Propargyl chloride, CH≡C–CH2Cl, was virtually inert towards NaH2AsO3, reacted to a very small extent with Na2HAsO3, but with Na3AsO3 gave a mixture of salts in which sodium 1-propynylarsonate, CH3–C≡C–AsO3Na26, predominated, the allenylarsonate, CH2 = C = CH–AsO3Na24, being present in (very) small amounts, while the isomeric 2-propynylarsonate CH≡C–CH2–AsO3Na25 was not detected. These results can be understood by attack of AsO33– on propargyl chloride to form 5 followed by base catalyzed prototropic rearrangements to 4 and to 6. Separation of 6 from 4 was tried as acids or salts without success. The purity of the product as acid 6 and lead salt of 6 was >80%. The reaction of propargyl chloride with Na2SO3 required dilute aqueous solutions (∼0.3 M) in order to give the salt CH≡C–CH2–SO3Na·4H2O, 11·4H2O, in ∼80% yields. More concentrated solutions (1 or 2 M) produced 11 and by-products from which CH2 = C = CH-SO3Na 10, but not CH3–C≡C–SO3Na 12, has been identified spectroscopically, while potassium 2-chloro-2-propenylsulfonate tetrahydrate CH2 = CCl–CH2–SO3K·4H2O, 13·4H2O, was isolated.
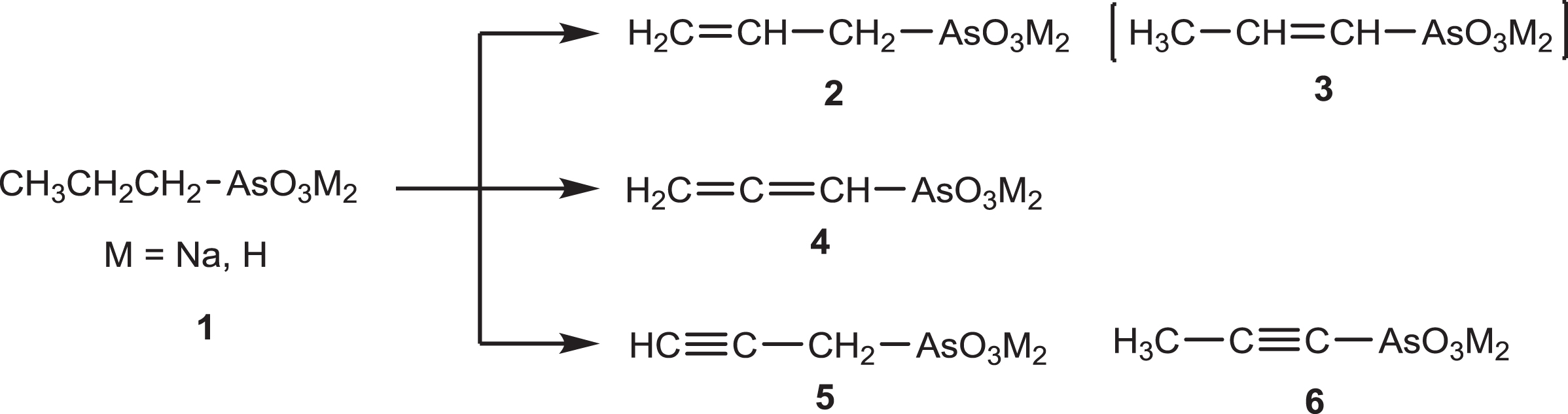
From the three carbon atoms arsonic acids or their salts, Fig. 1, the n-propyl and allylarsonic acids 1 and 2 have been known since 1922 [26]. The vinylarsonic 3 is not known because the simpler vinyl halides did not react with alkaline sodium arsenite (herein symbolized as Na3AsO3) [2]. The allenylarsonic acid or its salts 4 and the isomeric propynylarsonic acids or their salts 5 and 6 have not been prepared.
Conceivable saturated and unsaturated linear three carbon arsonic acids and sodium salts.
For the preparation of 5, the Meyer reaction [21, 24] between propargyl chloride or bromide and Na3AsO3 is the most obvious route. Propargyl chloride, being electrophilic, can react with a variety of nucleophiles such as AsO33–, SO32–, HO– to give propynylated products, e.g. 5, by an SN2 reaction and it will be of interest to compare the reaction rate of Na3AsO3 with propargyl halides with the rates of propyl halides and allyl halides which are slow and fast, respectively [26].
Because aqueous solutions of Na3AsO3 are strongly basic, isomerization of the first formed 5 to the allene 4 can take place, the extent of which will depend on the reaction time, temperature and the specific base [20]; in our case HO– or –AsO32–. The mechanism of the isomerization can be quite complicated [34]. The allene 4, in turn, may not be stable in alkaline solutions suffering [34] nucleophilic addition at the middle carbon giving, e.g. CH3COCH2AsO32–, prototropic rearrangement to 6, while aniotropic rearrangement, if occurs, should give 5.
Two close, but not analogous, reactions reported in the literature are the Arbuzov and the Nylen reactions with propargyl bromide.
The Arbuzov reaction with (MeO)3P at 90°C gave only 5% of the expected CH≡C–CH2P(O)(OMe)2 contaminated by the allenyl CH2 = C = CH–P(O)(OMe)2 and the isomeric CH3–C≡C–P(O)(OMe)2 phosphonates [9], while with (EtO)3P at 90°C the 1-propynyl derivative, CH3–C≡C–P(O)(OEt)2, was isolated [9]. Although a concise mechanism for the products obtained has not been offered, it seems that CH≡C–CH2–P(O)(OR)2 is formed by a normal SN2 Arbuzov reaction, the phosphoallene CH2 = C = CH–P(O)(OR)2 is probably formed by an SN2’ reaction (attack at an activated terminal CH≡C carbon) [10, 25], and CH3–C≡C–P(O)(OR)2 is formed (in the absence of a base) after hyperconjugation [34] of the acidic α-H in CH2 = C = CH–P+(OR)3Br– to give CH3–C≡C–P+(OR)3Br– and then follows the formation of the –P(O)(OR)2 group.
The Nylen reaction of sodium dialkyl phosphonate (sodium dialkylphosphite), (RO)2P–O–Na+, with propargyl bromide in THF was also dependant on the nature of the R group. When R = Me, the isolated product was CH3–C≡C–P(O)(OMe)2 in unspecific yield [9] with less than 1% contamination by CH≡C–CH2–P(O)(OMe)2. When R = Et, the main product in the mixture was, again, the 1-propynyl ester CH3–C≡C–P(O)(OEt)2.
These reactions revealed that the phosphoallene CH2 = C = CH–P(O)(OR)2 was formed but found in minute amounts. This fact can be explained by its high reactivity because it contains one double bond reactive towards electrophiles and another double bond reactive towards nucleophiles. The reason that the phosphoallene isomerizes to CH3–C≡C–P(O)(OR)2 in the absence of a base could not be found in the literature although it is known that complete isomerization can take place in the presence of a base [17] and, in this case, the explanation offered is the stability of a triple bond conjugated to –P(O)(OR)2 and the hyperconjugation of the triple bond to the –CH3 group [17].
The preparation of sulfonic acids or their salts can be accomplished by general methods [1] using S(VI) and S(IV) compounds one of which involves the reaction of alkyl halides with Na2SO3 (the Strecker reaction [33]). This reaction indicates that Na2SO3 is nucleophilic at sulfur and the SO32–, rather than HSO3– even if it predominates in solution, is the reacting species [1]. Thus, there is a similarity in the behavior of Na2SO3 and Na3AsO3 [31].
For three carbon atoms sulfonic acids and salts, Fig. 2 depicts the conceivable compounds, being analogous to Fig. 1. The sodium 1-propanesulfonate 7 is commercially available as monohydrate. The allylsulfonic acid as potassium, barium and lead salts 8 are known [27]. Although the vinylsulfonic acid, CH2 = CH–SO3H [6] is of industrial importance the acid 9 is seldom mentioned in the literature [13]. The much used for click chemistry sodium propargylsulfonate 11 was prepared from propargyl bromide in toluene and sodium sulfite in water/methanol (1:1 v/v) (see for example ref. [29, 39]). The allenylsulfonic acid 10 was prepared from allenyl thiol [28], while the acid 12 was prepared from 1-trimethylsilyl-1-propyne [4].
Conceivable saturated and unsaturated linear three carbon sulfonic acids and sodium and potassium salts.
In this paper we report on the reaction of the sparingly soluble in water and quite volatile (bp 58°C) propargyl chloride with aqueous solutions of NaH2AsO3, Na2HAsO3 and Na3AsO3 in order to see their nucleophilicities, especially for preparative purposes. These reactions, run in an alkaline environment, can give various products [34], due to the propensity of the initially formed 5 to suffer propargylic rearrangement to the allene 4 which, in turn, can prototropically rearrange (allenyl rearrangement) to 6. Thus, the reactivity of arsenites will be compared to that of P(III) nucleophiles towards propargyl halides filling in a literature gap. Because in the Meyer reaction the nucleophile AsO33– exists in concentrated aqueous of Na3AsO3, we tried the Strecker reaction of propargyl chloride with concentrated solutions of sodium sulfite. The 1H NMR spectra obtained were too complicated suggesting the presence of 10, 11, and 12, so we studied the reaction with aqueous Na2SO3 of various molarities, that revealed the formation of 10, 11, and 13 but not of 12. The sulfonates 11 and 13 have been isolated under proper conditions.
