Aiming at the preparation of the novel unnatural, non-isosteric sulfonolipids bearing one, two and three acyl groups 8, 9 and 10, their precursors hydroxyl-containing sulfonates have been prepared from a variety of hydroxyl-containing halogenides and epoxides using the Strecker reaction. Thus, the sulfonates 16 and 22 were prepared pure, while the sulfonate 27 could only be prepared as a by-product using 1,4-dibromo-2,3-butanediol 26 and in low yields. For these reactions, probable pathways leading to the isolated or spectroscopically identified products are proposed. Conclusions about the relative nucleophilicity of SO32- compared to AsO33 - (as well as HO- which is present in their aqueous solutions) were drawn based on the yields of the corresponding arsonic acids and sodium sulfonates. The IR (KBr) and 1H NMR (D2O) spectra of sulfonates (and in some cases of their sulfonic acids) are analyzed and discussed.
There is a variety of novel natural lipids, see ladderane phospholipids and other examples [18], as well as unnatural ones that have been synthesized [1] in order to throw some light on various aspects of cellular processes.
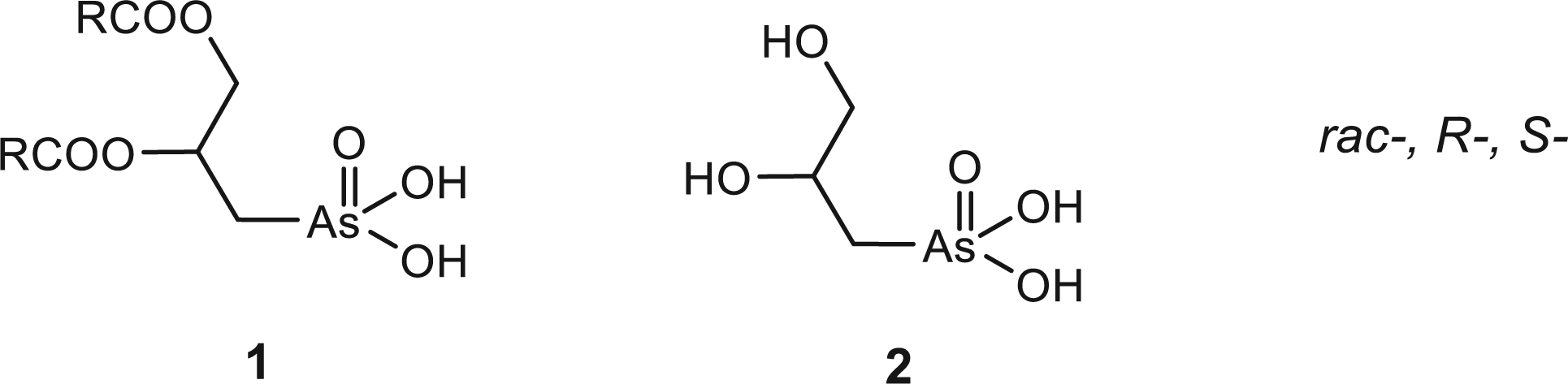
We prepared and studied a new class of unnatural lipids 1, called arsonolipids [34], in racemic and optically active forms which have interesting biophysical, biochemical and biological properties that have been reviewed [11].
Their preparations started from 3-chloro-1,2-propanediol and rac, R- and S-glycidols (20 and 21 in Fig. 1) that were reacted with Na3AsO3 giving the arsonic acid 2 which after reduction to an As(III) derivative, acylation and oxidation gave the arsonolipid 1.
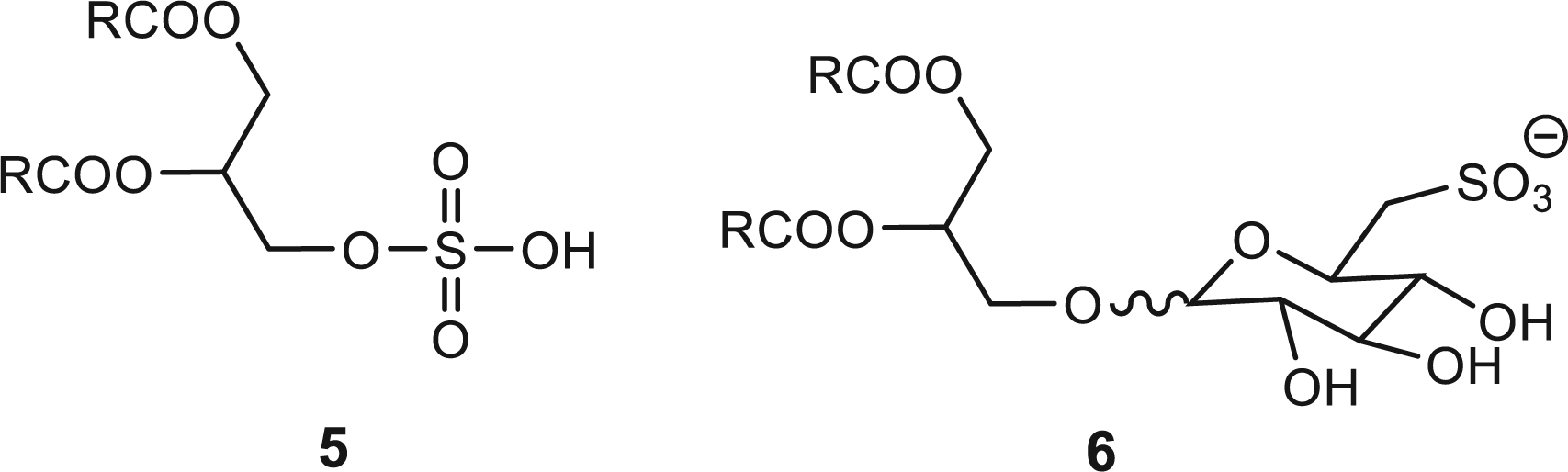
Starting compounds and the corresponding sulfonates isolated or identified in this work.
Sulfur is found in all living species. When sulfur is part of a lipidic moiety the molecules are called sulfolipids. The term sulfatide is used when the sulfur occurs as an ester 3, while the term sulfonolipid (by analogy with phosphonolipid) is reserved for molecules bearing the sulfur in the form of sulfonic acid 4 [9].
A sulfatide 5 has been prepared and used for studies on phospholipase A2 [25]. Although many sulfonolipids have been detected in Nature, the most abundant and best known is sulfoquinovosyldiacylglyerol [1,2-di-O-acyl-3-O-(6′-deoxy-6′-sulfo-α-D-glucopyranosyl-sn-glycerol] 6 [9], a key component of the photosynthetic mechanism of higher plants and other photosynthetic organisms. Both α anomer [6] and β anomer [17] have been synthesized.
Sulfonolipids having the acyl and the sulfonyl groups quite close, e.g. the isosteric [4] sulfonolipids 7 (R = C11H23, C13H27) have been prepared according to Equation (1) but no yields, properties or uses have been reported [10].
