
Abstract
The global searches of most stable geometrical structures of fluorine-lithium clusters for the series of LinF (n = 1–8) clusters were investigated by using the density functional theory. The stable geometrical structures, binding energies, dissociation energies, second-order energy differences, HOMO-LUMO gap energies and electronic features of LinF (n = 1–8) clusters were reported at the B3LYP/Lanl2dz level of theory. The relative energy results show that the odd number clusters are more stable than the even number cluster. The fluorine atom is generally at the apex position in the most stable structures, except Li5F and Li7F. In addition, the fluorine atom enhances the stability order of lithium clusters, however the stability decreases as the cluster size grows up.
Introduction
The superalkali or superhalogen clusters have gained immense importance in physics which are nano-sized materials between the atoms and the bulk with unique physical and chemical properties. The physical and chemical properties are different from those bulk materials and vary with the cluster sizes or impurity halogens doping. The halogen doped lithium clusters are therefore the subject of the field of cluster physics both experimentally and theoretically. The halogens are generally used to obtain superalkali clusters as an impurity-doped atom in the lithium clusters, for instance; LinBr (n = 1–8) [1], LinF (n = 2–4) [2], LinCl (n = 1–7) [3] and LinI (n = 3,5) [4]. These structures are called superalkali clusters providing that the ionization potential is lower than the corresponding metal atom and that are significant for the cluster based non-linear optical (NLO) materials or supersalts [5–10] Regarding the one fluorine atom-doped lithium clusters, a few systematic studies are available in the literature. Velickovic et al. have performed combined theoretical as well as experimental investigation on the ionization energies of LinF (n = 2–4) clusters [2]. Dustebek et al. have produced LinF (n = 2–6) by Knudsen-cell mass spectrometry and analyzed their ionization energies [11]. Srisvastava and Misra have investigated nonlinear optical behavior of LinF (n = 2–5) clusters [12].
In this study, we performed a systematic investigation of LinF (n = 1–8) clusters via DFT in order to reveal some physical and chemical properties of these clusters.
Computational details
All of the quantum chemical calculations presented here were carried out by using Gaussian 09 software program [13]. The molecular structure parameters of Lin (n = 2–9) clusters were used from our previous study [1]. The stable structures for LinF (n = 1–8) clusters were firstly produced implementing the same approach used for the lithium cluster, the difference is that a lithium atom was replaced by a fluorine atom one by one in each structure of the lithium clusters. The obtained structures were optimized at DFT/B3LYP/Lanl2dz level within Berny algorithm where the basis set includes the relativistic effective core potential for fluorine atom. In the second step, the vibrational calculations were calculated to verify ground state on the potential surfaces. The bond lengths and vibrational wavenumbers of LiF and Li2 dimers were calculated for the computational accuracy. The LiF molecule have a bond length of 1.600 Å with the vibrational wavenumber of 915.62 cm-1 that are in very good agreement with the experimental bond length of 1.564 Å and also vibrational wavenumber of 910 cm-1 for the LiF dimer [14]. The calculated bond length of 2.703 Å and the harmonic vibrational wavenumber of 343.34 cm-1 for the Li2 are also consistent with the experimental value of 2.673Å and 351 cm-1 [14]. Hence, the DFT/B3LYP/Lanl2dz theory level is reliable to explain the physical and chemical properties of the LinF (n = 1–8) clusters.
Results and discussion
Geometrical structures
The stable conformers of LinF (n = 1–8) clusters are given in Fig. 1 where (a) represent the lowest energetically structures for LinF. The symmetry, spin multiplicity, total electronic energy, relative energy, gapHL and Gibbs energy of LinF clusters are listed in Table 1.
