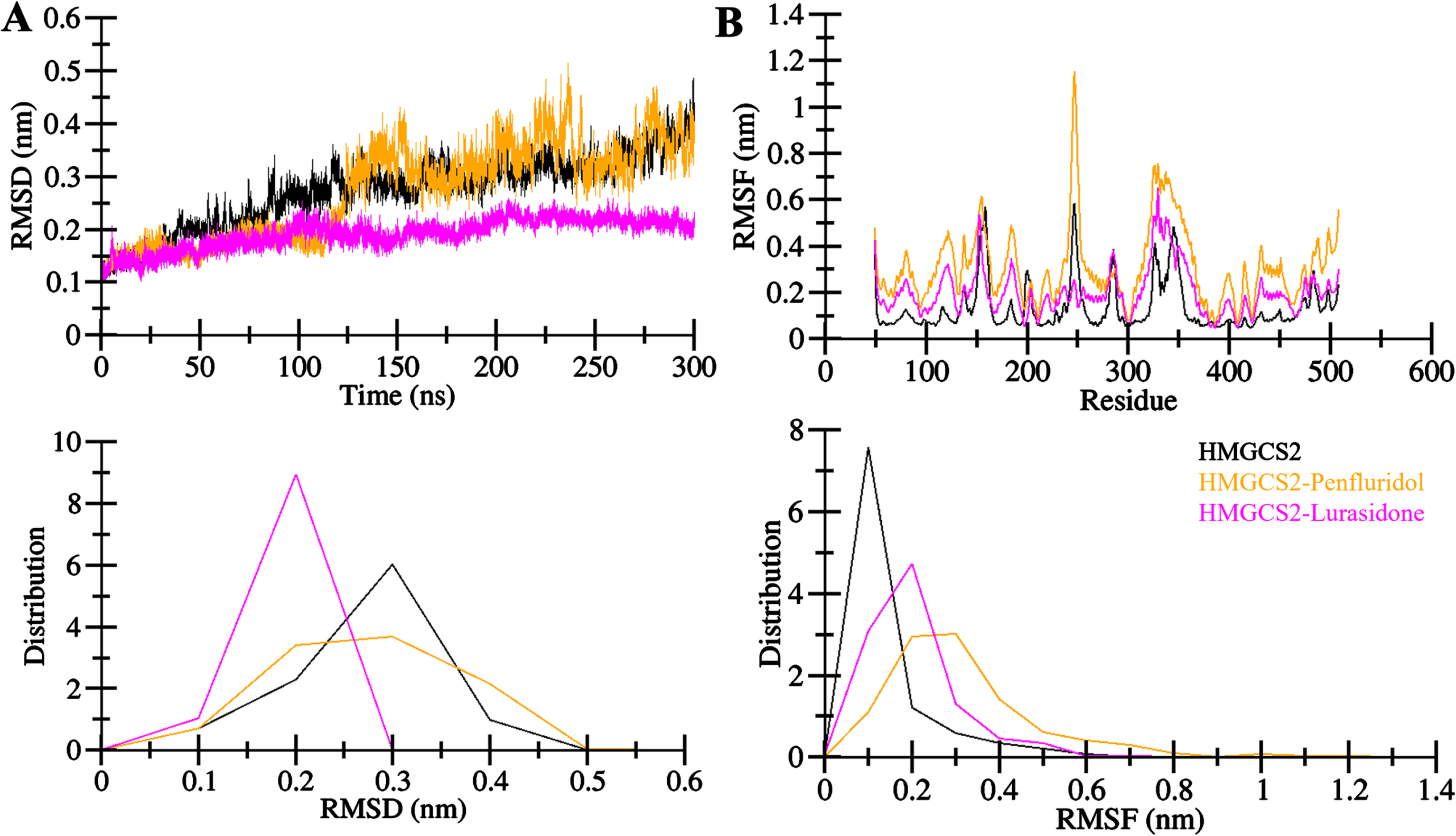
Structural dynamics in HMGCS2 upon Penfluridol and Lurasidone binding. A) RMSD plot of HMGCS2 in complex with Penfluridol and Lurasidone. B) RMSF plot of HMGCS2 and its complex with Penfluridol and Lurasidone. The lower panels show the distribution of the values as PDF.