Experimental
Materials and methods
Propargyl chloride (Janssen) and propargyl alcohol (Alfa Aesar) were used as received. Triphenyl trithioarsenite, (PhS)3As, was prepared from arsenic trioxide (Ferak) and thiophenol in methanol [30]. Silica gel 60 for column chromatography and silica gel 60 H for thin layer chromatography (TLC) were from Merck. Dowex 50W-X8 (H+ form) cation exchange (Bio Rad) and Dowex 1-X8 (acetate form) anion exchange (Bio Rad) resins [35] were used.
TLCs were run on microslides and visualization was effected by iodine vapors and/or by spraying with 35% sulfuric acid and charring. Infrared spectra were taken in KBr discs on a Perkin - Elmer, model 16PC, FT-IR spectrometer. NMR spectra (1H at 400 MHz, 13C at 100.6 MHz) were obtained on a Bruker, model DPX Avance, spectrometer using TMS or DSS (sodium 2,2-dimethyl-2-silapentane-5-sulfonate) as internal standards. The reaction samples taken were either diluted with D2O or freeze-dried and then dissolved in D2O and in both cases the solutions were alkaline. Electrospray mass ionization spectra (positive ion detection) were recorded on a Micromass - Platform LC spectrometer. Elemental analyses were obtained through the Center of Instrumental Analyses, University of Patras, Patras, Greece.
Reaction of propargyl alcohol with bases
With NaOH
In an NMR tube containing sodium hydroxide (1 mmol) in D2O (0.5 mL) was syringed in propargyl alcohol (58 μL, 1 mmol) and shaken to dissolve. The 1H NMR spectrum of the yellowish solution, run after 5 min, showed a singlet at 4.18 ppm and traces of a broad signal at 2.71 ppm, attributed to CH(or D)≡C–CH2OD. Addition of more propargyl alcohol (1 mmol) the yellow solution had the same 1H NMR spectrum.
With Na3AsO3
In an NMR tube containing a solution of arsenic trioxide (0.1 mmol), sodium hydroxide (0.6 mmol) and D2O (0.6 mL) was syringed in propargyl alcohol (0.2 mmol) and the tube mechanically tumbled for 3 h. 1H NMR spectra after 5 min and 3 h showed the main singlet at 4.19 ppm and traces of a singlet at 2.73 ppm, attributed to CH(or D)≡C–CH2OD.
Reaction of propargyl chloride with sodium arsenites
With NaH2AsO3
To a test tube containing arsenic trioxide (0.1 mmol), sodium hydroxide (0.2 mmol) and water (15 μL) so as to give NaH2AsO3 (0.2 mmol), was syringed in propargyl chloride (3 mmol) and stirred at room temperature (RT). After 18 h a viscous solution, smelling propargyl chloride, was obtained. Its TLC (MeOH/conc. NH3 4:1) showed two black spots (Rf 0.0 and 1.0) and its 1H NMR (D2O, DSS) spectrum showed prominent singlets at 2.68 ppm and 4.24 ppm attributable to CH(or D)≡C–CH2Cl and CH(or D)≡C-CH2Cl, respectively, a strong signal at 4.93 ppm and much smaller signals at 1.91 ppm (attributed to CH3–C≡C–AsO3Na26) and at 2.22, 3.17, 3.34, 3.56, 4.22, 5.49, 6.46, 6.84, 8.44 ppm that could not be assigned to specific compounds.
With Na2HAsO3
Under the same as above conditions, propargyl chloride (0.3 mmol) and Na2HAsO3 (0.2 mmol) in water (31 μL) after 18 h stirring at RT gave a viscous, odorous oil that by TLC (MeOH/conc. NH3 4:1) showed an additional spot (Rf 0.43) attributable to 4, 5 or 6. Its 1H NMR (D2O, DSS) spectrum now showed a stronger singlet at 1.91 ppm due to 6. Apart from the singlets at 2.68 and 4.24 ppm due to CH(or D)≡C–CH2Cl, fewer signals were now seen at 2.91, 4.21, 5.92, 6.18 ppm as well as at 4.94 (d, J 4.4 Hz), 5.49 (t, J 4.4 Hz) ppm (most likely due to the disodium salt 4).
With dilute Na3AsO3
In an NMR tube containing Na3AsO3 (0.2 mmol) in D2O (0.6 mL) (1 M in NaOH, 0.33 M in Na3AsO3) was syringed in propargyl chloride (0.2 mmol) and the tube mechanically tumbled for 48 h. The 1H NMR (D2O, DSS) spectrum after 5 min showed the presence of CH(or D)≡C–CH2Cl (2.69 and 4.24 ppm) and small singlets at 4.18 and 4.22 ppm that could not be assigned. From 3 to 48 h the 1H NMR spectra of the clear solutions did not show the presence of 6 at 1.91 ppm. The singlet at 4.18 ppm, increasing in intensity, was attributed to CD≡C–CH2OD, indicating hydrolysis of the chloride.
With concentrated Na3AsO3
In a small test tube was added arsenic trioxide (198 mg, 1 mmol), sodium hydroxide (240 mg, 6 mmol) and D2O (0.4 mL) (thus being 15 M in NaOH and 5 M in Na3AsO3). To the viscous solution, propargyl chloride (174 μL, 2.4 mmol) was added and stirred at RT for 6 days. A single phase was obtained after 28 h stirring. Then, samples were taken, diluted with D2O and analyzed by 1H NMR. Leaving apart the signals at 2.68 and 4.20 ppm due to CH(or D)≡C–CH2Cl, the main signal was at 1.90 ppm (doublet and finally multiplet) due to –CH3, –CH2D, –CHD2 and –CD3 of 6. Other small signals were seen at 2.17, 4.37, 4.40, 4.51, 4.90, 5.91, 5.92, 6.16, 6.18 and 6.38 ppm indicating the presence of by-products none of which could be identified with certainty.
Preparative reactions of propargyl chloride with alkaline sodium arsenite: formation of sodium allenylarsonate 4 and sodium 1-propynylarsonate 6
In a 5 mL round-bottomed flask was dissolved arsenic trioxide (495 mg, 2.5 mmol) and sodium hydroxide (600 mg, 15 mmol) in water (1.0 mL) to give a solution 13 M in NaOH and 5 M in Na3AsO3. To the vigorously stirred solution neat propargyl chloride (373 μL, 5 mmol) was added drop-wise (1.5 h). In the cases where excess NaOH or propargyl chloride have been used, their quantities have been adjusted accordingly. The heterogenous systems were stirred at RT for 17, 30 or 55 h, giving opalescent, very viscous solutions. Samples taken and analyzed by TLC (MeOH/conc. NH3 4:1) showed the “product” at Rf ∼0.5 and impurities at Rf ∼0. The dried samples had the following representative spectra. IR(KBr): 3382 vs, very broad, 2190 w, 1953 vw, 1654 m, 1438 w, broad, 822 vs, 624 ms, broad. 1H NMR (D2O, DSS): δ= 1.91 (s, 3 H, CH3) [for product 6], 2.69 (s, 0.27 H), and 4.24 (s, 2.56 H) [for CH(or D)≡C–CH2Cl], 4.94 (s, 0.68 H) and 5.49 (s, 0.07 H), [for by-product 4], and other signals at 2.19 (s, 0.05 H), 2.91 (s, 0.20 H), 5.67 (s, 0.03 H), 5.92 (s, 0.08 H), 6.18 (s, 0.08 H), 6.38 (s, 0.03 H).
These solutions were then worked up by acidification with aqueous HCl and by percolation through a strongly acidic cation exchange resin.