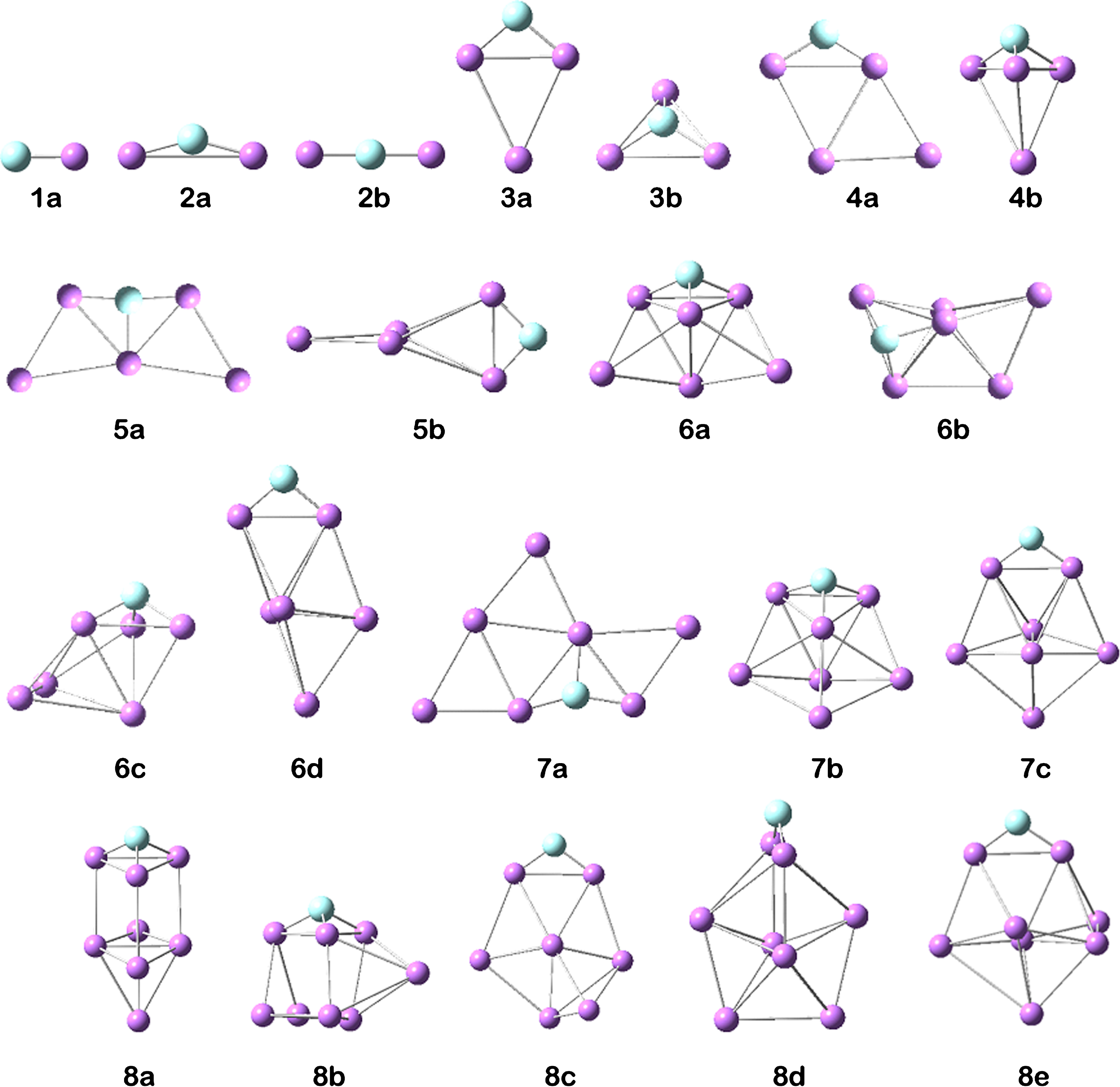
The illustrations of the conformers of LinF (n = 1–8) clusters; fluorine atoms are in light blue.
The isomer, symmetry, spin multiplicity (multi), total energy with zero point energy (Et), relative energy (ΔE), gapHL and Gibbs energy (ΔG) for the LinF clusters.