The root mean square fluctuations (RMSFs) analysis demonstrated a similar pattern in the simulation across all systems, HMGCS2, HMGCS2-Penfluridol, and HMGCS2-Lurasidone (Fig. 3B). The reduced and stable residual fluctuations upon Penfluridol and Lurasidone binding indicated enhanced stability in the complexes, particularly in the residues within the HMGCS2 binding pocket interacting with the ligands (Fig. 3B, upper). However, a few residues in the ligand bound HMGCS2 complexes exhibited higher fluctuations, with lower PDFs that suggest internal residual vibrations in the protein simulations (Fig. 3B, lower). Overall, RMSFs confirmed the ability of Penfluridol and Lurasidone to form stable complexes with HMGCS2 during MD.
The radius of gyration (Rg) is a useful metric to evaluate the compactness and folding of a protein structure [32]. The Rg distribution showed stable and consistent values between 2.21 nm and 2.31 nm for HMGCS2 in the presence of Penfluridol and Lurasidone throughout the simulation trajectory (Fig. 4A). Relative results indicated that the structural dynamics and folding of HMGCS2 remained constantly stable post-Penfluridol and Lurasidone binding. The HMGCS2-Lurasidone complex showed higher compactness during the simulation (Fig. 4A, upper). The PDF plot also supported these findings, showing a lower distributed pattern of Rg values for HMGCS2 in ligand-bound states compared to the free state (Fig. 4A, lower). The analysis of solvent-accessible surface area (SASA) further contributed to understanding the protein folding degree. SASA analysis indicated that the HMGCS2 structure remained relatively stable, and more compact in the presence of Penfluridol and Lurasidone (Fig. 4B, upper). The distribution pattern of SASA values mirrored that of Rg, indicating a lower equilibration pattern across complex systems without compromising overall folding and compactness. The PDF plot for SASA confirmed this consistent pattern in HMGCS2 and its complexes with Penfluridol and Lurasidone (Fig. 4B, lower). Overall, the analysis provided a comprehensive understanding of the dynamic behavior, stability, and structural details of HMGCS2 in both free and ligand-bound states with Penfluridol andLurasidone.