The sulfonolipids 8, 9 and 10 can be prepared from a suitable 1-halo-2-alkanol or epoxide and sodium sulfite (the Strecker reaction [31]) producing sulfonated intermediates that on acylation can provide the three target sulfonolipids.
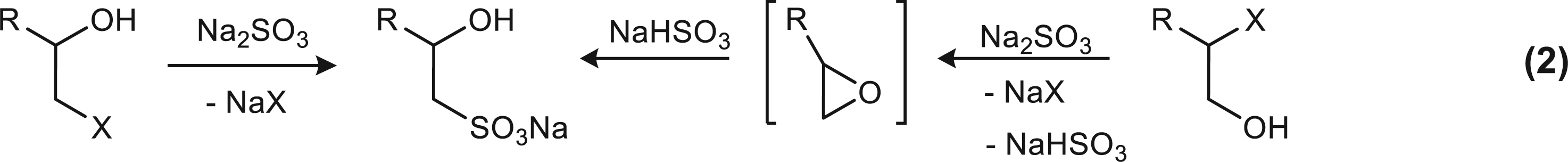
The Strecker reaction uses alkyl halides for the preparation of sulfonates with short [12] and long carbon chains [24]. The conversion of the salts to acids [20, 36] is complicated but a cation exchange resin can be used. In the literature there are reports on the Strecker reaction with 1-halo-2-alkanol [22, 30] and 2-halo-1-alkanol [22] which afforded 2-hydroxy-1-sulfonates as shown in Equation (2) where in one case epoxide formation is postulated [22].
The Strecker reaction on epoxides can be effected by Na2SO3 and NaHSO3. As early as 1868, Erlenmeyer obtained sodium isethionate 16 (Fig. 1) from ethylene oxide and Na2SO3 in an autoclave [5]. Studies revealed that the ring opening is due to SO32- and not to HSO3- [26, 38] and a free radical mechanism does not operate [26]. However, there are reports on the reactivity of NaHSO3. Thus, excess NaHSO3 reacted with 1,2-epoxyoctane giving 2-hydroxyoctane-1-sulfonate [22] and propylene oxide giving 2-hydroxypropane-1-sulfonate [30]. The products obtained from 1,2-epoxy compounds are expected to be the 2-hydroxy-1-sulfonates by attack of SO32- at the terminal position which is less sterically hindered and SO32- is a bulky nucleophile [38]. However, there are reports with, e.g., 1,2-epoxyoctane where the isomeric 1-hydroxyoctane-2-sulfonate has been produced (15% with 1 M Na2SO3) [26] but others report the production of 2-hydroxyoctane-1-sulfonate in water (69%) [22] or in microemulsions containing a phase transfer catalyst [8]. Finally, it has been demonstrated that an epoxide (1,2-epoxyoctane) was more reactive than 1-halo-2-alkanol with NaHSO3 giving the 2-hydroxy-1-sulfonate [22].
In this paper we describe the Strecker reaction with the substrates and the produced sulfonates (isolated or identified) shown in Fig. 1. The sulfonates 12 and 14 are known but we prepared them to be used for synthesis of other novel synthetic sulfonolipids.
Experimental
Materials and methods
Allyl chloride and bromide 11 (Merck) were redistilled. 4-Bromo-1-butene 13, 2-chloroethanol (ethylene chlorohydrin) 15, 3,4-epoxy-1-butene (butadiene monoxide) 17, 3-chloro-1,2-propanediol 20, rac-glycidol 21, 3,4-dihydroxy-1-butene 23 and 1,4-dibromo-2,3-butanediol 26 were from Aldrich and used as received. Crystalline sodium sulfite (Riedel-de Haën) was used as received. Crude 3,4-epoxybutane-1,2-diol 24 was prepared from 3-butene-1,2-diol 23 by epoxidation with m-chloroperbenzoic acid in dichloromethane [7, 16] having the same 1H NMR as that reported [16] and 4-chlorobutane-1,2,3-triol 25 was prepared by ring opening of 24 by concentrated hydrochloric acid contaminated by trans-3,4-dihydroxytetrahydrofuran 29 (see Fig. 6) [16]. Dowex 50W-X8 (H+ form) cation exchange resin (Bio-Rad) and silica gel 60 H (Merck) for thin layer chromatography (TLC) were used for chromatographies. 95% Ethanol (referred to as ethanol) was used for extractions and the drying of the sulfonates has been done in vacuo over P2O5 (referred to as drying).
Removal of the water from the Strecker reaction was usually done on a rotary evaporator at ca 70°C. TLCs were run on microslides and visualization was effected by spraying with 35% sulfuric acid and charring. Infra red spectra were taken in KBr discs on a Perkin-Elmer, model Spectrum RX I, FT-IR spectrometer. NMR spectra (1H at 600 MHz, 13C at 151 MHz) were obtained on a Bruker, model Avance III HD, spectrometer using TMS or DSS (sodium 2,2-dimethyl-2-silapentane-5-sulfonate) as internal standards. Elemental analyses were obtained through the Center of Instrumental Analyses, University of Patras.
Preparation of sodium allyl sulfonate, 12
To a solution of sodium sulfite (12.60 g, 0.1 mol) in water (50 mL) in a 250 mL round- bottomed flask was added neat allyl chloride 11 (12.0 mL, 0.13 mol) and the emulsion stirred at room temperature (RT) for 3 days. A clean solution was obtained after 2 days. Evaporation gave a solid that was dried in vacuo (21.90 g; expected product 14.40 g, NaCl 5.85 g). Methanol (100 mL) was added, boiled and filtered (fluted paper) hot, whereupon the product crystallized, and the filter with the NaCl and crystallized product was washed with boiling methanol (50 mL). Concentration to ca 60 mL, leaving at RT overnight, filtration (sintered glass) and washing with cold methanol (10 mL) gave the product (9.34 g) that was dried in vacuo for 3 days (weight loss 300 mg) and at 100°C for 12 h (weight loss 23 mg) to give the anhydrous product 12 (9.02 g, 63%) as a white solid very soluble in methanol. Melting point: 239–242°C shrinks and at 254°C decomposes; lit. [33]: 239–241°C. IR (KBr), : 3484 m, broad, 3086 vw, 3020 vw, 2981 vw, 2928 vw, 1426 w, 1400 vw, 1265 s, 1222 vs, 1190 vs, 1060 vs, 995 w, 935 w, 915 m, 797 m, 665 s, 534 s. 1H NMR (D2O, DSS): δ (ppm): 3.64 (d, 3J = 7.8 Hz, 2 H, CH2DSO3Na), 5.36 (d, 3JBC (trans) = 17.2 Hz, 1 H, CHAHB = CHCCH2D), 5.38 (d, 3JAC (cis) = 10.0 Hz, 1 H, CHAHB = CHCCH2D), 5.92 (m, 1 H, CHAHB = CHCCH2D).