Acidification by aqueous hydrochloric acid: isolation of 1-propynylarsonic acid 6 (purity >80%)
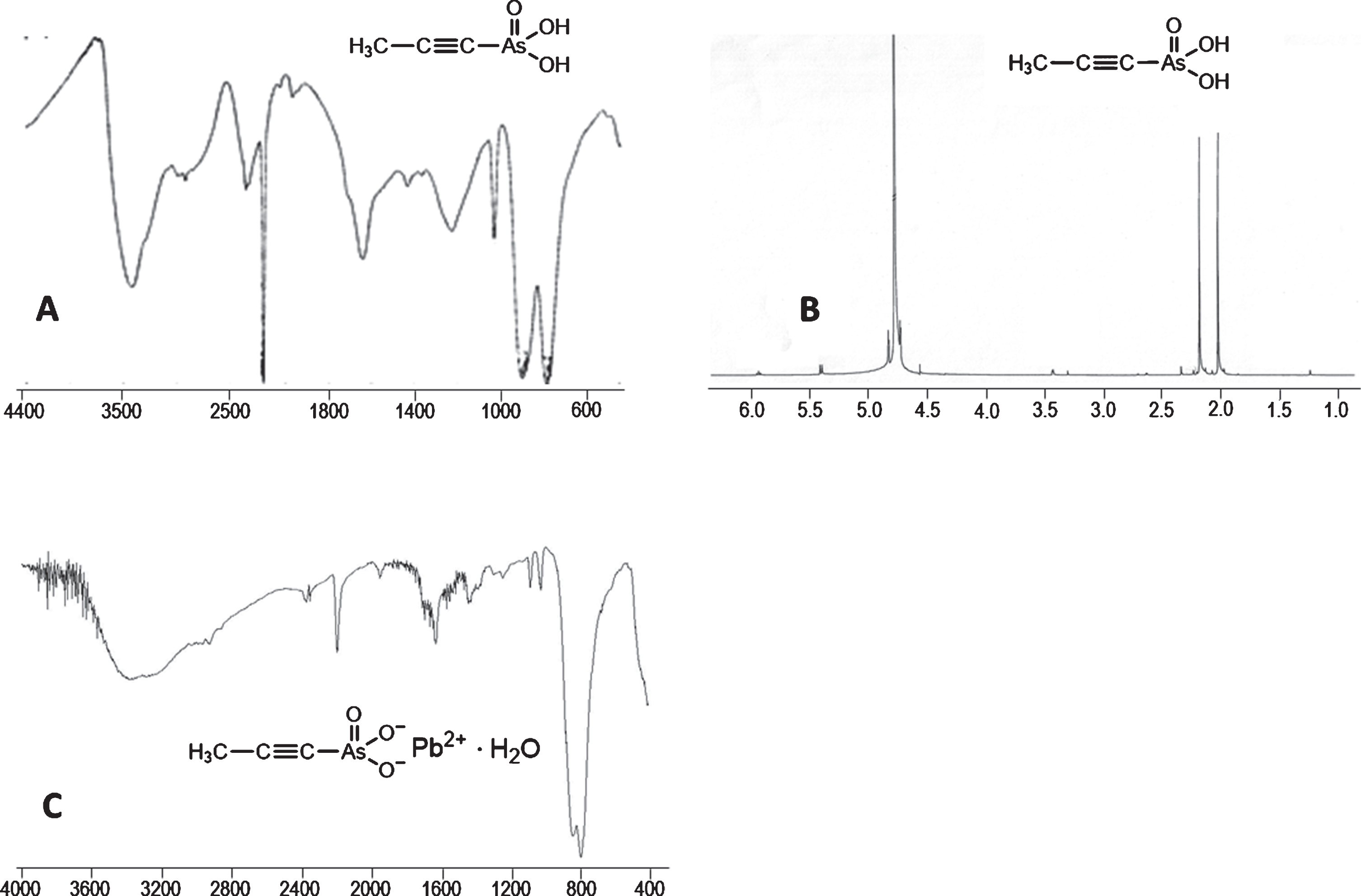
To the viscous solution from the reaction of Na3AsO3 (5 mmol) with propargyl chloride (7.5 mmol) for 55 h, was added drop-wise, while stirring, concentrated hydrochloric acid till pH 2-3. Its quantity was less than calculated (0.83 mL). A precipitate (NaCl + As2O3) was formed and TLC (MeOH/conc. NH3 4:1) of the supernatant showed a black spot at Rf 0.55. Evaporation and drying in vacuum over P2O5 gave a solid (1.47 g) (expected product(s) + NaCl 1.70 g) implying losses of CH≡C–CH2–groups and having the following bands in the IR (KBr) spectrum: 3424 vs, broad, 2200 ms, 1958 vw, 1640 s, 1234 w, broad, 1030 w, 896 vs, 796 vs. Extractions at RT (so as not to extract too much As2O3) with methanol (4×3 mL) extracted the acids 4 and 6 (788 mg, expected 820 mg) leaving a beige solid (630 mg, expected NaCl 880 mg). This beige solid after extraction with water, left As2O3 (by IR) (61 mg, 12% recovered). From the solid (788 mg) more As2O3 and NaCl were removed by dissolving it in methanol and filtering. Evaporation of the methanol gave 720 mg of a solid that on dissolution in minimum amount of methanol (5 mL) and portion-wise addition of acetone (20 mL) precipitated a new solid. After 12 h at RT, centrifugation gave a white precipitate (347 mg) that by IR (KBr) had a very weak band at 2198 cm–1 and no pure product could be isolated from it. The supernatant (containing 366 mg of acids 4 and 6) was evaporated, suspended in methanol (1.5 mL) and acetone (10 mL) was added while stirring. Centrifugation gave a solid (28 mg) that was discarded because it containd As2O3. The new supernatant, after evaporation, was treated with acetone (7 mL). A solid (61 mg) was obtained that was discarded too. Finally, the yellowish acetone supernatant (did not form a solid at –20°C) was evaporated and dried in vacuum to give a colorless, viscous oil (283 mg) that on trituration with acetone-ether, centrifugation and drying in vacuum gave the impure product 6 as a foam (247 mg, 30%). By TLC (MeOH/conc. NH3 4:1) one spot at Rf 0.55 was detected. It was soluble in MeOH, Me2CO but insoluble in Et2O. M.p.: at 104–107°C foams and at 130–132°C the foam decomposed. IR (KBr): 3418 s, a little broad, ∼2900 m, ∼2800 m, broad, 2202 vs, sharp, ∼1970 vw, 1638 ms, 1228 m, broad, 1032 m, sharp, 902 vs, 786 vs, Fig. 3A. 1 H NMR (D2O): δ= 2.02 (s, 3 H, CH3) [for acid 6], 2.12 [for (CH3)2CO], 5.39 (d, J 6.8 Hz, 0.37 H) and 5.93 (t, J 6.8 Hz, 0.04 H) [for acid 4], and unknown impurities at 1.23 (s), 2.30 (s), 3.30 (s), 3.42 (t), Fig. 3B, from which >80% purity was estimated for acid 6.
Impure products isolated from the reaction of propargyl chloride with alkaline sodium arsenite. A: The IR (KBr) spectrum of the acid 6 shows a very strong band for disubstituted –C≡C–at 2202 cm–1 compared to the –AsO3H2 bands at 902 and 786 cm–1. B: The 1H NMR (D2O) spectrum of acid 6 shows the –CH3 protons at 2.05 ppm, Me2CO and traces of impurities. The signals from the acid 4 could not be assigned in the region 4.80–6.00 ppm. C: The IR (KBr) spectrum of the water-insoluble lead salt 6·H2O showed a medium band for –C≡C–at 2188 cm–1 and a very weak band for C=C=C at 1942 cm–1. The -AsO32 - band at ∼800 cm–1 was split.
Acidification with a cation exchange resin: isolation of impure lead 1-propynylarsonate monohydrate, CH3–C≡C–AsO3Pb·H2O
The orange, viscous solution, obtained from the reaction of Na3AsO3 (5 mmol) plus 10% excess NaOH with propargyl chloride (5 mmol) for 30 h at RT, was diluted with water (50 mL), applied onto a Dowex 50W-X8 (H+ form) resin and eluted with water (500 mL). The first 100 mL (having pH 2-3) were evaporated (rotary, 50°C) to give a semi-solid (648 mg; expected 820 mg of acid 6). Its 1H NMR (D2O, DSS) spectrum showed the acid 6 at 2.10 ppm as the main product and many other signals due to by-products and indicating that some by-products have been removed and new ones appeared during chromatography. Extraction of non-polar organic by-products with Et2O (5 mL) gave a light orange oil (32 mg) that was discarded. The well dried residue was extracted with acetone (4×3 mL) by stirring for 15 min each time leaving a residue of pure (by IR) As2O3 (131 mg, 27% recovery). The acetone extracts on evaporation and drying in vacuum over P2O5 gave a very viscous, light orange oil (540 mg) whose IR (neat) spectrum showed the expected for the acids 6 and 4 bands at 2204 s, 1966 vw, 900 vs, 800 vs cm–1, while its 1H NMR spectrum (D2O, DSS) showed the product acid 6 at 2.10 ppm and other minor impurities, from which As2O3 in principle may still be present due to co-extraction.
The light orange oil was worked up in order to obtain a pure product: acid 6 or salts of acid 6, or the reduction products CH3–C≡C–As(SPh)2 or (CH3–C≡C–AsO)x.
By anion exchange resin
The reaction products as acids 4 and 6 in water (from 5 mmol Na3AsO3 and 5 mmol propargyl chloride) was chromatographed using a strongly basic anion exchange resin (Dowex AG 1-X8, acetate form) eluting with water and 1 M acetic acid [35]. All fractions obtained, after evaporation and drying in vacuum had solids weighing from 2 to 10 mg thus indicating that the product 6 (expected 820 mg) has been decomposed.
By treatment with bases and metal salts
As will be described in Section 3.2 only the treatment with Pb(AcO)2·3H2O afforded a water-insoluble solid.
The crude mixture of acids 4 and 6 (833 mg, theoretically 5.1 mmol) in water was filtered through celite. To the clear filtrate a solution of lead acetate trihydrate (1.925 g, 5.1 mmol) in water (5 mL) was added and the suspension stirred at RT for 1 h. Centrifugation and washing the solvate with water (3×10 mL) by stirring at RT for 15 min each time gave a solvate that after drying in vacuum over P2O5 gave a white soft powder (1.587 g, 81% as lead 1-propynylarsonate monohydrate) insoluble in DMSO. M.p: at ∼220°C darkens to beige and the color did not change up to 300°C. Calculated for C3H3O3AsPb·H2O (Mr 387.18): C 9.31, H 1.30%; found C 9.72, H 1.11%. IR (KBr): 3370 m, broad, 2188 mw, 1938 vw, 1624 mw, 1078 w, 1020 w, 834 vs, 788 vs, Fig. 3C.