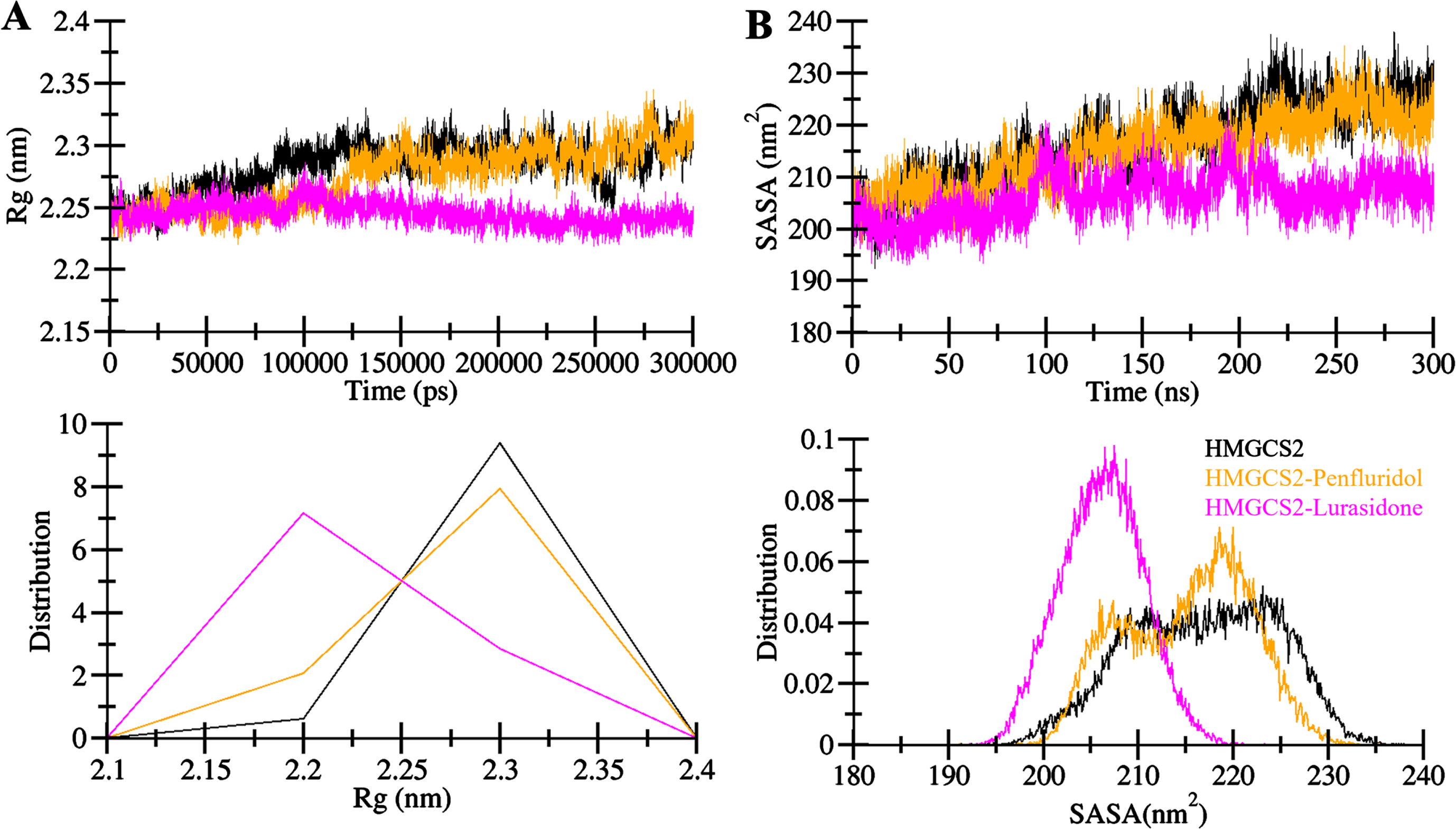
Structural compactness of HMGCS2 upon Penfluridol and Lurasidone binding. A) The Rg and B) SASA plot of HMGCS2 with Penfluridol and Lurasidone binding.
Dynamics of hydrogen bonds
The stability and conformational changes in a protein structure are significantly influenced by intramolecular hydrogen bonds [33]. Hydrogen bonds play a pivotal role in maintaining the overall folding and conformational arrangement of the protein structure [34]. The examination of intramolecular hydrogen bonds provides valuable insights into the protein’s conformational changes [35]. In this study, we calculated the time evolution of the number of intramolecular hydrogen bonds formed within HMGCS2. This analysis allowed us to assess the constancy of intramolecular bonding in HMGCS2 before and after the binding of Penfluridol and Lurasidone. Figure 5 depicts the fluctuation in the number of intramolecular hydrogen bonds within HMGCS2 throughout the MD simulations. The plot reveals that the hydrogen bonds formed within HMGCS2 were persistent, contributing to the stability of the protein structure (Fig. 5A). A slight increase in intramolecular hydrogen bonds within the HMGCS2-Penfluridol and HMGCS2-Lurasidone complexes suggests enhanced compactness compared to the free HMGCS2 structure, aligning with the findings from the dynamic study. The PDF representation of intramolecular hydrogen bonds in all three systems further supports the observation of fair constancy during the simulation (Fig. 5B). This analysis highlights the role of Penfluridol and Lurasidone in stabilizing the intramolecular hydrogen bonding pattern within HMGCS2 which results in higher compactness of the protein structure.