Preparation of sodium but-3-ene-1-sulfonate, 14
In a wide mouth 100 mL round-bottomed flask was dissolved sodium sulfite (13.86 g, 0.11 mol) in water (50 mL), yellowish 4-bromo-1-butene 13 (10.2 mL, 0.1 mol) was syringed in and the heterogenous system was refluxed (oil bath, 135°C) for 17 h to give a yellowish solution. Evaporation gave an off-white solid that was transferred to a 500 mL conical flask, boiled with ethanol (300 mL) and filtered (sintered glass) hot to remove excess Na2SO3. The ethanolic filtrate after cooling at +4°C overnight gave white flakes. Filtration and drying gave a solid (13.00 g) that was recrystallized from ethanol (200 mL) and after cooling at +4°C overnight gave the monohydrated product 14 (11.03 g, 63%) as a white solid, very soluble in water, soluble in methanol, insoluble at RT but moderately soluble in hot ethanol and insoluble in acetone, ethyl acetate and methyl cyanide. Melting point: at 245°C darkens, at 247°C shrinks, becomes translucent and brownish and thereafter foams. Calculated for C4H7O3SNa·H2O (Mr 176.16): C 27.27, H 5.15%; found: C 27.16, H 4.81%. IR (KBr), : 3598 w, 3526 w, 3400 w, 3080 w, 2980 w, 2926 w, 1644 w, 1618 w, 1450 w, 1418 w, 1284 m, 1220 vs, 1066 vs, 996 w, 914 m, 800 w, 746 m, 644 w, 602 m. 1H NMR (D2O, DSS): δ 2.48 (dddt, 2 H, 3JDE = 7.8 Hz, 3JCD = 7.8 Hz, 4JAD = 1.2 Hz, CHAHB = CHCCH2DCH2ESO3Na), 2.99 (t, 2 H, 3JDE = 7.8 Hz, CH2ESO3Na), 5.07 (ddt, 1 H, 3JAC (cis) = 10.2 Hz, 4JAD = 1.2 Hz, CHAHB = CHCCH2D), 5.15 (ddt, 1 H, 3JBC (trans) = 17.4 Hz, 4JBD = 1.8 Hz, CHAHB = CHCCH2D), 5.91 (m, 1 H, CHAHB = CHCCH2D). The chemical shifts reported for compound 14 in literature [14] are shifted by 0.16 ppm apparently due to reference error.
From the ethanolic filtrates more product (3.13 g, 18%) was isolated. Total yield 81%.
Preparation of crude but-3-ene-1-sulfonic acid
A mixture of 14 and NaBr (3.78 g) in water (20 mL) was applied onto a cation exchange resin and eluted with water (200 mL). Evaporation gave a light orange oil which turned brown at the end of evaporation. The brown oil (1.83 g) was dried giving a brown mobile oil (1.07 g). Its IR (neat) revealed too much water and its 1H NMR (D2O, DSS) revealed an impure product, δ: 2.48 (m, 2 H, CH2CH2SO3 H), 2.98 (m, 2 H, CH2CH2SO3 H), 5.10 (dd, 2 H, CH2 = CH), 5.90 (m, 1 H, CH2 = CH). The impurity (∼ 40%) had the following δ: 1.72 (d, 3 H, CH3), 2.20 (m, 2 H, CH2CH2SO2-O), 3.10 (m, 2 H, CH2CH2SO2-O), 4.37 (m, 1 H, CHCH3) and was tentatively assigned as 5-methyl-1,2-oxathiolane-2,2-dioxide [29] despite small shift variations comparing to the literature values, since in literature CDCl3 is used as solvent instead of D2O.
Preparation of sodium 2-hydroxyethane-1-sulfonate, 16
In a 100 mL round-bottomed flask was dissolved sodium sulfite (12.60 g, 0.1 mol) in water (50 mL), 2-chloroethanol 15 (6.7 mL, 0.1 mol) was syringed in and the emulsion was stirred at RT (in order to diminish the evaporation of ethylene epoxide, b.p. 11°C) till a clear, colorless solution was obtained (5 days). The solution was transferred to a 250 mL conical flask and the water was evaporated while stirring on a hot plate to give a wet white solid (evaporation on a rotary evaporator was problematic because of frothing). To the wet solid ethanol (150 mL) was added, boiled and filtered (hot sintered glass and celite) hot. (The solid on the filter was washed with boiling ethanol (50 mL) and the cooled filtrate deposited 69 mg of product). The hot ethanolic filtrate, containing the product and ethylene glycol, on cooling at RT solidified. After leaving at RT overnight, filtration and trituration with warm ethanol (100 mL) gave, after drying, the product 16 (5.60 g, 34%) as non-hygroscopic white flakes, soluble in water and methanol, sparingly soluble at RT and moderately soluble in hot ethanol and insoluble in acetone. Melting point: at 189°C turns light orange, at 202°C shrinks, at 220°C bubbles and at 245°C turns orange-brownish. Calculated for C2H5O4SNa·H2O (Mr 166.13): C 14.46, H 4.25, S 19.30%; found: C 14.58, H 4.57, S 19.03%. IR (KBr), : 3424 vs, broad, 2944 w, 2894 w, 1654 m, 1398 m, 1318 m, 1208 vs, 1190 vs, 1168 vs, 1074 vs, 1060 vs, 952 w, 852 w, 752 vs, 616 s, 590 s, 538 vs, 474 s. 1H NMR (D2O, DSS): δ 3.14 (t, 3J = 6.3 Hz, 2 H, CH2SO3Na), 3.94 (t, 3J = 6.3 Hz, 2 H, CH2OH).
All the ethanolic filtrates were combined, evaporated to dryness giving 3.00 g of a white solid that was triturated with hot ethanol (30 mL) to extract ethylene glycol. After cooling at RT, filtration gave more product (2.07 g, 12%). Total yield 46%.
Reaction of 3,4-epoxy-1-butene 17 with Na2SO3: isolation of sodium 4-hydroxybut-1-ene-3-sulfonate, 19
To a solution of sodium sulfite (3.13 g, 25 mmol) in water (10 mL), 3,4-epoxy-1-butene 17 (2.0 mL, 25 mmol) was syringed in whereupon heat evolved. The emulsion was stirred at RT for 24 h, the clear solution was cooled (ice-water), acidified to pH 6 with concentrated hydrochloric acid, evaporated and dried in vacuo to give a solid (7.03 g). The solid was boiled with ethanol (150 mL) and filtered (sintered glass) hot. The ethanolic filtrate on cooling precipitated a solid that after drying was virtually pure anhydrous sulfonate 19 (801 mg, 18%). Melting point: at 242°C becomes translucent and at 243°C shrinks and bubbles. Its IR (KBr) and 1H NMR (D2O, DSS) spectra are reported in the sequel.
