
Abstract
The anharmonic force field and spectroscopy constants for phosphaethene are determined using density functional theory with B3PW91 and B3LYP functions employing cc-pVTZ and cc-pVQZ basis sets, respectively. Molecular structure, dipole moment and rotational spectroscopic constants are compared with obtainable experimental data. Most of the DFT results are close to observations for centrifugal distortion constants both in A-reducted and S-reducted form. The equilibrium states rotational constants computed by B3LYP/cc-pVQZ level of theory are in good agreement with observations, about only 0.07% higher than experimental ones. Vibrational spectroscopy constants and force constants are predicted by B3LYP. Vibrational frequencies, S-reduction quartic centrifugal distortion constants and rotational constants for several isotopomers of phosphaethene (13CH2PH, CD2PH, CH2PD, and CD2PD) are also calculated at the same levels. The isotopic effects of substitution by atom D are stronger than substitution by atom 13C for some rotational spectroscopic parameters.
Introduction
The molecules containing C = N or C = P double bond play an important role in organic chemistry [1]. Their geometry structures and chemical characters are affected by the nitrogen or phosphorus atom. Like their nitrogen analogs, the C = P double bond is unstable too [2]. Although the CX2 = NY and CX2 = PY (X, Y = H, F, Cl, Br) are two series of the unstable molecules, these molecules are interesting from the geometry structural, NMR, infrared spectroscopic, and the theoretical points of view [2–19]. The simplest CX2 = PY molecule, CH2PH, one of the unstable molecules, has been studied extensively [3–8, 19].
Some attentions have been paid on molecular geometry and rotational spectroscopic constants that a lot of experimental investigations have been reported for CH2PH. In 1976, Kroto et al. reported their first detecting of CH2PH in pyrolysis products of (CH3)2PH [3]. Five years later, they carried out an accurate analysis of microwave spectrum for CH2PH [4, 5] and derived rotational constants and five quartic centrifugal distortion coefficients. They also determined dipole moment by measuring two Stark shifts (101 ← 000 and 303 ← 212 transitions) for CH2PH. In 1981, Brown et al. improved the microwave spectra with some measurements in the region 5–125 GHz for phosphaethene [6]. They confirmed the molecular structure by least-squares fit for CH2PH and CD2PH. They confirmed dipole moment components which were obtained by the analysis of the four Stark effects (101 ← 000, 202 ← 101, 212 ← 303, and 312 ← 313 transitions). They also measured the rotational and distortion constants for 13CH2PH, 12CD2PH, and 12CD2PD isotopes. Their experimental values were reproduced by some ab initio calculations. In 1985, the isovalent molecules (H2CNH, H2SiNH, H2CPH, and H2SiPH) were investigated employing large-scale MRD-CI method by Bruna et al. [8]. They calculated the dipole moments, charge distributions, and the molecular geometries in ground states for these four molecules. The vertical excitation energies were also reported as predictions. In 1988, Dyall et al. measured the spin-rotation constants and compared them with the theoretical values from COLUMBO and GAUSSIAN82 programs employing the Hartree-Fock method [9]. Some of these studies were experimental, in which all the CH2PH were obtained by pyrolysis. Until Lacombe and his coworkers synthesized phosphaethene molecule by the VGSR method in 1988, [10] which made the measurement of rotational spectroscopic constants much easier. In 2006, Margulès et al. used this synthesize method to reinvestigate the rotational spectrum by measuring the submillimeter-wave spectrum for phosphaethene [18]. They measured 119 new lines in submillimeter region 525–650 GHz. The measurement of ground state rotational constants and quartic centrifugal distortion constants was improved significantly. They also reported the equilibrium structure calculated at the high level of theory, CCSD(T), using cc-pVnZ and cc-pwCVQZ basis sets, respectively. In 2009, Woon and Herbst reported the molecular geometries and the dipole moments using the RCCSD(T) theory with basis set aug-cc-pVTZ for a number of neutral molecules [19].
With the evolvement of analytic second derivatives, DFT predictions of ro-vibrational spectroscopic constants are reliable and cost-effective [20–26]. But as stated above, these theoretical studies [8, 19] just calculated molecular geometry, spin-rotation constants, and the dipole moment for CH2PH. However, those works did not give the calculated anharmonic force field and spectroscopic constants which are useful for detection of CH2PH in planetary atmosphere or interstellar space [18] Thus the goals of this work are, first, to calculate spectroscopic constants with DFT method and check them with experimental ones; second, to discuss the isotopic effects on some rotational spectroscopic constants and vibrational frequencies for 5 isotopomers.