The most stable conformer of Li2F (Fig. 1.2a) is an isosceles triangle with the C2v symmetry where the fluorine atom is at the apex position and the average bond length of Li-F is 1.678 Å. A triangular structure with the C2v symmetry is reported for the Li2F [2, 12]. The fluorine-centered linear conformer (Fig. 1.2b) is less stable ca. 0.91 eV. The lowest energy structure of Li3F is also planar rhombus one with C2v symmetry. In this structure, the fluorine atom is at the apex and the average bond length of Li-F is 1.707 Å. The other stable triangular pyramidal conformer (Fig. 1.3b) is less stable by 0.51 eV. The planar rhombus having Cs symmetry (Fig. 1.4a) is the more stable structure of Li4F and the average bond length of Li-F is 1.708 Å. The low-lying conformer of this cluster have same stability with the a very small energy difference (Fig. 1.4b). This small energy difference (0.001 eV) indicate that Li4F have two more stable structure which will be difficult to seperate. The boat form with the doped fluorine atom (Fig. 1.5a) is turned out as the ground state of Li5F among the optimized structures and the average bond length of Li-F is 1.751 Å. The other conformer (Fig. 1.5b) is less stable by 0.735 eV compared to the ground state structure. The lowest energy configuration of the Li6F is a trigonal pyramid with the fluorine atom at the top, and the average bond length of Li-F is 1.754 Å. The other conformers (Fig. 1.6b-d) is less stable ca. 0.25–0.49 eV. For Li7F cluster, three stable conformers were obtained. The conformer (Fig. 1.7a) has the lowest energy value and the conformers 7b and 7c given in Fig. 1 are less stable by 0.027 eV and 0.054 eV. The lowest energy structure of the Li7F cluster has the average bond length of 1.757 Å for the Li-F. A doping of fluorine atom to the Li9 cluster results in an antiprism as the lowest energy geometry of Li8F. In this cluster, five stable conformer were obtained which 8a is the most stable conformer. The other conformers (Fig. 1.8b-e) is higher in energy than the lowest energy structure. The Li-F average bond length of 1.8a conformer is also found 1.798 Å.
The lowest energy structure to relative energy results of each cluster, are also supported from the Gibbs energy results of each clusters. Regarding the bond length of Li-F, the bond length becomes longer with the increasing of coordination number of fluorine atom. Lithium clusters when doped a fluorine atom, the geometry transition from 2-D to 3-D occurs for doped lithium clusters at Li6F. This transition is due to the formation of cage critical points [1]. The doped fluorine atom also modifies the Lin clusters for considered region except the Li4. It is also worth noticing that while the fluorine atom is at the apex position have similar molecular geometric configuration in the lowest energy structures for the LinCl [3] and LinBr [1], except Li5F and Li7F clusters.
The relative stability of clusters is discussed through the binding energy per atom (E