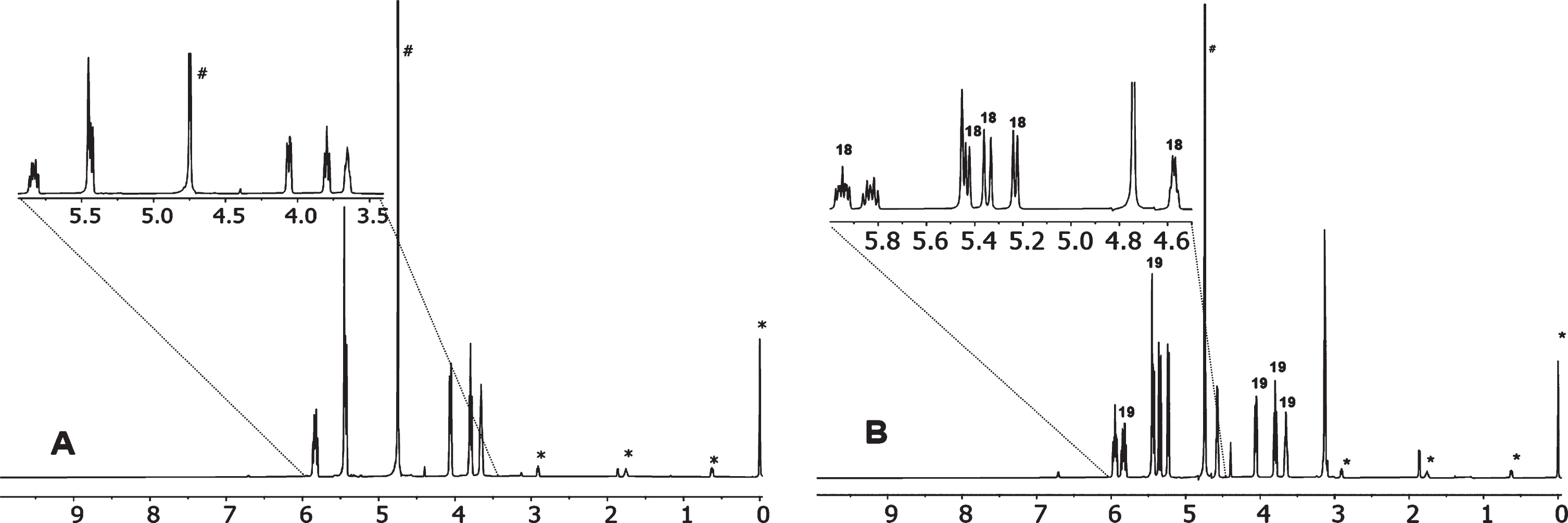
The ethanolic filtrate from the precipitated 19, after cooling at –20°C overnight precipitated a sticky solid that on drying gave a solid (2.17 g) proved to be a mixture of 18 and 19 by 1H NMR, Fig. 2, and from the new ethanolic filtrate a semi-solid (1.22 g), probably containing 3,4-dihydroxy-1-butene 23, was obtained after evaporation and drying, that was not analyzed. From the mixture of 18 and 19, slightly impure 19 could be isolated by recrystallization from ethanol.
1H NMR (D2O, DSS) spectrum (A) of pure 19 and (B) of a mixture of 18 and 19 (* signals from DSS, # signal from solvent, 18 signals from molecule 18).
The NaCl on the filter, contained organic reaction products, was washed with boiling ethanol (4×50 mL). The filtrates on cooling at RT overnight precipitated pure 19 (1.12 g, 26%) as a white solid. Total weight 1.917 g, 44%. The final ethanolic filtrate on evaporation and drying gave a white solid (687 mg) that was not analyzed. The NaCl remained on the filter (1.54 g, expected 1.46 g) by IR (KBr) contained very little 19.
For pure 19: Melting point: at 247–248°C shrinks and at 249°C bubbles. Calculated for C4H7O4SNa (Mr 174.15): C 27.59, H 4.05, S 18.41%; found: C 27.48, H 4.23, S 18.01%. IR (KBr), : 3329 s, broad, 2079 w, 2940 w, 2845 w, 1350 w, 1229 vs, 1182 vs, 1085 m, 1054 s, 1028 m, 1011 w, 920 w, 665 s. 1H NMR (D2O, DSS): δ 3.65 (m, 1 H, CHCSO3Na), 3.79 (dd, 1 H, 3JAC = 9.0 Hz, 2JAB = –11.4 Hz, 1 H, CHAHB(OH)CHCSO3Na), 4.06 (dd, 1 H, 3JBC = 4.2 Hz, 2JAB = –11.4 Hz, 1 H, CHAHB(OH)CHCSO3Na), 5.44 (dd, 1 H, 3JED (cis) = 9.6 Hz, CHEHF = CHD), 5.44 (dd, 1 H, 3JFD (trans) = 18.0 Hz, CHEHF = CHD), 5.83 (m, 1 H, CHEHF = CHDCHCSO3Na).
Knowing the shifts of pure 19, the shifts of 18 can be found by 1H NMR (D2O, DSS) Fig. 2. Thus, for 18 the spectrum has δ: 3.13 (m, 2 H, CHAHBSO3Na), 4.57 (dt, 1 H, 3JCD = 6.0 Hz, 3JAC = 6.0 Hz, 3JBC = 6.0 Hz, CHC(OH)CHAHBSO3Na), 5.23 (d, 1 H, 3JED (cis) = 10.2 Hz, CHEHF = CHD), 5.35 (d, 1 H, 3JFD (trans) = 16.8 Hz, CHEHF = CHD), 5.95 (ddd, 1 H, 3JFD (trans) = 16.8 Hz, 3JED (cis) = 10.2 Hz, 3JCD 6.0 Hz, CHEHF = CHDCHC(OH)).
Preparation of sodium 2,3-dihydroxypropane-1-sulfonate, 22, from 3-chloro-1,2-propanediol, 20
To a solution of sodium sulfite (2.52 g, 20 mmol) in water (10 mL) in a 50 mL round-bottomed flask, 3-chloro-1,2-propanediol 20 (1.7 mL, 20 mmol) was syringed in and the emulsion stirred at RT for 3 days whereupon a clear solution was obtained. Freeze-drying gave a jelly-like solid which was stirred overnight with ethanol (40 mL) to give a powdery suspension. The suspension was boiled, filtered (sintered glass) hot and washed with boiling ethanol (25 mL). The filtrate (containing the product 22 and glycerol) on cooling crystallized and after filtration, washing with ethanol and drying gave the monohydrated product 22 (1.01 g, 26%) as a white mass, very soluble in water, sparingly at RT and moderately soluble in hot ethanol, sparingly at RT but soluble in hot methanol and insoluble in acetone. It was a bit hygroscopic giving a hard solid. Melting point: at 160°C becomes translucent, at ca 170°C shrinks and transforms to a white mass, at ca 220°C bubbles and at 225°C turns light orange. Calculated for C3H7O5SNa·H2O (Mr 196.15): C 18.37, H 4.62, S 16.34%; found: C 18.28, H 4.28, S 16.40%. Its IR (KBr) and 1H NMR (D2O, DSS) are described in Section 2.8.