By reductions to As(III) compounds
A solution of a mixture of acids 4 and 6 (74 mg, 0.45 mol) in methanol (1 mL) reacted exothermally with neat thiophenol (184 μL, 1.80 mmol) giving an oil that solidified in 15 min. TLC (petroleum ether) of the supernatant showed a spot for the dithioarsonites, R-As(SPh)2, at Rf 0.20 and a weak spot at Rf 0.46 for PhSH/PhSSPh. After centrifugation and washing with methanol (3×1 mL) the yellow supernatant gave a brownish semi-solid (60 mg, ∼40%). Its IR (neat): 3056 m, 2174 ms, 1934 w, 1576 ms, 1472 s, 1434 s, 1068 m, 1022 ms, 738 vs, 686 vs indicated the presence of disubstituted C≡C at 2174 cm–1 and of allenic C=C=C at 1934 cm–1. From the precipitate (184 mg) by extracting with boiling methanol (5×1 mL), cooling at RT, centrifugation, evaporation and drying of the extracts gave a yellowish semi-solid (85 mg) whose IR (neat) indicated the product CH3–C≡C–As(SPh)2 contaminated by PhSSPh.
The reaction of propargyl chloride with dilute aqueous sodium sulfite: isolation of sodium 2-propynylsulfonate tetrahydrate, CH≡C–CH2–SO3Na·4H2O, 11·4H2O
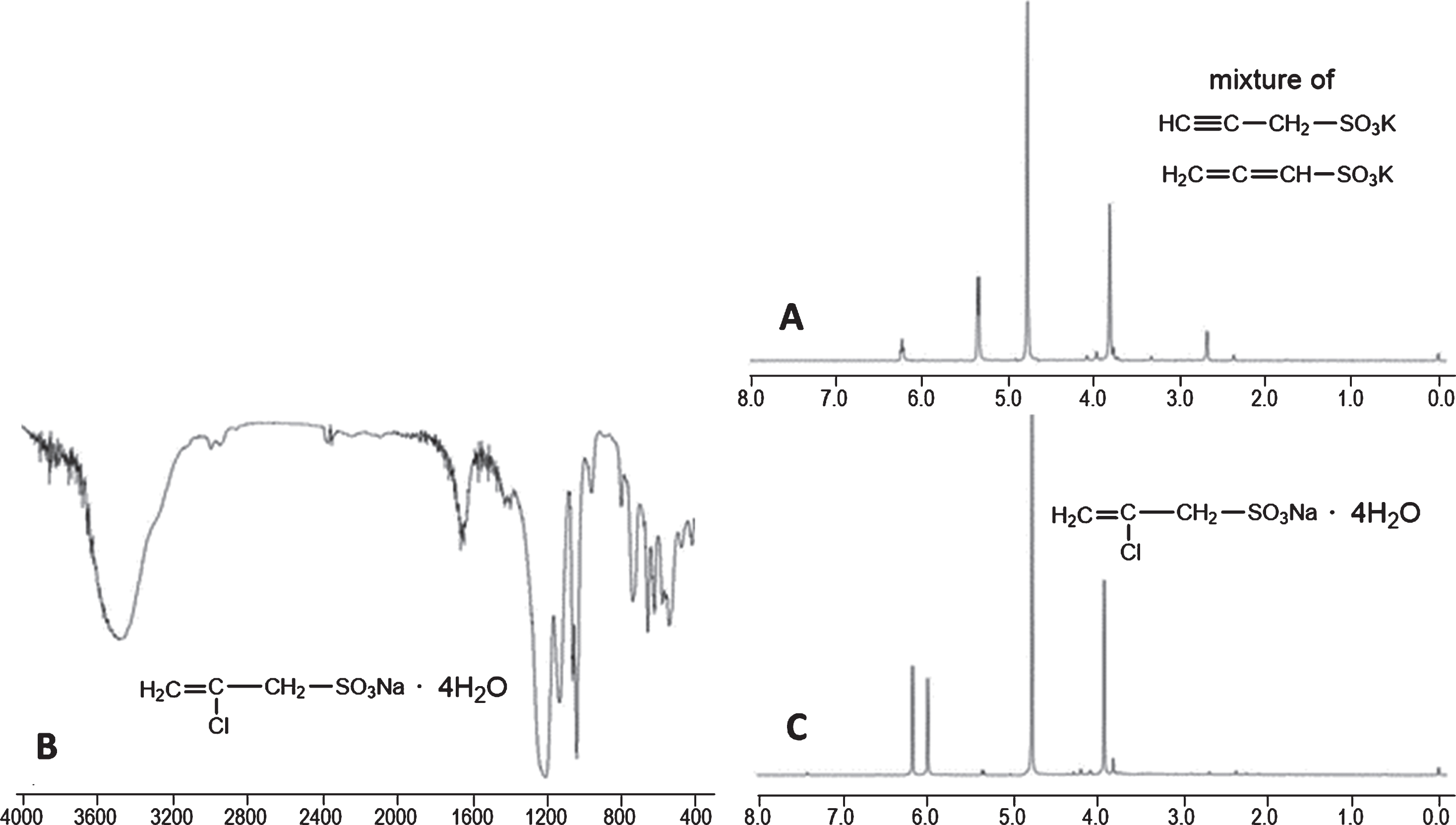
In a 250 mL round-bottomed flask containing boiled water (60 mL), nitrogen was bubbled to de-aerate and solid sodium sulfite (2.520 g, 20 mmol) was dissolved to give 0.33 M Na2SO3. Addition of the light orange propargyl chloride (1.50 mL, 20 mmol) and stirring at RT for 24 h gave a clear, yellowish solution a sample of which, after drying and dissolution in D2O showed two prominent signals at 2.71 (t, J 2.4 Hz, 0.43 H, CH(or D)≡C–CH2–SO3Na indicating ∼60% exchange CH≡C–to CD≡C–) and at 3.83 (d, J 2.4 Hz, 2 H, CH(or D)≡C–CH2–SO3Na). The solution was freeze-dried for 2 days to give a white soft solid (3.674 g, expected anhydrous sodium salt 11 plus NaCl: 4.010 g), whose 1 H NMR (D2O, DSS) was similar to the above one indicating that no new by-products arose during the drying. The solid, which should contain NaCl and Na2SO3/Na2SO4, was suspended in methanol (120 mL), stirred at RT for 2 h and the fine suspension was filtered through celite. The filtrate, after drying (weighing 3.384 g) was suspended in boiling methanol (100 mL), cooled at RT and centrifuged to give a solid (68 mg) that by 1H NMR (D2O, DSS) and IR (KBr) was probably impure sodium salt of 13, Fig. 2. Evaporation of the methanolic supernatant and drying in vacuum over P2O5 gave the sodium salt of 11 (3.294 g, 116% as anhydrous 11; 77% as 11·4H2O). It is soluble in water and DMSO and insoluble in boiling Me2CO or MeCN. M.p.: at ∼117°C sweats (removal of waters), at ∼200°C darkens to light orange, at ∼240–260°C turns light brown and does not change up to 300°C. Calculated for C3H3O3SNa·4H2O (Mr 214.17): C 16.82, H 5.18, S 14.97%; found C 16.76, H 4.82, S 14.96%. IR (KBr): 3564 vs, 3494 vs, 3280 vs, 2970 m, 2932 m, 2128 w, 1622 s, 1408 w, 1266 vs, 1248 vs, 1204 vs, 1158 vs, 1058 vs, 774 s, 706 s, 662 s, 616 vs, 534 s, 516 s, 474 s, 418 s, Fig. 4A (see also ref. [39]). 1H NMR (D2O, DSS): δ= 2.69 (t, J 2.8 Hz, 0.39 H corresponding to 61% exchange of CH≡C to CD≡C), 3.83 (d, J 2.8 Hz, 2 H, CH2-SO3Na) (see also ref. [29, 39]) and traces of impurities at 5.36 (d, J 6.4 Hz, 0.04 H, CH2 = C=CH-SO3Na) and 6.24 (t, J 6.4 Hz, 0.02 H, CH2 = C=CH-SO3Na) and at 3.93 (s), 5.03 (s), 5.15 (s) attributable to CH2 = CCl-CH2SO3Na 13, Fig. 4B. 13C NMR (D2O, DSS): δ= 42.48 (CH2), 74.63 (CH≡), 75.60 (CH≡C–).
Products of the reaction of propargyl chloride with 0.33 M Na2SO3. A: The IR (KBr) spectrum of the tetrahydrated sodium salt of 11 showed a very strong C-H stretching for CH≡C at 3280 cm–1 while the C≡C band at 2128 cm–1 was very weak. B: The 1H NMR (D2O, DSS) spectrum of 11·4H2O showed CH2 protons as a doublet (J 2.8 Hz) at 3.82 ppm while the CH proton was at 2.69 ppm as a triplet (J 2.8 Hz) and the integration revealed 61% exchange with deuterium. Traces of the sodium salt of 13 were seen as singlets at 3.93, 5.03 and 5.15 ppm, while the sodium salt of 10 showed signals at 5.36 (d, J 6.4 Hz) and 6.24 (t, J 6.4 Hz).
The reaction of propargyl chloride with more concentrated aqueous solutions of sodium sulfite: isolation of potassium 2-chloro-2-propenylsulfonate tetrahydrate, CH2 = CCl–CH2–SO3K·4H2O, 13·4H2O, and identification of potassium allenylsulfonate 10 and potassium 2-propynylsulfonate 11
Running the reaction of propargyl chloride with 1 M (for 48 h) and 2 M (for 30 h) sodium sulfite in water, samples diluted with D2O showed the presence of CH(or D)≡C–CH2SO3Na 11 but additional singlets were seen at 5.36, 5.37, 6.00, 6.18 and a multiplet at 6.25 ppm. Two more singlets at 1.27 and 1.92 ppm were seen in the 2 M case only, thus indicating that the more concentrated the solution of Na2SO3 the more by-products are formed. In both cases the sodium salt of 12 was not present.