b
), dissociation energy (ΔE) and second-order energy differences (Δ2E) considering the lowest energy structures. The expressions used in the calculations are as follows:
where E is the total molecular energy including the zero-point energy for the related system. The binding energy per atom, dissociation energy, second-order energy differences and gapHL for LinF (n = 1–8) clusters is given in Fig. 2.
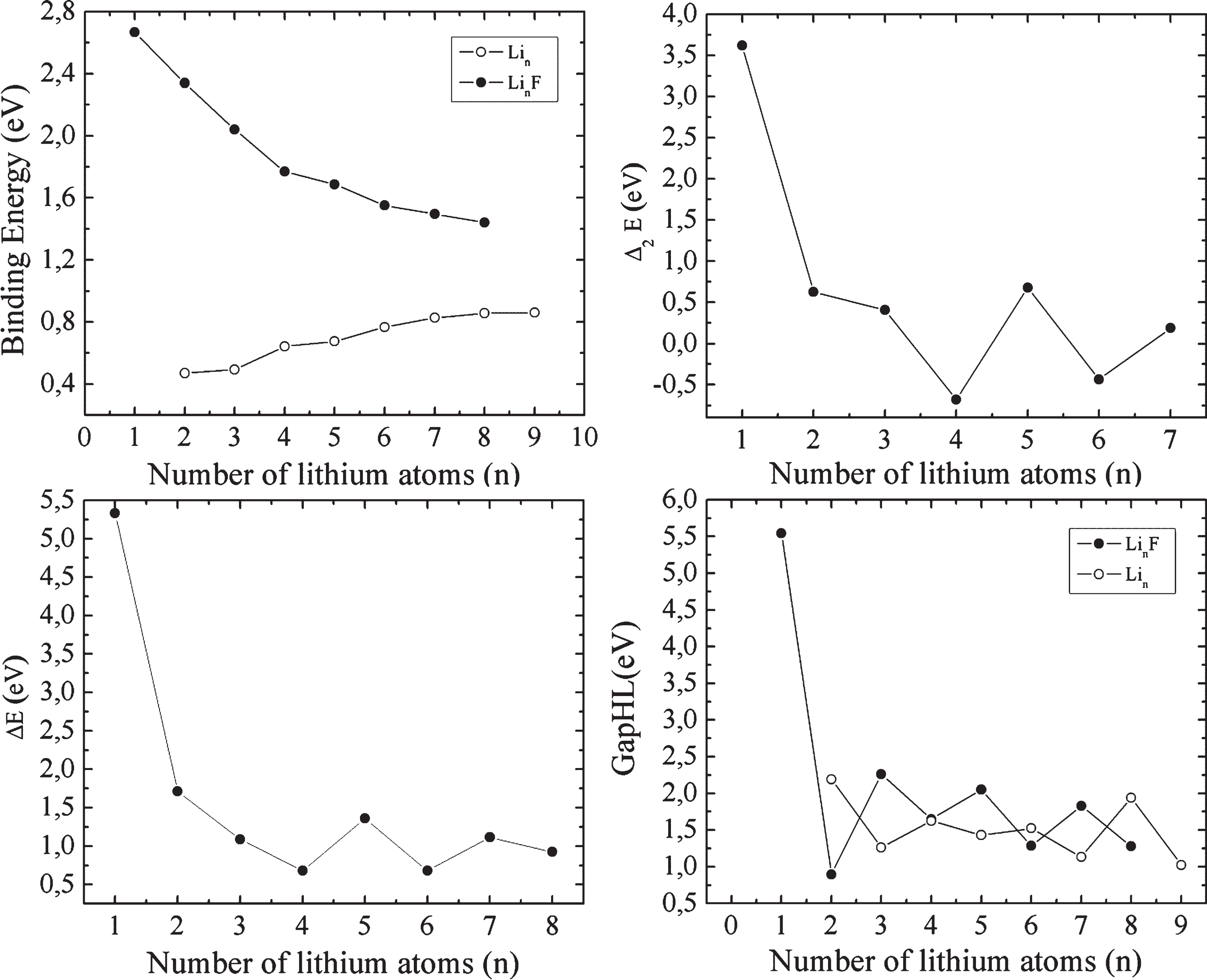
The binding energies per atom, the second-order energy difference, the dissociation energy and the HOMO-LUMO gap for LinF (n = 1–8) clusters.
The binding energy of LinF (n = 1–8) is higher than the lithium clusters indicating that the doped fluorine atom enhances the stability of lithium clusters. However, the doped clusters become more reactive as the cluster size grows up since the binding energy decreases. The decrease in the binding energy can be due to the surface effect. For Lin (n = 1–9) clusters, the stability increases smoothly up to n = 7 and then increase slowly. Another feature from binding energy curves is the initial difference of both systems that exhibits the strong binding of small doped clusters [1].
The second-order energy differences (Δ2E) and the dissociation energy (ΔE) determines the relative stability of the clusters. The second-order energy difference (Δ2E) and dissociation energy (ΔE) of LinF (n = 1–8) clusters is plotted as a function of the cluster sizes in Fig. 2. The higher peaks are associated with odd number clusters. The clusters are therefore more stable than others (except LiF and Li2F). The higher stability results from the electron pairing effect.
The HOMO-LUMO gaps are also calculated for the lowest energy structures of LinF (n = 1–8) and Lin (n = 2–9) clusters. The odd number of doped clusters possesses higher stability than the even number clusters in Fig. 2 and that is in agreement with the result of second-order energy differences (Δ2E) and the dissociation energy (ΔE). Moreover, eight valance electrons of the LiF leads to a maximum value of gapHL, hence the cluster can be magic number cluster.
The fluorine-doped lithium clusters have higher stability than the lithium clusters, as well as chlorine-doped clusters and bromine-doped clusters that is from the more effective core potential of fluorine atom. The stability order of fluorine-doped lithium clusters decreases while increase of the lithium atom number or the volume of molecular bulk. The second order energy difference, dissociation energy and GapHL calculations indicate that LinF (n = 3,5,7) clusters are more stable than the LinF (n = 4,6,8). However, LiF is the most stable than the other clusters. The fluorine atom is generally at the apex position in the LinF clusters except Li5F and Li7F clusters. The relative energy results show that doped fluorine atom modifies the Lin clusters for considered region except the Li4 and the lithium cluster geometry makes the transition from two dimension to three dimension at Li6F.