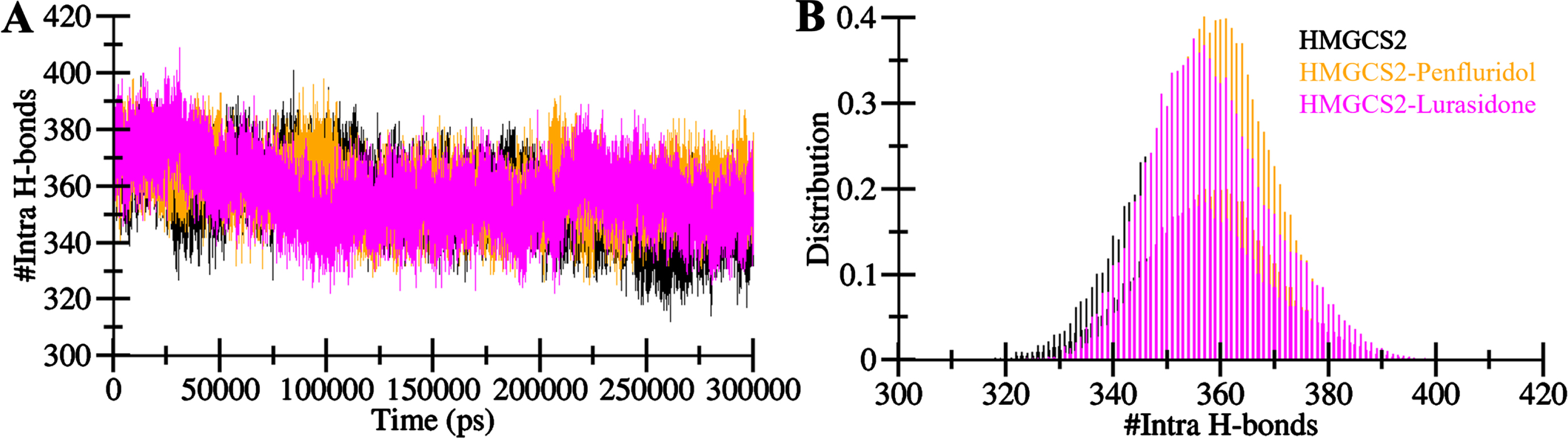
Intramolecular hydrogen bonds in HMGCS2. A) Visualization of intra-HMGCS2 hydrogen bonds. B) Probability density function (PDF) depicting the distribution of intramolecular hydrogen bonds within HMGCS2.
Further, to estimate the stability of the protein-ligand complexes of HMGCS2 with Penfluridol and Lurasidone, the investigation extended to the time evolution of intermolecular hydrogen bonds (Fig. 6). The count of hydrogen bonds formed within the Penfluridol-HMGCS2 was 1–6 with less stability and 1–2 with higher stability (Fig. 6A, upper panel). For Lurasidone-HMGCS2 complex, 1 to 3 hydrogen bonds were formed, with stability of 1 hydrogen bond (Fig. 6B, upper panel). Notably, the PDF plot exhibited consistently high probabilities for at least one hydrogen bond formation in both complexes (Fig. 6, lower panel). The formation of enduring docked complexes relies on the presence of intermolecular hydrogen bonding between the protein and ligands. The findings indicated that Penfluridol and Lurasidone retained their initial docking conformations on HMGCS2 throughout the simulation, facilitated by a stable network of intermolecular hydrogen bonds. Overall, the intermolecular hydrogen bonds corroborate the stability and robustness of the docked complexes of HMGCS2 with Penfluridol and Lurasidone indicating the effective interaction and sustained binding of these ligands to HMGCS2 over the simulation duration.