Computational methods
All the quantum-chemical calculations presented below are carried out with the GAUSSIAN03 [27], and the density functional theory [28] is used. Two methods are employed: the first one is B3LYP denoted by Becke’s three-parameter functions [29] and Lee-Yang-Parr functions [30]. The second one is B3PW91 denoted by Becke’s [29] and Perdew-Wang 91’s [31] functions.
Optimized geometries of CH2PH are calculated using analytic gradients. Then, the associated harmonic force field is calculated analytically at optimized geometry in Cartesian coordinate. The harmonic spectroscopic constants are derived by the usual manner [32, 33] The anharmonic spectroscopic data are obtained by theoretical force fields using DFT [34, 35]. No orbital has been kept frozen during these calculations.
The electronic configuration describes the distribution of electrons in atomic orbitals. Considering the descriptions of ground electronic configuration for P, H, and C are 1s22s22p63s23p3, 1s1 and 1s22s22p2, respectively, two correlation consistent polarized valence basis sets are used. One is cc-pVTZ (triple-zeta) [36]. The other is cc-pVQZ (quadruple-zeta) [36]. The quadruple-zeta basis set maybe sufficient in calculating spectroscopic properties, because basis set extension beyond cc-pVQZ does not improve calculation significantly in such applications. In this work below, VTZ and VQZ are the abbreviated descriptions of cc-pVTZ and cc-pVQZ, respectively.
Results and discussion
The spectroscopic constants, geometry structure and full quartic force field for CH2PH are contained in Tables 1–8. The calculated results are compared with the available previous, empirical or experimental data [3–8, 19]. While it is usual to compare them with experimental data and predict some spectroscopic ones [37].
The computed equilibrium structures of CH2PH and experimentally derived results [5, 18] are shown in Table 1. The molecule structure of CH2PH optimized within the constraint of C s symmetry is illustrated in Fig. 1. By comparing our calculated equilibrium geometries to the experimental results, [18] a good agreement can be found: most of them agree within uncertainties. Exactly, the discrepancies of calculated results can be found: 0.0001Å (–0.0106Å in the worst case) for bond lengths, –0.0384° (–0.5188° in the worst case) for bond angles. Geometry structures are nearly converged at the VTZ basis set: [38, 39] improvements from VTZ to VQZ shorten the bond lengths (about 0.003Å) and increase the bond angles (about 0.1°). The calculated results of HPC bond angle are relatively too large (by about 0.39°). It can be explained that the P and C atoms are close to both the mass center and the a principal inertial axis (see Fig. 1), which makes it extremely difficult to measure precise structure using rotational spectroscopy [40].
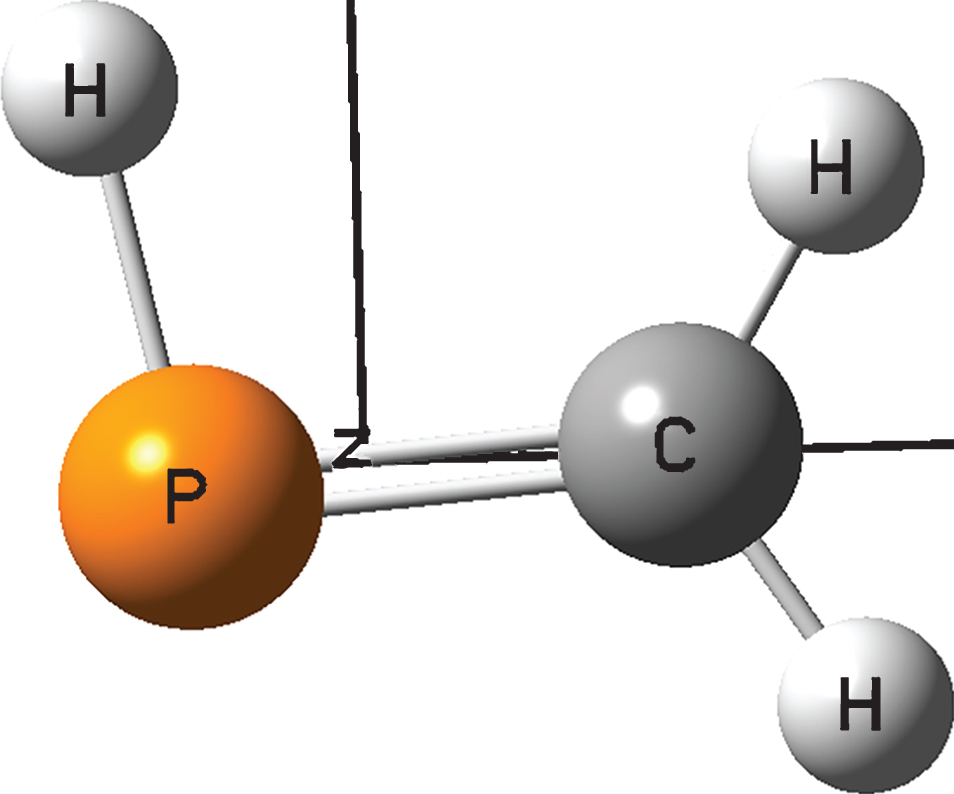
Molecular structure of CH2PH. The horizontal and vertical axes are the a and b principal inertial axes, respectively.