Working up the ethanolic filtrate and washings, more product (435 mg, 11%) was obtained. Total yield 37%. From the filtrate an oil (11 mg) was obtained and assuming that it is glycerol then 40% of 20 was converted to glycerol.
Preparation of racemic sodium 2,3-dihydroxypropane-1-sulfonate, 22, from rac-glycidol, 21
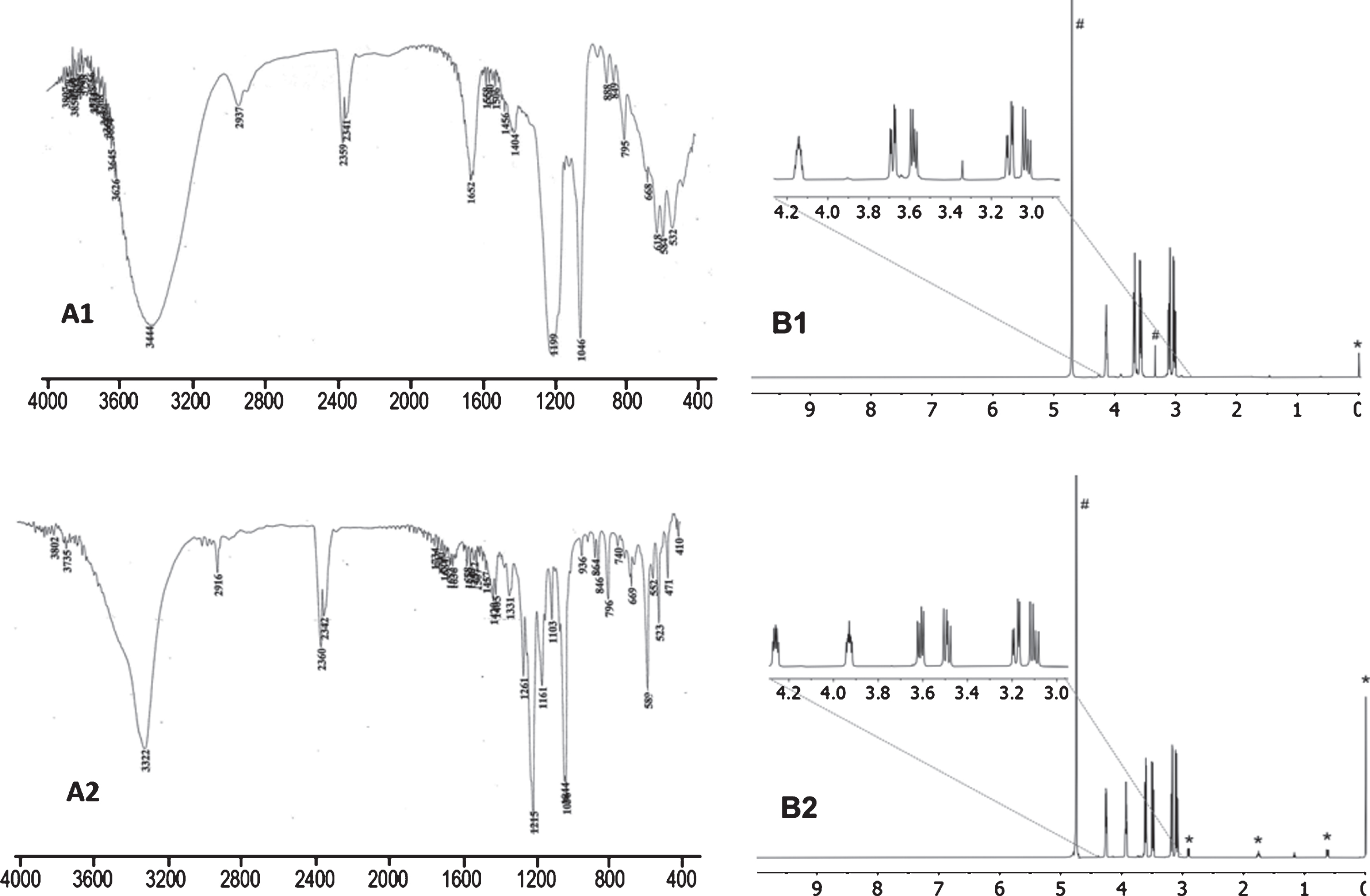
In a long neck 250 mL round-bottomed flask sodium sulfite (12.60 g, 0.1 mol) was dissolved in water (50 mL) and rac-glycidol 21 (6.6 mL, 0.1 mol) was added drop-wise (1 h). Heat was evolved after 5–10 drops of 21 have been added. After stirring at RT overnight and cooling at 0°C (ice-water), concentrated hydrochloric acid (8.3 mL) was added drop-wise for neutralization (pH 6). Concentration removed 30 mL water and the opalescent solution was transferred to a 500 mL conical flask. Addition of ethanol (200 mL), boiling and filtering hot (hot sintered glass and celite) left a gummy solid in the flask that was washed with boiling ethanol (2×50 mL). The opalescent ethanolic filtrates on cooling at RT deposited an oil and after 24 h the ethanol was decanted (see below) and the oil dried to give a white hard solid. Trial recrystallizations gave either a fluffy solid (H2O/EtOH) or gums (H2O/Me2CO, EtOH) or powder (MeOH). Therefore, to the dried solid, methanol (50 mL) was added and stirred at RT till a powder was obtained (ca 1 h). The suspension was centrifuged, the solid was washed with methanol and dried in vacuo to give the monohydrated product 22 (12.70 g, 65%). Melting point: as in Section 2.7. IR (KBr), : 3400 vs, broad, 2940 w, 2885 w, 1646 s, 1406 m, 1196 vs, broad, 1046 vs, 888 w, 794 m, 618 vs, 582 vs, 532 vs, Fig. 3(A1). 1H NMR (D2O, DSS): δ 3.03 (dd, 1 H, 2JED = –14.4 Hz, 3JEC = 7.8 Hz, CHC(OH)CHDHESO3Na), 3.11 (dd, 1 H, 2JED = –14.4 Hz, 3JDC = 4.2 Hz, CHC(OH)CHDHESO3Na), 3.58 (dd, 1 H, 2JAB = –12.0 Hz, 3JBC = 6.6 Hz, CHAHB(OH)CHC(OH)), 3.68 (dd, 1 H, 2JAB = –12.0 Hz, 3JAC = 4.2 Hz, CHAHB(OH)CHC(OH)), 4.14 (m, 10 line signal, 1 H, CHC(OH)CHDHESO3Na), Fig. 3(B1).