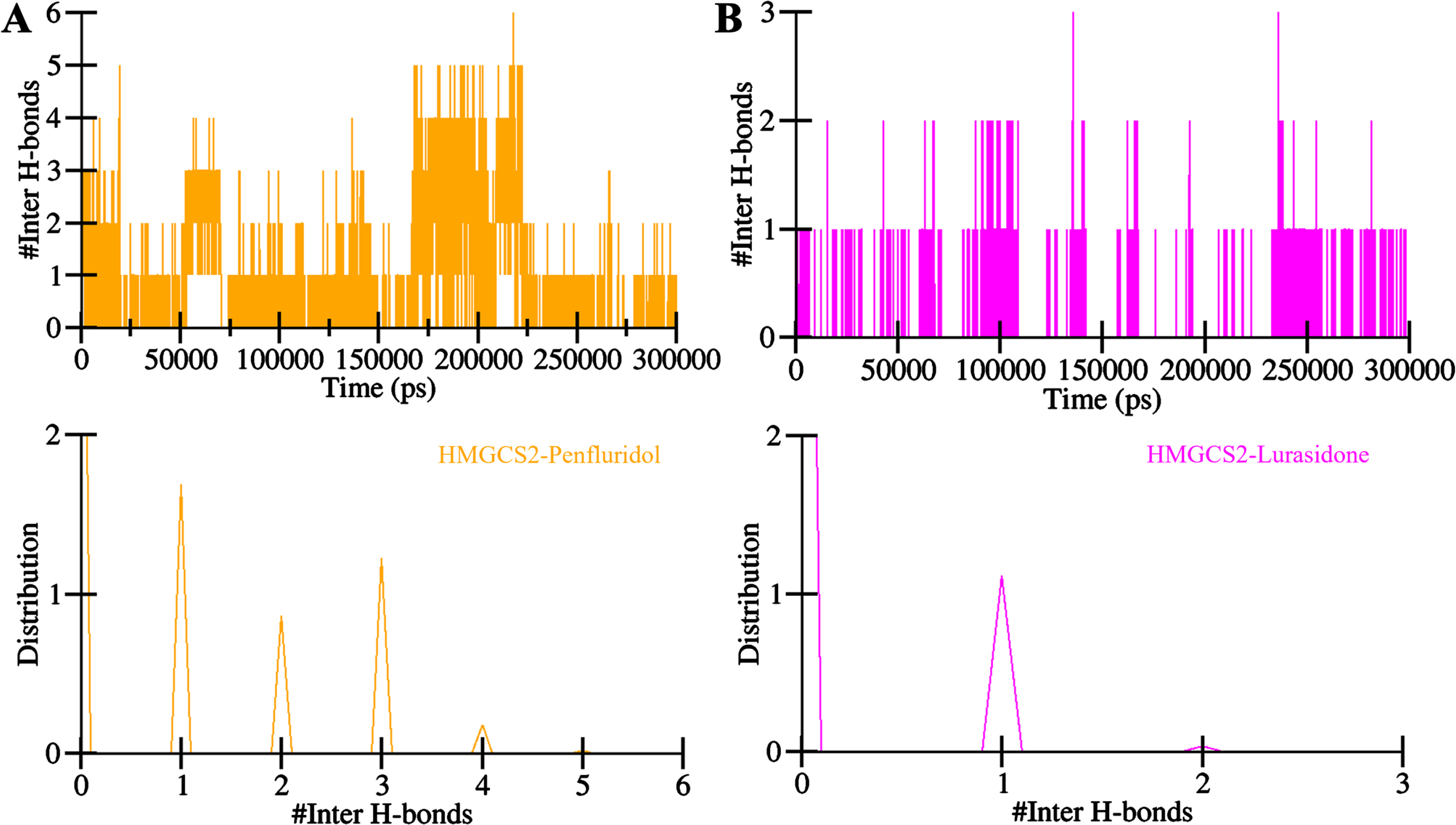
Intermolecular hydrogen bonding plots of HMGCS2 with A) Penfluridol and B) Lurasidone.