In order to effect the separation of the sodium salts 10, 11 and 13, Fig. 2, they were converted to their acids either by using aqueous 12 M HCl or by the cation exchange resin, followed by formation of salts that could yield a pure compound by fractional crystallization.
Treatment of a methanolic suspension of sodium salts 10, 11 and 13 plus NaCl with the calculated amount of 12 M aqueous hydrochloric acid followed by evaporation, drying in vacuum and extraction with methanol gave after evaporation and drying a white solid, composed of the acids 10, 11 and 13. Alternatively, cation exchange resin chromatography of the mixture from the 2 M Na2SO3 reaction, collection of the first 150 mL of the eluting water, evaporating (rotary, 50°C) and drying in vacuum, the acids 10, 11 and 13 were obtained as an orange-brown viscous oil. The 13C NMR (D2O, DSS) spectra of the acid mixtures showed 9 lines (some of which were split) as follows. For acid 11: 41.97, 42.01, 42.05 (CH≡C–CH2–SO3 H), 74.02, 74.09 (CH≡C–CH2–SO3 H), 74.79 (CH≡C–CH2–SO3H). For acid 10: 81.83, 81.89, 81.96 (CH2 = C = CH–SO3H), 97.77, 97.79 (CH2 = C = CH–SO3 H), 207.01 (CH2 = C = CH–SO3 H). For acid 13: 49.91, 49.95, 50.00 (CH2 = CCl–CH2–SO3H), 121.94 (CH2 = CCl–CH2–SO3H), 141.40 (CH2 = CCl–CH2–SO3H). The 1H NMR (D2O, DSS) spectra were similar, featuring the signals from the acids 10, 11 and 13 as follows. For the main product 11: 2.70 (t, J 2.4 Hz, 0.73 H corresponding to 27% exchange CH(to D)≡C–CH2–SO3H), 3.82 (d, J 2.4 Hz, 2.00 H, CH≡C–CH2–SO3 H). For the by-product 10: 5.36 (d, J 6.6 Hz, 0.94 H, CH2 = C = CH–SO3 H), 6.25 (t, J 6.6 Hz, 0.41 H, CH2 = C = CH–SO3 H). For the by-product 13: 3.93 (s, 0.36 H, CH2 = CCl–CH2–SO3H), 6.00 (s, 0.18 H, CHH = CCl–CH2–SO3H), 6.18 (s, 0.17 H, CHH = CCl–CH2–SO3H). The only difference was that in the spectrum from 2 M Na2SO3 the signals due to the acids 10 and 13 have been approximately 30 and 350% increased compared to those of the acid 11, thus indicating that the more concentrated Na2SO3 solution produced greater amounts of by-products, and the acid 12 did not seem to have been formed during the acidification.
In order to prepare separable compounds from the acids 10, 11 and 13, pyridine and triethylamine gave oils after neutralization in acetone solutions and adding ether, while from methanolic solutions nothing precipitated after addition of LiOH. However, successful separation was achieved using KOH.
To an orange solution of the mixture of acids (194 mg, 1.62 mmol) in methanol (1 mL) was added a solution of potassium hydroxide (91 mg, 1.62 mmol) in methanol (3 mL). After stirring at RT for 30 min, centrifugation and washing with methanol (2×2 mL) gave a supernatant and a beige solvate. The supernatant was evaporated and the solid was recrystallized using less methanol. The 1 H NMR spectrum in D2O indicated the presence of the potassium salt of 11 at 2.69 and 3.83 ppm the integration of which revealed 60% exchange of CH≡C to CD≡C and the potassium salt of 10 at 5.35 (d, J 6.0 Hz, 2 H, CH2 = C = CHSO3K) and 6.24 (t, J 6.0 Hz, 1 H, CH2 = C = CHSO3K), Fig. 5A. In the 13C NMR spectrum in D2O, six lines were seen: those at 38.42, 70.44 and 71.18 ppm are due to CH≡C–CH2–SO3K 11 and those at 78.29, 94.20 and 203.44 are due to CH2 = C = CH–SO3K 10. Thus, from the supernatant, containing the potassium salts of 10 and 11, no pure compound could be isolated.
Products of the reaction of propargyl chloride with 1 and 2 M Na2SO3. A: 1H NMR (D2O, DSS) spectrum of an inseparable mixture of potassium salts of 11 (at 3.83 and 2.69 ppm, the latter indicating 60% exchange of CH≡C for CD≡C) and of 10 [at 5.35 (d, J 6.0 Hz) and 6.24 (t, J 6.0 Hz)]. B: The IR (KBr) spectrum of the potassium salt 13·4H2O shows very weak stretching CH2 band at ∼3000 cm–1, while its 1H NMR (D2O, DSS) spectrum C showed only three singlets at 3.93, 6.00 and 6.18 ppm, and a trace of an impurity at 3.82 ppm.
The beige solvate, after drying in vacuum, gave a beige solid (104 mg) that was impure by 1H NMR (D2O, DSS). It was purified by washing with methanol (2×2 mL) at RT followed by trituration with boiling methanol (3 mL) and overnight stirring at RT. The product, the potassium salt 13, (71 mg, 16% as 13·4H2O) was insoluble in methanol but soluble in water. M.p.: from 125 to 200°C shrinks slightly, at 230°C shrinks faster and turns orange-brown, at 265°C turns brown and at 280–290°C turns blackish-black. Calculated for C3H4ClO3SK·4H2O (Mr 266.74): C 13.51, H 4.54, S 12.02%; found C 13.55, H 4.49, S 12.47%. IR (KBr): 3482 s, broad, 1650 w, 1198 vs, 1126 s, 1056 s, 1032 vs, 954 w, 796 w, 734 m, 654 m, 618 m, 538 m, 472 m, 420 m, Fig. 5B. 1 H NMR (D2O, DSS): δ= 3.93 (s, 2 H, CH2SO3K), 6.00 (s, 1 H, CHH = CCl–), 6.18 (s, 1 H, CHH = CCl–) and a trace of a doublet at 3.83 ppm; Fig. 5C. 13C NMR (D2O, DSS): δ= 52.81 (CH2SO3K), 124.77 (CH2 = CCl–), 144.28 (CH2 = CCl–). The electrospray ionization (positive mode) mass spectrum in MeOH/H2O 1:1 showed the presence of chlorine: 317.16 (100%) and 319.17 (35%) as well as at 213.24 (60%) and 215.26 (22%).
Results and discussion
The electrophilicity of propargyl chloride
An overview of the reactions of propargyl halides has been published [34] and revealed the absence of reactions with the alkaline aqueous solutions of Na3AsO3. The reaction of propargyl chloride (b.p. 58°C) with solid NaOH at RT for 3 days gave the chloroallene CH2 = C = CH–Cl (b.p. 45°C) and not propargyl alcohol [19] indicating that under the reported conditions the propargyl rearrangement [34] was faster than the nucleophilic attack of HO– at the –CH2Cl carbon. Thus, propargyl chloride under basic conditions can be ionized to -:C≡C–CH2Cl [11, 12], isomerized to CH2 = C = CHCl [19] and can give substitution products CH≡C–CH2–Nu with nucleophiles [34]. Additional by-products can be produced from the chloroallene [34], and from propargyl alcohol [34], if formed. We found that a solution of propargyl alcohol in D2O in the presence of NaOH or Na3AsO3 quickly (<5 min) exchanged the acetylenic hydrogen, CH≡C–, for deuterium, CD≡C–. Therefore a successful preparation of 4, 5 and 6, Fig. 1, will depend on the reaction conditions and the nucleophilicity of the As(III) species.
The nucleophilicity of As(III) was first checked with triphenyl trithioarsenite, (PhS)3As [30], extending its studies done on fatty acyl derivatives [16,36, 16,36]. Tumbling an NMR tube with equimolar quantities of propargyl chloride and (PhS)3As in CDCl3 the singlets at 2.52 and 4.12 ppm became a triplet (J 2.4 Hz) and a doublet (J 2.4 Hz) after 15 min, while after 3 h a new singlet appeared at 1.25 ppm and a more complicated pattern was seen in the aromatic region, indicating that (PhS)3As had an activity but the products could not be identified.
Due to equilibria of Equation (1) [31], the reaction of propargyl chloride with the various sodium arsenites can reveal the most reactive one. Thus, the 1H NMR spectrum from the reaction of propargyl chloride with NaH2AsO3 in H2O after 18 h showed the presence of a small signal at 1.91 ppm, attributed to the sodium salt of 6, and other, not assignable to particular compounds, signals. With Na2HAsO3 in H2O, the 1.91 ppm signal had increased and the sodium salt of 4 has also been produced. However, with 0.33 M Na3AsO3 the arsonate 6 was not seen indicating that dilution, Equation (1), gave the inert H3AsO3 [31]. The picture changed when 5M Na3AsO3 in D2O was used, where the product 6 started appearing after 18 h tumbling.