(A1) IR (KBr) and (B1)1H NMR (D2O, DSS) spectra of 22 obtained from either 20 or 21. (A2) IR (KBr) and (B2)1H NMR (D2O, DSS) spectra of 27 obtained as a by-product using Na2SO3 and excess 26 (* signals from DSS, # signals from solvent).
The methanolic filtrates were evaporated and dried to give a solid (2.30 g) that was triturated with methanol (25 mL) overnight to give pure product (910 mg, 5%). Total yield 70%.
The decanted ethanol was evaporated and dried to give a semi-solid that was triturated with boiling ethanol (25 mL) leaving a gum. After cooling at +4°C overnight the gum transformed to a hard solid. Centrifugation, washing with ethanol gave a slightly impure (by 1H NMR) product 22 (2.63 g, 14%). The impurities were glycerol (at 3.52 and 3.77 ppm) and other compounds: signals at 3.62, 3.66, 3.91 and 4.23 ppm.
Reaction of 3,4-epoxybutane-1,2-diol, 24, with sodium sulfite
The crude product 24 from 23 (11.2 mmol) in water (8 mL) was added to a nearly saturated solution of sodium sulfite (1.41 g, 11.2 mmol) in water (7 mL) and the solution stirred at RT for 4 h. Neutralization (pH 6) with concentrated hydrochloric acid and concentration gave a solution (ca 4 mL) that on adding methanol (50 mL) a gum and a suspension formed. Re-evaporation and drying gave a gum plus a very viscous oil that was analyzed by 1H NMR (D2O, DSS) and found to contain two main products which were not 27 or 28. The existence of absorptions at the region 3.0–3.3 ppm indicates that these two compounds should be sulfonates and possible structures are described in Section 3.4.
Reaction of 4-chlorobutane-1,2,3-triol, 25, with sodium sulfite: identification of the sulfonate 27
To an impure sample of 25 (1.15 g, assumed to be 8.2 mmol pure 25) diluted with water (6 mL) in a 100 mL round-bottomed flask was added solid sodium sulfite (1.19 g, 9.43 mmol) and the solution stirred at RT for 6 days. TLC (CHCl3/MeOH 2:1) showed that 25 had reacted (Rf 0.70) and two spots at Rf 0.53 and 0.67 appeared. Evaporation and drying gave a translucent semi-solid (2.43 g). Addition of ethanol (50 mL), boiling and filtration gave a solid that was Na2SO4·xH2O [by IR (KBr)]. The ethanolic filtrate after evaporation and drying gave an oil (1.17 g) that by TLC (CHCl3/MeOH 2:1) gave only one spot Rf 0.67 (pure 27 gives a faint spot at Rf 0.30; Section 2.11). The 1H NMR (D2O, DSS) spectrum of the oil showed a signal at 4.3 ppm seen in sulfonate 27. The 13C NMR (D2O, DSS) spectrum showed the presence of -CH2Cl at 45.8 ppm due to 25 and dominant singlets at 72.8 and 76.2 ppm due to trans-3,4-dihydroxytetrahydrofuran, 29 (see Fig. 6).
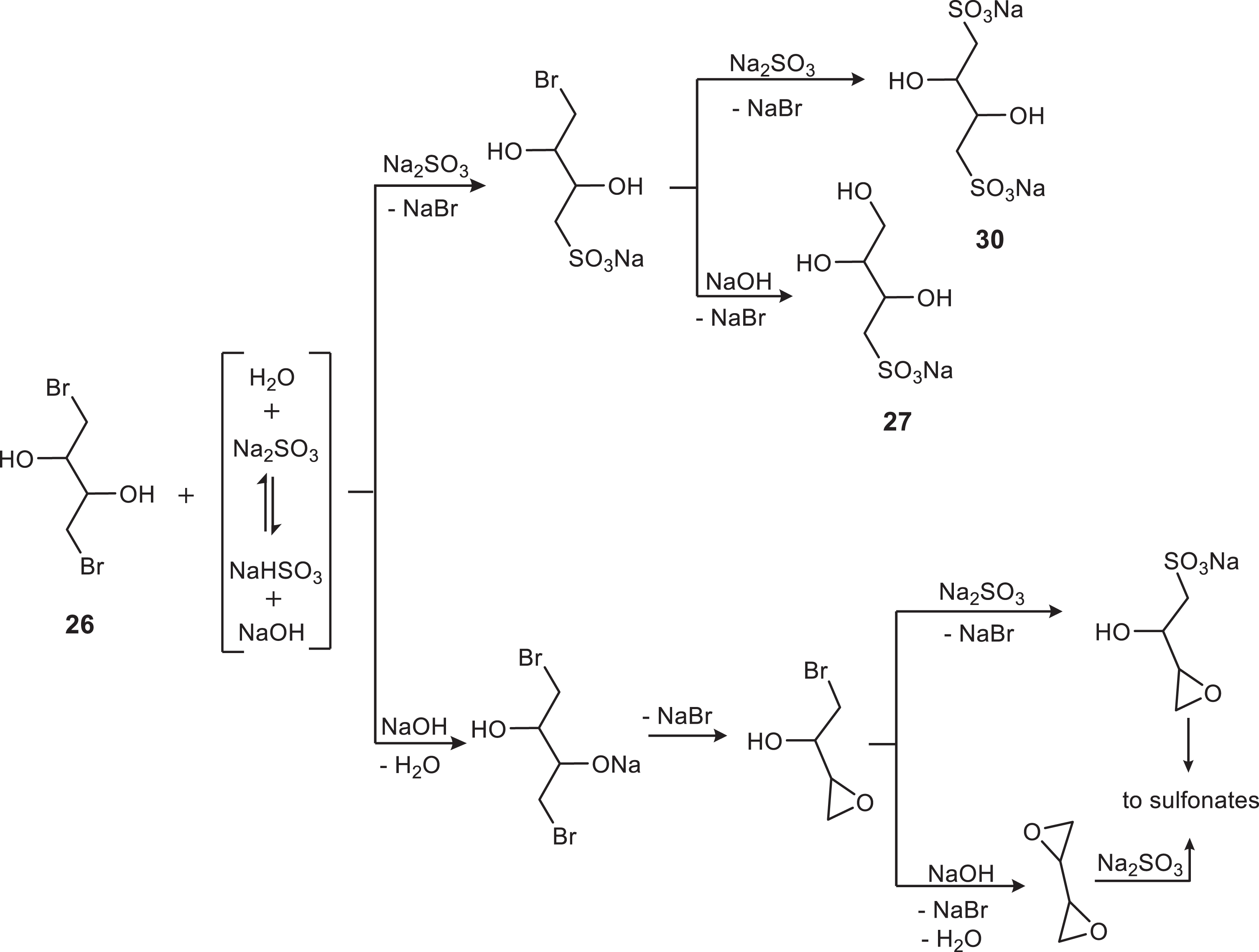
Preparation of sodium 2,3,4-trihydroxybutane-1-sulfonate, 27, from 1,4-dibromo-2,3-butanediol, 26
To a solution of sodium sulfite (4.10 g, 32.6 mmol) in water (18 mL) in a 100 mL round-bottomed flask was added a solution of the dibromide 26 (5.24 g, 21.1 mmol) in methanol (9 mL). The suspension formed at once was stirred at RT for 5 days, giving a heavier suspension. After evaporation and drying, the somewhat sticky solid (14.38 g) was transferred to a conical flask with boiling ethanol (100 mL) while stirring to break the lumps, boiled and filtered (sintered glass). The solid on the filter was washed with boiling ethanol (3×100 and 2×20 mL) to extract any co-precipitated product 27 leaving the bis sulfonate 30 (see Fig. 7) (3.91 g, 82% as anhydrous) as a non-hygroscopic powder, the properties of which will be described elsewhere. From the combined ethanolic filtrates, on standing a fluffy solid precipitated. It was filtered, washed with warm ethanol (2×10 mL) to extract NaBr and dried to give the product 27 (600 mg, 9%) as a white non-hygroscopic powder, pure by TLC (CHCl3/MeOH 2:1) faint spot on charring at Rf 0.30. Melting point: at 225°C slight shrinking and becomes translucent and at 227–228°C shrinks and bubbles. Calculated for C4H9O6SNa (Mr 208.16): C 23.08, H 4.36, S 15.40%; found: C 22.81, H 3.98, S 15.02%.