Principal component and free energy landscape analyses
To explore the collective motions and conformational sampling of HMGCS2, as well as its complexes with Penfluridol and Lurasidone, PCA was employed from the simulated trajectories. The trajectory through the Cα atoms of all three systems (HMGCS2, HMGCS2-Penfluridol, and HMGCS2-Lurasidone) in the essential subspace is presented in Fig. 7. The plot illustrates that the HMGCS2-Penfluridol complex occupies almost the same essential subspace as free HMGCS2 (Fig. 7A). In contrast, the HMGCS2-Lurasidone complex showed a dense cluster of conformations landscape confined to a smaller subspace compared to the HMGCS2 and HMGCS2-Penfluridol complex. Specifically, the HMGCS2-Lurasidone complex aligns closely with free-HMGCS2 in both eigenvectors (EVs) (Fig. 7B). Notably, the reduced flexibility in the essential subspace covered by the HMGCS2-Lurasidone complex suggests increased stability during the simulations.
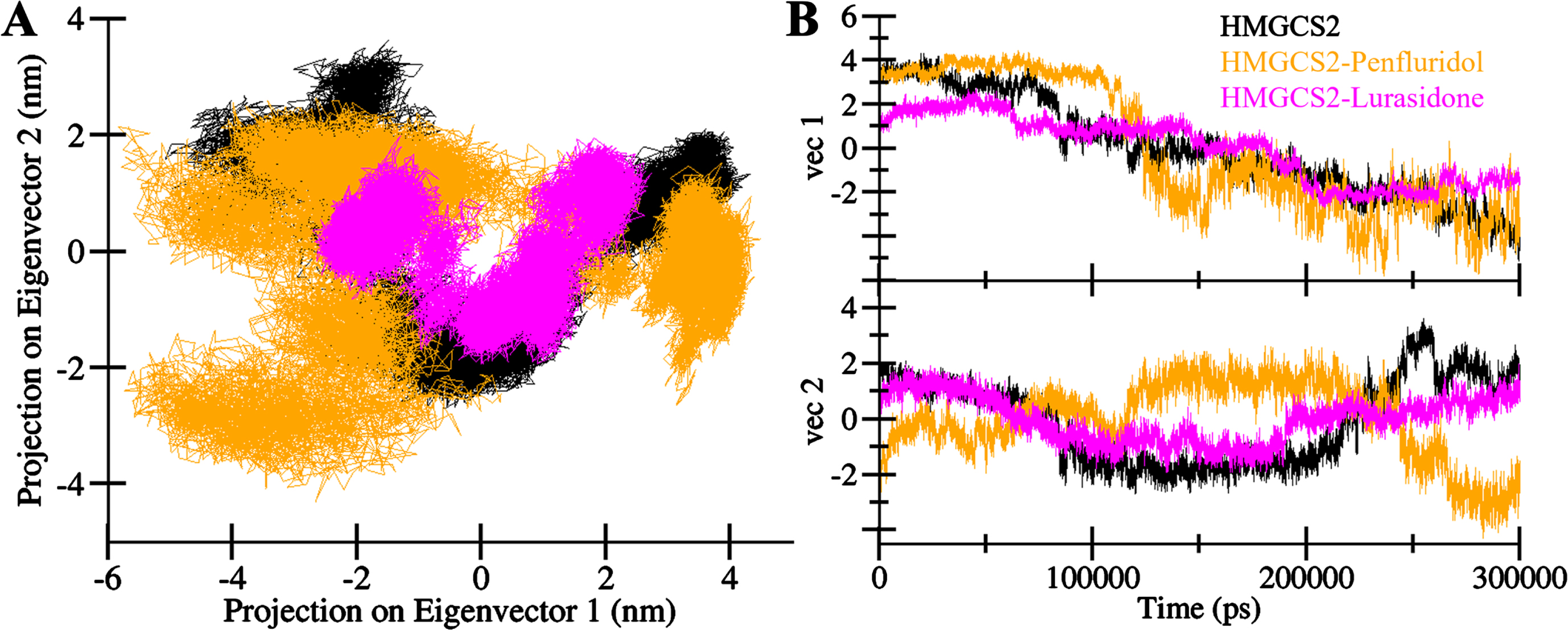
Principal component analysis. A) 2D projection of HMGCS2, HMGCS2-Penfluridol and HMGCS2-Lurasidone. B) Time evolution of the trajectories.