The above experiments indicated a) that the active nucleophile was the trianion AsO33–, existing in concentrated solutions, and, in order to react with the water sparingly soluble propargyl chloride, vigorous stirring is necessary [7], b) that AsO33– was as good a nucleophile as HO–, c) that the initially formed substitution product 5 rearranged prototropically to the allene 4, finally giving the observed product 6, and d) that the proton movement from –CH2-AsO32– to form the terminal methyl group is through the solution because in D2O deuterium was incorporated giving, e.g., -CHD2. The intermediate allene 4 was identified by 1 H NMR based on literature values. Thus, for CH2 = C = CH–P(O)Y2 (Y = different groups) the CH2 and CH resonate in the regions 5.00–5.62 and 5.42–6.21 ppm, respectively [18], while for CH2 = C = CH–P(O)(OR)2 resonate at 4.97 and 5.34 ppm [9]. As for the rate of isomerization 4 ⟶ 6, we note that the isomerization of CH2 = C = CH–P(O)(OEt)2 in EtONa/EtOH to CH3-C≡C-P(O)(OEt)2 required 12 h stirring at RT followed by stirring at 100 °C for 8 h [17]. Thus, in the case of propargyl chloride and Na3AsO3, a one phase system may indicate reaction of the chloride but not the complete rearrangement of 4 to 6.
The reaction of propargyl chloride with alkaline Na3AsO3: The isolation of impure 6 as an acid or a salt
Propargyl chloride reacted with Na3AsO3 in concentrated solutions using 1:1 molar ratio, excess propargyl chloride or excess NaOH. In all cases very viscous aqueous solutions were obtained the TLC of which showed a black spot at Rf ∼0.50. Samples examined by IR (KBr) showed a weak band at 2190 cm–1 due to disubstituted –C≡C–. In the literature the triple bond of CH3-C≡C-P(O)(OR)2 was found at 2208–2217 cm–1 [9, 40], while in CH≡C–CH2–P(O)(OR)2 the band was found at 2090–2123 cm–1 [9, 40]. Thus the presence of 6 and not 5 was established. A very weak band at 1953 cm–1 indicates the allenyl compound 4 by analogy with the values 1961 and 1976 cm–1 for CH2 = C = C = P(O)(OR)2 (R = Et [40] and R = Me [9]), respectively. The 1 H NMR (D2O) spectrum showed the presence of the sodium salt 6, the allenylarsonate 4, CH(or D)≡C-CH2Cl and other compounds that could not be identified; the isomer 5 and CD≡C-CH2OD probably were not present.
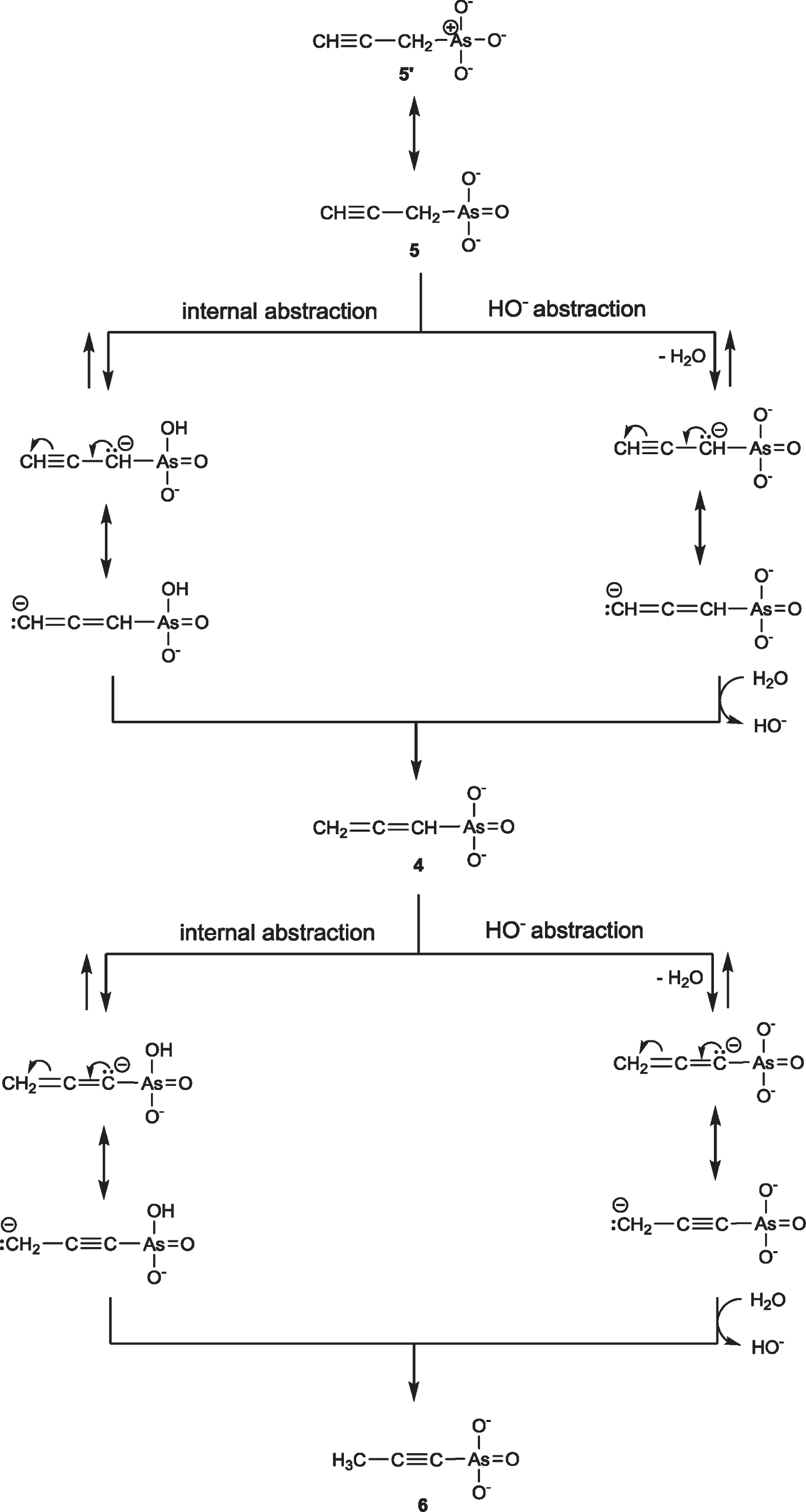
The formation of 6 is proposed to proceed via the production of 5 and 4, Fig. 6. An SN2′ displacement of -Cl by attack of AsO33– at the CH≡ carbon, although it can give the allenylarsonate 4 in just two steps and avoiding the formation of 5, probably does not operate with this primary alkyl halide, while such an SN2′ displacement is questionable even in the case of tertiary alkyne halides [34]. An SN2 reaction of AsO33 - to propargyl chloride to give 5′ indicated that AsO33– is a good nucleophile but the reaction is slower compared to that of allyl bromide (2-3 h) and compares better to the reaction of n-propyl bromide (24–48 h) [26]. From 5 the detected allenylarsonate 4 can be formed by a prototropic propargyl rearrangement [34] and reacts by a second prototropic (allenyl) rearrangement [34]. In Fig. 6, the abstraction of the α-H by HO- should be favored at the initial stages where the concentration of HO-, due to Equation 1, is higher, while the internal abstraction, being possible because of the pKa2 ∼8 of arsonic acids [8], at later stages of the reaction. That the –AsO32– group can abstract the α-H from 5 to give 4 is corroborated from the Arbuzov and Nylen reactions [9] which, in the absence of HO–, gave poor yields of phosphonates as was mentioned in the Introduction. Moreover, the formation of allenyl and α-propynyl phosphonates [40] indicates that the –P(O)(OR)2 group can abstract the α-H, although to a smaller extent compared to the –AsO32– group. That 4 should be an intermediate to 6 is also corroborated from the report that allenylphosphonates can be isomerized to CH3–C≡C–P(O)(OR)2 in the presence of an alkoxide [17].
The reaction of propargyl chloride with alkaline Na3AsO3 will give 5′ and 5 by an SN2 reaction followed by the formation of 4 by a prototropic rearrangement (propargylic rearrangement) and by a second prototropic (allenyl) rearrangement 6 will be produced. The α-H abstraction can be effected by the basic –AsO32– group or by HO– present due to hydrolysis of –AsO32– and AsO33–.
The classical method for isolating aliphatic arsonic acids from the reaction of an alkyl halide and Na3AsO3 is to acidify to pH 2-3 with concentrated hydrochloric acid, filter the NaCl and As2O3 (from the Na3AsO3 that had not reacted) and recrystallize the arsonic acid [26]. Following this strategy we continuously found that the HCl consumed was less than the calculated and the NaCl and As2O3 were not precipitated quantitatively. After a laborious procedure with extractions, the free acid 6 was obtained as a foam in 30% yield, containing acid 4. It foamed at 104–107°C and decomposed at 130–132°C, these values being quite close to the melting of the allylarsonic acid 2 at 129°C [26]. Because the product was impure, elemental analyses were not performed. Its IR (KBr), Fig. 3A, although having somewhat broad bands showed a very strong but sharp band for disubstituted –C≡C–bond at 2202 cm–1 and a very weak band at ∼1970 cm–1 for C=C=C. The arsonic group –AsO3H2, showed a medium band at ∼2900 cm–1, due to δ(As–OH) [37], a very strong band at 902 cm–1 (due to ν(As = O)) and at 786 cm–1 (due to ν(As–O)). A band at 3418 cm–1 indicates hydroscopicity of the acid 6. The 1H NMR (D2O), Fig. 3B, showed the methyl protons of the acid 6 at 2.02–2.10 ppm, compared to 1.91 ppm of the sodium salt of 6. Apart from the presence of acid 4 and Me2CO, many small signals were seen and the purity of acid 6 was estimated to be >80%. Thus, this classical method afforded an impure product and the absence of traces of co-extracted As2O3 (804 cm–1 in the IR) was not certain.