Molecular equilibrium geometry structure and electric dipole moment of CH2PH
aH t and H c are the hydrogen atoms in trans and cis, respectively.
Turning attention to the dipole moment, μ, the B3LYP results agree within 0.04 Debye between the experimental values [5] which were determined by Stark shifts. From the experimental and calculated dipole moment data, it is possible to obtain the rotational angle of principal inertial axes systems for CH2PH molecule [40]. The angles formed by
The equilibrium and ground states rotational constants for 5 isotopic species in Table 2 are compared with experimental data [5, 18]. The relationship between ground state rotational constants and equilibrium ones are: [41]
Rotational constants of equilibrium and ground states for phosphaethene (MHz)
Isotopes: (1) 12CH2PH, (2) 13CH2PH, (3) CD2PH, (4) CH2PD, (5) CD2PD. aSemi-experimental results for equilibrium rotational constants.
Where the summation covers all normal modes,
Theoretical fundamental frequencies ν I are obtained from related harmonic ones ω I by including anharmonic constants x ij (see Table 7) according to: [34]
As the B3LYP predictions of fundamental vibrational frequencies are very accurate and cost-effective, [21, 43] Table 3 contains nine fundamental ν I and harmonic vibrational wave numbers ω I for phosphaethene using B3LYP/VQZ level. It can be found that the anharmonicity effect is quite small for fundamental frequencies corresponding to the bending modes, especially for the value of ν7, which is about 4 cm–1 lower than ω7. While for the stretching vibrations, the anharmonicity effects appear to be larger, except for CP stretching mode. The harmonic values of PH (ω3), the CH2 symmetric (ω2) and asymmetric (ω1) frequencies are about 82, 122, and 143 cm–1 bigger than ν3, ν2, and ν1, respectively. The ω I and ν I for some isotopologues are also given in Table 3. Comparing the frequencies of CH2PH with 13CH2PH, it can be found that, for small mass differences the frequencies of the isotopic molecules are close to those of the ordinary molecule. And the shifts of ω1, ω2, ω4, ω5, ω8 between CH2PH (or 13CH2PH) and CD2PH (or CD2PD) are remarkable. The same thing has happened to ω3 on going from CH2PH to CH2PD. That’s because the atom H in question has a large amplitude in these normal modes [42]. Table 3 also shows that inverse isotopic shift is found in ω3 (13CH2PH). The shift can be attributed to a change in the specific kinematic characteristics of the vibrations [12]. As far as we know, these parameters have no related experimental or theoretical values. The calculated results provide the reliable predictions of the fundamental and harmonic vibrational wave numbers for phosphaethene.
Harmonic and fundamental vibrational wave numbers calculated at B3LYP/VQZ level of theory for phosphaethene (cm–1)
Table 4 presents A-reduction centrifugal distortion constants and observed [5, 18] ones for CH2PH. The discrepancies between observed [18] ground-state quartic values and calculated equilibrium ones are less than about 7% both at B3PW91/VQZ and B3LYP/VQZ levels. The available experimental sextic centrifugal distortion constants are for Φ K , Φ JK , Φ KJ , and φ jk , while the computed values of Φ J , φ j , and φ k are regarded as predictions. The calculated results of Φ JK and φ jk are close to experimental results [18] with deviations below 3.0 and 0.67 Hz, respectively. However, the deviations of Φ KJ and Φ K are about 102 and 103 Hz, respectively. It may be caused by numerical unstability of the equations employed to obtain the centrifugal distortion constants [20].