Preparation of the unsaturated aliphatic sulfonates 12 and 14
The allyl sulfonate is known as a salt either potassium [23] or sodium [19, 33]. The first was prepared in warm water using allyl iodide, while for the sodium salt excess allyl chloride was used in water at 33–70°C [19] or ethanol-water mixture at reflux yielding 12 in 75% yield [33]. We prepared 12 in water at RT (to minimize the production of allyl alcohol) in yields > 60%. Using allyl bromide the yield was 62% but, due to the solubility of NaBr in alcohols (16 mL EtOH or 6 mL MeOH per gram NaBr), required repeated recrystallizations for complete removal of NaBr compared to the effective removal of NaCl due to its insolubility in alcohols. The salt 12 was obtained anhydrous only when dried at 100°C [23] while drying in vacuo over P2O5 gave a hydrated form and this may be the reason why some sulfonates reported here are monohydrates.
The sodium salt 14 was obtained impure from the commercially available bromide 13 and Na2SO3 in water at 70°C [37] or at reflux [14, 21] and used as such for further transformations to give novel inhibitors of mitogen-activated protein kinases [14, 37] and other compounds [21]. We prepared the sulfonate 14 in water under reflux in 81% yield.
Because the conversion of sulfonates to sulfonic acids is a tedious process, we tried the conversion of 14 containing NaBr to their acids using a cation exchange resin, eluting with water. Removal of water under reduced pressure should remove hydrobromic acid as an azeotrope (b.p. 74.12°C at 100 mm Hg; contains 49.80% HBr [3]). Concentrating the colorless solution, it gradually turned light orange and then brown. It is known that lower alkyl sulfonic acids darken quickly even in vacuo [36, 39], while long chain ones are white crystalline solids [20]. The brown oil was analyzed spectroscopically (Section 3.5). An impurity was found in the 1H NMR spectrum tentatively identified as 5-methyl-1,2-oxathiolane-2,2-dioxide [29]. This sultone has been prepared by cyclization of 3-hydroxybutane-1-sulfonic acid at 140–150°C/0.4 mm Hg [2]. However, in our case this compound was formed by protonation of the methylene carbon of the CH2 = CH- group followed by attack of -SO3- at the secondary CH3CH+- carbocation. It is not known if this reaction takes place on the cation exchange column or during the evaporation of water (rotary, 70°C). Protonation of a -CH = CH2 group by chlorosulfuric acid to give a secondary carbocation has been postulated in another case [32].
Preparation of hydroxy sulfonates 16, 18 and 19
The sulfonate 16 was prepared as monohydrate from 15 at RT in 46% total yield. The reaction was slower compared to those with allyl halides 11 and the lower yields can be due to either volatility of ethylene oxide (b.p. 11 °C) or to base-catalyzed hydrolysis of the epoxide and/or 15 to ethylene glycol. Probably, hydrolysis of the epoxide, Fig. 4, is more important.
Proposed routes for the preparation of sodium sulfonates 16 and 22 from 15 and 20, respectively. Note that the intermediate epoxide is formed from 1-chloro-2-alkanol and from a 2-chloro-1-alkanol, Equation (2) [22].
The monosubstituted ethylene oxide 17 whose reaction with Na2SO3 is of interest because the SO32- [26, 38] nucleophile can attack at CH2 or CH carbons of a substituted epoxide [26, 38]. At this point it should be noted that HSO3- is nucleophilic as well giving 85% 2-hydroxypropane-1-sulfonate from propylene oxide [30]. There were many suggestions as to why a nucleophile attacks the CH2 or CH carbons [26], but using a combination of perturbation theory and the hard and soft acid and base principle it was suggested [13] that towards 17 a soft nucleophile is more likely to attack the = CH2 carbon (under orbital control) leading to 1,4-addition with ring opening, while a hard nucleophile will attack (under charge control) the most positive centers which are the epoxide carbons (i.e. attack at CH will give 1,2-addition; attack at CH2 will be an SN2 reaction).
While we could not isolate any arsonic acid or salt from the reaction of 17 with Na3AsO3 [16], the reaction of 17 with Na2SO3 in water at RT for 24 h gave 19 in 44% total yield and the isomeric 18 was detected by 1H NMR but could not be isolated. Thus, SO32- is a quite strong nucleophile but weaker than HO-.
Preparation of sodium 2,3-dihydroxypropane-1-sulfonate, 22
This sulfonate can be prepared from the chloride 20 or the epoxide (glycidol) 21. The reaction of 20 with aqueous Na2SO3 was slow despite the finite solubility of 20 in water. Due to the presence of the very soluble 22 and the by-product glycerol, the removal of water was done not on a rotary evaporator but by freeze drying (at 20 mmol scale) or by evaporation on a hot plate (at 0.1 mol scale). However, in both cases the solid residue kept significant amounts of water. At 20 mmol scale the yield was 37%, while at 0.1 mol scale it was 24–28% due to incomplete extraction of the product from the hydrated solids (Na2SO3, NaHSO3, NaCl) with boiling ethanol. The dry product 22 absorbed water slowly from air giving a hard solid but it was not deliquescent. From the mechanism of the reaction, Fig. 4, it is evident that the low yield of 22 is due to substantial hydrolysis of 20 to glycerol probably mainly via the epoxide. Evidence for formation of an epoxide intermediate is provided by the analogous reaction of 20 with Na3AsO3 [27] and from the fact that both 2-bromo-1-alkanol and 1-bromo-2-alkanol when reacted with Na2SO3 gave 2-hydroxyalkane-1-sulfonate [22].