In another strategy, we first removed the NaCl by a cation exchange resin and the semi-solids obtained were purified by trituration with Et2O (removing organic compounds) and extracting the impure acid 6 with acetone (leaving As2O3: at least ∼30% recovered in various runs).
The impure acid 6, could not be purified by an anion exchange resin because all fractions, obtained with 1 M aqueous AcOH elution, contained only traces of solids indicating decomposition of acid 6. Arsonic acids are known to decompose under drastic conditions by acids and bases [8], but certain aliphatic arsonic acids can decompose under very mild conditions, e.g. by acylating agents [38] or by reduction with thiophenol [15]. In the case of the acid 6 decomposition on the anion exchange resin can be effected by the acetate anion of the 1 M acetic acid eluting solvent, giving the unknown and presumably unstable in 1 M aqueous acetic acid CH3–C≡C–OCOCH3 and resin-bound AsO3H2–, based on the observation that anionic (e.g. RO-, X-, RS- and neutral (e.g. R3N, R3P, (RO)3P) nucleophiles can displace the initially bound halogen in R-C≡C-X compounds [3, 22].
The impure acid 6, obtained from the cation exchange resin, could not be purified by salt formation. Thus, when its aqueous solutions were treated with pyridine followed by addition of acetone an oil was formed, with LiOH and Ba(OH)2·8H2O the solids obtained after addition of acetone were either impure or could not be characterized, and with aqueous BaCl2·2H2O and addition of acetone BaCl2·2H2O (by IR [23]) was precipitated. With aqueous Ba(AcO)2 the solution when treated with acetone gave a water-insoluble solid of unknown constitution and adding more acetone to the supernatant a water-soluble solid was obtained that showed in the IR (KBr) a very strong band at 838 cm–1, indicative of –AsO32– and/or –AsO3H- groups [37]. Its 1H NMR (D2O) spectrum showed a singlet at 1.90 ppm due to the barium salt 6 and a singlet at 1.94 ppm due to CH3COO-, but their integration (3:1.5) indicated that the solid was a mixture of (CH3–C≡C–AsO3)2Ba and CH3–C≡C-AsO3HBaOCOCH3. Finally, Pb(AcO)2·3H2O in water gave a precipitate the supernatant of which contained some lead salt of 6 and other unknown impurities. The product, CH3–C≡C-AsO3Pb·H2O, was a soft white solid, insoluble in H2O and DMSO, did not melt up to 300°C and its IR (KBr and Nujol) showed a medium weak band at 2188 cm–1 for disubstituted –C≡C– and a very weak band at 1942 cm–1 due to allenic C=C=C, compared to very strong bands at 834 and 786 cm–1, Fig. 3C. In the IR (KBr) spectrum shown in Fig. 3C, two things should be noticed: a) the –C≡C– band of the lead salt of 6 was medium weak compared to the –C≡C– band of the acid 6 compared to the –AsO32– and –AsO3H2 bands, respectively, and b) the –AsO32– band of the lead salt of 6 showed the symmetric and degenerate AsO3 vibrations [32] at 834 and 788 cm–1, while in other neutral arsonic salts (even when hydrated) one band was seen absorbing in the region 800–860 cm–1 [37]. Thus, from aqueous solutions containing the impure acid 6 a pure barium or lead salts of 6 could not be isolated.
Thiophenol reduced the mixture of acids 4 and 6 in methanol [30] giving the corresponding diphenyl dithioarsonites, R-As(SPh)2, and the disulfide PhSSPh, but no separation could be achieved by selective extraction. Finally, reduction of these arsonic acids by ascorbic acid in methanol in the presence of a catalytic amount of iodine [14] followed by silica gel column chromatography no pure product RAs(OH)2 or (RAsO)x could be isolated.
The reaction of propargyl chloride with a dilute (0.33 M) solution of sodium sulfite: isolation of CH≡C–CH2–SO3Na·4H2O, 11·4H2O
The pH of an aqueous solution of Na2SO3 is ∼9 [41] due to the equilibria of Equation (2). Because the pKa1 and pKa2 of H2SO3 are 1.76 and 7.21, the dominating species should be the monoanion HSO3–, which should be as nucleophilic towards alkyl halides as H2AsO3– or HAsO32– are (Section 3.1).
The reaction of propargyl chloride with an equimolar quantity of 0.33 M Na2SO3 in D2O in a tumbling NMR tube revealed the presence of 11 as CD≡C–CH2SO3Na at 3.83 ppm and CD≡C–CH2Cl at 4.23 ppm during the period of 3–48 h. Singlets initially (5 min) seen at 2.70 and 3.00 ppm disappeared after 3 h tumbling. Thus, a clean reaction was observed and this was adjusted to a preparative scale.
In the literature [29, 39] the anhydrous(?) sodium salt 11, Fig. 2, was prepared from propargyl bromide (in toluene) and 26% mol excess sodium sulfite in methanol/water (1:1 v/v) in yields 69–92%. We run the preparation of 11 from propargyl chloride using equimolar quantities of sodium sulfite in de-aerated water. After 24 h reaction at RT, the water was removed by freeze-drying where no rearrangement in the system was noticed by 1 H NMR spectra before and after freeze-drying. In order to separate the product 11 as sodium salt from NaCl and Na2SO3/Na2SO4 extraction with methanol effectively removed these insoluble solids. From the supernatant the sodium salt of 11 as a tetrahydrate was isolated in 77% yield based on the Na2SO3 and purity of > 95%. Its IR (KBr) spectrum, Fig. 4A, showed a very weak band for a monosubstituted triple bond at 2128 cm–1 and the very strong H–C≡C stretching band at 3280 cm–1. Also the band at ∼3500 cm–1 was split due to free and intramolecularly bound waters. Our IR spectrum did not match with that reported for the presumably anhydrous sodium salt 11 [39].
The 1H NMR spectrum of the sodium salt 11 in D2O, Fig. 4B, showed the CH2–SO3Na protons as a doublet (J 2.4 Hz) at 3.82 ppm and the CH≡C proton as a triplet (J 2.4 Hz) at 2.69 ppm with the integrations corresponding to 61% exchange of CH≡C to CD≡C indicating that K = [CD≡C–] ÷ [CH≡C–] = 1.56. The other very small signals in Fig. 4B can be assigned to the sodium salts of 10 and 13. The chemical shifts found for the sodium salt 11 in D2O were different from those reported: 4.08 and 2.99 ppm [39] and 3.69 and 2.56 ppm [29], most likely due to not buffered solutions. The 13C NMR spectrum in D2O showed three lines as expected at 42.48, 74.63 and 75.60 ppm, while in the literature [39] they were found at 39.6, 71.9 and 72.6 ppm.
The reaction of propargyl chloride with more concentrated (1 or 2 M) solutions of sodium sulfite: isolation of CH2 = CCl–CH2–SO3K·4H2O, 13·4H2O
Exploratory experiments with 1 or 2 M Na2SO3, where the HO– concentration is expected to be higher than in dilute solutions, Equation 2, revealed that a significant amount of by-products were produced including the sodium salt of 13, Fig. 2. However, the sodium salt of 12 was not present indicating that a more alkaline solution is required for the prototropic rearrangement 10 ⟶ 12 (see also ref. [17]).
In preparative experiments, the crude reaction mixtures containing the expected sodium salts of 10–13, were converted to their acids by either 12 M HCl or cation exchange resin. Formation of suitable salts might then lead to the separation of the component sulfonates.
From the mixture of acids 10, 11 and 13 in methanol, treatment with methanolic KOH precipitated the potassium salt of 13 as a beige solvate leaving in the supernatant the potassium salts of 10 and 11, having the NMR spectrum of Fig. 5A, that could not be separated. The 13C NMR singlets of 11 as potassium salt were seen at 38.42, 70.44 and 71.18 ppm, while for the sodium salt of 11 the singlets were at 42.48, 74.63 and 75.60 ppm but in the acid 11, however, split singlets were seen at 41.97/42.01/42.05, 74.02/74.09, and 74.79 ppm. For the potassium salt of 10 singlets were seen at 78.29, 94.20 and 203.44 ppm, while the free acid 10 showed split singlets at 81.83/81.89/81.96, 97.77/97.79, and 207.01 ppm.
The beige solvate after trituration with boiling methanol and drying gave the potassium salt of 13 as a tetrahydrate (by elemental analyses), which slowly decomposed and charred at 280–290°C. The IR (KBr) spectrum showed absence of bands due to –C≡C–, CH≡C–, and C=C=C. The dominant bands were due to the –SO3– group, Fig. 5B. The 13C NMR spectrum showed the presence of three carbons and the electrospray ionization (positive mode) revealed the presence of chlorine (–Cl). Accordingly the three singlets in the 1H NMR spectrum, Fig. 5C, should be assigned to the CH2SO3–, and the two CH2 = protons of 13.
The mechanism of formation of 13 in the concentrated solutions of Na2SO3 can be explained by a nucleophilic attack of Cl– at the central carbon of 10 which acquired a positive charge [34] due to the electron withdrawal effect of the –SO3– group. This reaction can, therefore, take place when enough 10 and NaCl is produced. In the literature [5] the chloride CH2 = CCl–CH2–SO2–Cl has been isolated from the reaction of POCl3 with a mixture of the sodium salts of 10 and 11 and hypothesized that the chloride probably arose from a direct attack of HCl to 10 and/or 11 or their corresponding sulfonyl chlorides. The question why an analogous reaction did not take place in the arsenite system after the formation of the sodium salt of 4 (having a quite strong electron withdrawing –AsO32– group) can be answered by the fact that in this case the alkalinity was very high (pH > 14) and the prototropic rearrangement to 6 was greatly favored (see also ref. [17]).