Equilibrium quartic and sextic centrifugal distortion constants (A-reduction) of CH2PH
As the asymmetric top parameter, κ, of CH2PH is –0.972 at B3LYP/VTZ level of theory, we also give the S-reduction centrifugal distortion constants along with observed ones [6, 18]. From Table 5, one can see that the DFT calculations of quartic centrifugal distortion constants are close to Refs. [6] and [18]. These results confirm that DFT calculations can give accurate S-reduction centrifugal distortion constants, and permit us to analysis the isotopic effects. Consideration of the calculated and experimental results show that the isotopic shifts of the substitution by D are particularly larger than the substitution by 13C for D J , D JK , and D K , the same as the situation of rotational constants above. The substitution by the D atoms decreases all the five quartic centrifugal distortion constants compared with CD2PH or CH2PD.
Equilibrium quartic centrifugal distortion constants (S-reduction) of phosphaethene
Isotopes: (1) 12CH2PH, (2) 13CH2PH, (3) CD2PH, (4) CH2PD, (5) CD2PD.
From the previous data, it appears that the B3LYP results are reliable. Thus the vibration-rotation interaction constants and anharmonicity constants are shown in Tables 6 and 7 using B3LYP method for 12CH2PH. To the best of our knowledge, there is no relative previous or experimental result to be given until now. These data can provide a prediction for
Vibration-rotation interaction constants calculated at B3LYP method for CH2PH (MHz)
Vibrational anharmonicity constants x ij calculated at B3LYP method for CH2PH (cm–1)
Although anharmonicity constants, x ij , are mass-dependent, [16] the experimental ones are not yet available. Table 7 just lists the B3LYP values for 12CH2PH. From Table 7 it is found that the differences of x ij are small with basis set extension from VTZ to VQZ. The prediction of x ij can be used to confirm the location of some overtone and combination bands [13]. It is hoped that the calculated prediction in Table 7 will be meaningful in future experimental work for CH2PH.
Table 8 contains cubic and quartic force constants of CH2PH using B3LYP/ VQZ level. Table 8 shows that some cubic force constants including the CH2 symmetric stretching (F222 = –1339.12 cm–1), the PH stretching (F333 = 1463.05 cm–1), and the coupling of the CH2 asymmetric and symmetric stretching (F211 = –1383.79 cm–1) appear obviously large. Similarly, some diagonal quartic force constants involving CH2 asymmetric stretching (F1111 = 570.43 cm–1), and PH stretching (F3333 = 954.18 cm–1) are found to be quite significant.
Cubic and quartic force constants calculated at the B3LYP/VQZ level of theory for CH2PH (cm–1)a
aAll terms are calculated but only values over 50 cm–1 are given for the force constants.
The molecular equilibrium structure, spectroscopic constants, and full quartic force fields for phosphaethene are obtained at B3PW91 and B3LYP methods with VTZ and VQZ basis sets, respectively. By comparing calculated data with obtainable experimental ones, it is found that B3PW91 and B3LYP methods can reproduce the reliable spectroscopic constants and equilibrium geometries suitable. Furthermore, the rotational spectrum constants for the five isotopomers are also obtained. The isotopic effects for the quartic centrifugal distortion constant D K and rotational constants A e , A0 are obvious going from CH2PH to CD2PD. Finally, some spectroscopic constants are predicted and the inverse isotopic shift is found in ω3 (13CH2PH). The predictions for these spectroscopic constants, such as fundamental and harmonic vibrational wave numbers, cubic and quartic force constants, anharmonicity constants, vibration-rotation interaction constants are expected to guide future high resolution experimental work for phosphaethene. And these spectroscopic constants are useful for detection of phosphaethene in planetary atmosphere and interstellar space.
Footnotes
Acknowledgments
This work was supported by National Natural Science Foundations of China (Nos.51562032 and 61565013). The calculated data were partially performed on Shuguang Super Computer Center of Ludong University.