Further, FELs from the PCA matrices were constructed to depict the energy minima and conformational landscape of HMGCS2, HMGCS2-Penfluridol, and HMGCS2-Lurasidone complexes using two principal components (PCs). The contoured maps for FELs are elucidated in Fig. 8. Deeper blue regions in the FELs signify conformations with lower energy near-native states. The FEL analysis indicates that Penfluridol and Lurasidone binding with HMGCS2 induces slight alterations in the magnitude and position of the phases within 2–3 stable global minima. HMGCS2 in the free state primarily exists in 2–3 global minima expanding to 3–4 basins. Similarly, HMGCS2-Penfluridol and HMGCS2-Lurasidone complexes adopt an almost similar number of states, with smaller global minima and 2–3 local basins with varying populations and magnitudes (Fig. 8B-C). Collectively, the results of HMGCS2 in complex with Penfluridol and Lurasidone demonstrate stability with minimal conformational switching observed during the 300 ns simulations. The close alignment of the complexes within the essential subspace and the alterations in the FELs suggest that Penfluridol and Lurasidone binding influences the energy landscape of HMGCS2 which contributes to enhanced stability and structural integrity.

Free energy landscape plots of (A) free HMGCS2, (B) HMGCS2-Penfluridol, and (C) HMGCS2-Lurasidone.
DISCUSSION
HMGCS2 emerges as a promising drug target in AD which offers a potential avenue for therapeutic intervention. Exploiting drug repurposing methodologies proves instrumental in contemporary drug discovery efforts that present a cost-effective and expedited route to identify novel treatments for AD and other challenging conditions. In this study, we employed an integrated approach to drug repurposing to identify potential repurposed drug molecules that can act as potential HMGCS2 inhibitors. Our approach commenced with the screening of the DrugBank database with HMGCS2 inhibitory potential. Initially, a structure-based molecular docking strategy was employed to prioritize molecules that demonstrated a preference for binding to the binding site of HMGCS2. The selected molecules underwent predictions for anti-AD activity using the PASS server. Interaction analysis was then conducted to pinpoint molecules that were specifically bound to critical residues within the HMGCS2 active site. We conducted extensive 300 ns MD simulations on HMGCS2 and its docked complex with Penfluridol and Lurasidone to evaluate the stability and structural dynamics. The MD results revealed high stability of HMGCS2 in the presence of Penfluridol and Lurasidone throughout the simulation period. However, the lack of experimental validation in this study is a potential limitation that emphasizes the need for future investigations to confirm and further elucidate the proposed inhibitory activity of Penfluridol and Lurasidone. Despite this limitation, our study provides a robust indication that Penfluridol and Lurasidone hold promise as potential leads for the development of HMGCS2 inhibitors.
AUTHOR CONTRIBUTIONS
Anas Shamsi (Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Supervision; Validation; Visualization; Writing – original draft); Mohammad Furkan (Investigation; Methodology; Writing – original draft); Mohd Shahnawaz Khan (Data curation; Formal analysis; Methodology; Writing – review & editing); Dharmendra kumar Yadav (Formal analysis; Funding acquisition; Methodology; Validation; Writing – review & editing); Moyad Shahwan (Data curation; Formal analysis; Investigation; Methodology; Writing – original draft).
Footnotes
ACKNOWLEDGMENTS
MSK acknowledge and extend their appreciation to the Researchers Supporting Project Number (RSP2024R352), King Saud University, Riyadh, Saudi Arabia for funding this study. A.S. is grateful to Ajman University, UAE for supporting this publication.
FUNDING
MSK acknowledge and extend their appreciation to the Researchers Supporting Project Number (RSP2024R352), King Saud University, Riyadh, Saudi Arabia for funding this study. A.S. is grateful to Ajman University, UAE for supporting this publication.
CONFLICT OF INTEREST
Anas Shamsi is an Editorial Board Member of this journal but was not involved in the peer-review process of this article nor had access to any information regarding its peer-review.
The authors have no conflict of interest to report.
DATA AVAILABILITY
The data supporting the findings of this study are available within the article and its supplementary material.