Conclusions
Propargyl chloride reacted only with concentrated aqueous solution of Na3AsO3 giving sodium 1-propynylarsonate 6 plus very small amounts of sodium allenylarsonate 4, Fig. 1, and other unknown by-products. The sodium salt of 2-propynylarsonic acid 5 was not detected indicating that 5 suffered propargyl and allenyl rearrangements due to the high alkalinity in the reacting system. Efforts at isolation of pure acid 6 or pure lead salt of 6 were not successful. Reduction of the arsonic acids 4 and 6 with thiophenol and by ascorbic acid/iodine followed by selective extractions or chromatography were also not successful in isolating a pure As(III) derivative of 6.
The reaction of propargyl chloride with dilute, ∼0.3 M, sodium sulfite produced the desired sulfonate 11·4H2O in good yields and purities, while with concentrated (1 or 2 M) solutions of Na2SO3 by-products are formed. One by-product, 10, has been identified spectroscopically, while the second, 13, has been isolated as a potassium salt. In all cases sodium 1-propynylsulfonate 12 was not formed indicating that the alkalinity of the systems was not enough to initiate an allenyl rearangement.
Footnotes
Acknowledgments
We thank Professor G. M. Tsivgoulis of this Chemistry Department for the interpretation of some spectra encountered in this study.
References
1.
AndersenK.K., Sulfonic acids and their derivatives, in Comprehensive Organic Chemistry, (
BartonD. and OllisW.D., editors), 1979, Pergamon Press, Oxford, vol. 3, pp. 331–362.
BeltrameP., GavezzottiA. and SimonettaM., Reaction of halogenoacetylenes with sulfur nucleophiles studied by Extended Hückel Molecular Orbital theory, J S C Perkin II (1974), 502–507.
4.
BouchmellaK., DutremezS.G., GuérinC., LongateJ.-C. and
DahanF., Guanidinium alkynesulfonates with single-layer stacking motif: Interlayer hydrogen bonding between sulfonate anions change the orientation of the organosulfonate R group from “alternate side” to “same side”, Chem Eur J16 (2010), 2528–2536.
5.
BradamanteS., Del ButteroP. and
MaioranaS., Reactons of prop-2-yne-1-sulphonyl chloride with α-morpholinostyrene, J C S Perkin I (1973), 515–615.
6.
DistlerH., Zur Chemie der Vinylsulfosäure, Angew Chem77 (1965), 291–311.
7.
DixonH.B.F., The biochemical action of arsonic acids especially as phosphate analogues, Adv Inorg Chem44 (1997), 191–227.
8.
DoakG.O. and
FreedmanL.D., Organometallic Compounds of Arsenic, Antimony and Bismuth, Wiley, N. York, 1970, Chapter II, pp. 17–62.
9.
GordonM. and GriffinC.E., Phosphonic acids and esters. XIII. Nylen and Arbuzov reactions with propargyl bromide, J Org Chem31 (1966), 333–334.
10.
GriffinC.E. and
MitchellT.D., Phosphonic acids and esters. IX. Thermal reactions of trialkylphosphites with nonactivated acetylenes, J Org Chem30 (1965), 1935–1939.
11.
HartzlerH.D., Carbenes from derivatives of ethylcarbinols. The synthesis of alkenylidenecyclopropanes, J Am Chem Soc83 (1961), 4990–4996.
12.
HennionG.F. and
NelsonK.W., The kinetics of the hydrolysis of acetylenic chlorides and their reaction with primary and secondary amines, J Am Chem Soc79 (1957), 2142–2145.
13.
IhataO., FujitaM. and KuribayashiH., Copolymer containing functional group of sulfonic acid or its salt, and process for producing said polymer, US Patent 7816474 (2010) to Sumimoto Chemical Company Limited, Tokyo, Japan.
14.
IoannouP.V., On the direct reduction of arsonic acids to arsenoso compounds. Mechanisms and preparations, Appl Organometal Chem14 (2000), 261–272.
15.
IoannouP.V., AfroudakisP.A. and SiskosM.G., Preparation of 2-picolylarsonic acid and its reductive cleavage by ascorbic acid and by thiophenol, Phosphorus, Sulfur, and Silicon177 (2002), 2773–2783.
16.
IoannouP.V. and TsivgoulisG.M., The nucleophilicity of M(SPh)3 (M=P, As) and L-As(SPh)2 (L=Ph, 2-O2N-Ph) towards fatty acyl chlorides, RCOCl, Main Group Chem10 (2011), 177–185.
17.
IoninB.I. and
PetrovA.A., Prototropic isomerization of phosphonic esters containing acetylenic, dienic, and enynic groups, J Gen Chem USSR34 (1964), 1165–1170.
18.
IoninB.I., Ignat’evV.M. and LebedevV.B., Investigation of organophosphorus compounds by the NMR method analysis of A2BX spectra of propadienylphosphonic derivatives, J Gen Chem USSR37 (1967), 1774–1782.
19.
JacobsT.L., McClenonJ.R. and MuscioO.J.Jr., The stereochemistry of allene dimerization, J Am Chem Soc91 (1969), 6038–6041.
20.
KrookM.A., Propargyl chloride, in Encyclopedia of Reagents for Organic Synthesis, (
PaquetteL. A. editor), 1995, Wiley, Chichester, vol. 6, pp. 4315–4317.
21.
MeyerG., Ueber einige anomale Reactionen, Chem Ber16 (1883), 1439–1443.
22.
MillerS.I. and DicksteinJ.I., Nucleophilic substitution at acetylenic carbon. The last holdout, Acc Chem Res9 (1976), 358–363.
23.
MillerF.A. and WilkinsC.H., Infrared spectra and characteristic frequencies of inorganic ions, Anal Chem24 (1952), 1253–1294.
24.
PietchR., Untersuchungen zur Umsetzung von Alkylbromiden mit Trinatriumarsenit, Monatsch Chem96 (1965), 138–146.
25.
PudovikA.N., Zh Obshch Khim20 (1950), 92; cited by C. E. Griffin and T. D. Mitchell.
26.
QuickA.J. and AdamsR., Aliphatic arsonic and arsinic acids, and aliphatic-aromatic arsinic acids, J Am Chem Soc44 (1922), 805–816.
27.
RadA.v., Ueber Allylsulfonsäure und einige ihrer Salze, Ann Chem Pharm161 (1872), 218–224.
28.
ReissigH.-U. and ZimmerR., Synthesis of linear allenes from other allenes, Sci Synth44 (2007), 301–352.
29.
RyuE.-H. and ZhaoY., Efficient synthesis of water-soluble calixarenes using click chemistry, Org Lett7 (2005), 1035–1037.
30.
ServesS.V., CharalambidisY.C., SotiropoulosD.N. and IoannouP.V., Reaction of As(III) oxide, arsenous and arsenic acids with thiols, Phosphorus, Sulfur, and Silicon105 (1995), 109–116.
31.
ServesS.V., SotiropoulosD.N., IoannouP.V. and DixonH.B.F., On the mechanism of the Meyer reaction with epoxide and 2-haloalcohols as substrates, Phosphorus, Sulfur, and Silicon90 (1994), 103–109.
32.
SimonA. und
SchumannH.-D., Raman- und Infrarotspektroskopische Untersuchungen an Alkylderivaten der Arsensäure. I. Die Schwingungsspektren einiger Alkalialkanarsonate, Z Anorg Allg Chem393 (1972), 23–38.
33.
StreckerA., Ueber eine neue Bildungsweise und die Constitution der suflosäuren, Ann Chem Pharm148 (1868), 90–96.
34.
TaylorD.R., The chemistry of allenes, Chem Rev67 (1967), 317–359.
35.
TsivgoulisG.M. and IoannouP.V., A reinvestigation of the synthesis of arsonolipids (2,3-diacyloxypropylarsonic acids), Chem Phys Lipids152 (2008), 113–121.
36.
TsivgoulisG.M. and IoannouP.V., The nucleophilicity of M(SPh)3 (M=P, As) and PhAs(SPh)2 towards carboxylic acid anhydrides, Main Group Chem10 (2011), 51–62.
37.
TsivgoulisG.M., SotiropoulosD.N. and IoannouP.V., 1,2-Dihydroxypropyl-3-arsonic acid: A key intermediate for arsonolipids, Phosphorus, Sulfur, and Silicon57 (1991), 189–193.
38.
TsivgoulisG.M., SotiropoulosD.N. and IoannouP.V., Decomposition of alkylarsonic acids by acylating agents, Phosphorus, Sulfur, and Silicon55 (1991), 165–168.
39.
WeinhartM., GrögerD., EndersS., DerneddeJ. and HaagR., Synthesis of dendritic polyglycerol anions and their efficiency towards L-selectin inhibition, Biomacromolecules12 (2011), 2502–2511.
40.
WelchC.M., GonzalesE.J. and GuthrieJ.D., Derivatives of unsaturated phosphonic acids, J Org Chem26 (1961), 3270–3273.
41.
WindholzM., The Merck Index, 9th Edition, 1976, Merck and Co, Rahway, N. J., USA.