The analogous reaction of 20 with Na3AsO3 gave 45% of the arsonic acid 2 [34] or 57% of the dilithium salt of 2 [15] that can be compared to the 37% yield of 22 that most likely indicates that the formation of epoxide dominates and that the nucleophiles AsO33 -, SO32- and HO- have about the same reactivity towards 20.
If the nucleophiles AsO33 - and SO32- are more reactive than HO- towards an epoxide, then the reaction of glycidol 21 with Na3AsO3 and Na2SO3 should give higher yields of arsonates and sulfonates. Indeed, the reaction of 21 with Na3AsO3 gave 75–79% of 2 with rac, S or R stereochemistries [28], while with Na2SO3 the yields of 22 depended on the reaction conditions. Thus, when glycidol was added very slowly (3 h at 0.1 mol scale) to a cold (ca 0°C) suspension of aqueous Na2SO3 the total yield was 43%, while when the reaction was done at RT (Section 2.8) the yields were 65–70%. Also, if the oil remained after extraction with ethanol and evaporation was not dry enough, lower yields of 22 were obtained (35–40%) while improvement of these yields to an extent of 65–70% was observed when the oil was thoroughly dried.
The mechanism of the reaction is shown in the lower part of Fig. 4, and apart from glycerol, other by-products can be formed by base-catalyzed oligomerization of glycidol that can be observed in the 1H NMR spectrum of impure 22.
Preparation of sodium 2,3,4-trihydroxybutane-1-sulfonate, 27, from 24, 25 and 26
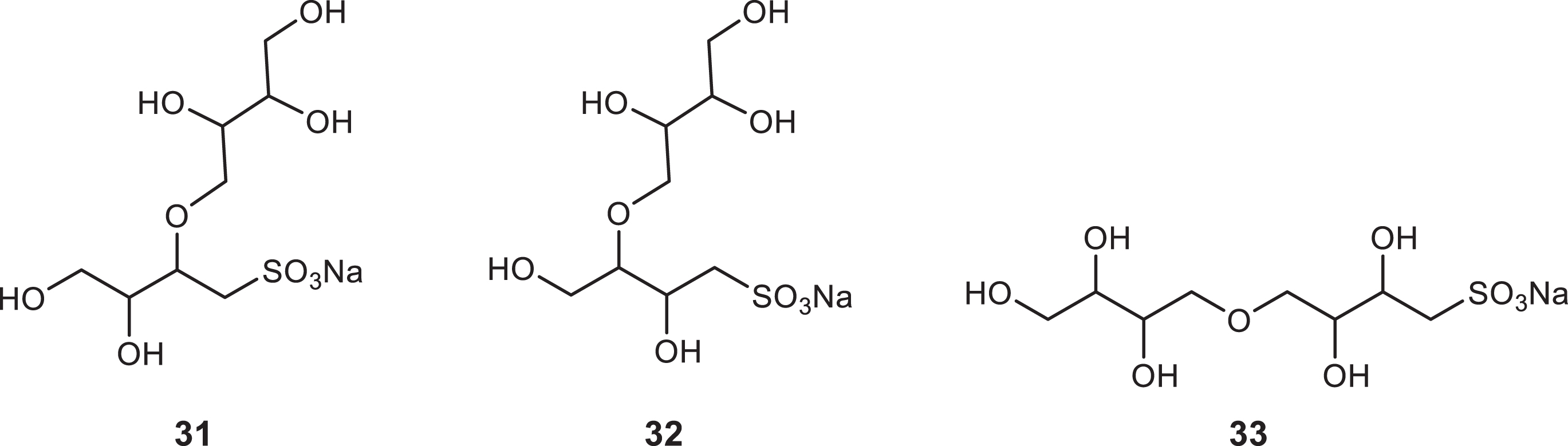
The epoxy diol 24 was prepared slightly impure from 23 [7, 16] and was used for the preparation of 2,3,4-trihydroxybutylarsonic acid by reaction with Na3AsO3 (utilizing cation and anion exchange resins for purification) (46% yield) [16]. The reaction of 24 with Na2SO3 gave as main products two different sulfonates that were not 27 or 28. Partial analysis of the very complex 1H NMR spectrum revealed that these two sulfonates should both bear the structural unit -CHAHB-SO3Na. It is possible that after the initial nucleophilic attack of the SO32- to the epoxide ring, the produced alkoxy anion reacts with another epoxide either directly or after migration to another hydroxyl group giving the two of the three possible by-products 31, 32, 33, Fig. 5.
Probable by-products from the reaction of the epoxy diol 24 with Na2SO3. Two of them have been identified by 1H NMR. The reaction did not afford the sulfonates 27 and 28.
In an effort to obtain pure 27 or 28 we used the chloride 25 obtained by ring opening of 24 by concentrated hydrochloric acid [16]. The impure 25 with Na3AsO3 gave ca 30% of 2,3,4-trihydroxybutylarsonic acid utilizing cation and anion exchange resins for purification [16], while with Na2SO3 the slow (6 days at RT) reaction on working up gave Na2SO4 and an oil in which, by 1H NMR, 27 (free from 28) was identified, while in the 13C NMR spectrum the chloride 25 and the cyclic 29 were detected. These results indicate that SO32- attacked the CH2Cl carbon and the epoxide 24 probably was not formed as shown in Fig. 6. This mechanism differs from that of Fig. 4 in that the -CH2OH in 25 was more reactive.
The reaction of 25 with aqueous Na2SO3 produced 29 and traces of 27. The isomeric sulfonate 28 was not detected, so the reaction did not involve the epoxide 24.
The sulfonate 27 was formed, along with the bis sulfonate 30, as a by-product of the reaction of excess 26 with Na2SO3. Because 30 was practically insoluble in boiling ethanol, the isolation of 27 was easy but the yields were low (8–12%). The properties of 30 will be described elsewhere (GMT et al. accepted to MGC). In this reaction several probable intermediates can be formed, Fig. 7, none of which were detected or isolated. From Fig. 7 it seems that the production of 27 and 30 most probably involves direct SN2 displacements rather than epoxide or diepoxide intermediates. The 1,3-butadiene diepoxide is known to react with Na3AsO3 giving mono and bis arsonic acids [35].
From the reaction of excess dibromide 26 with aqueous Na2SO3 the monosulfonate 27 and the bis sulfonate 30 have been isolated.
Spectral data of the sulfonates
The IR spectra were obtained in KBr discs using oven-dried KBr and minute amounts of samples due to very strong -SO3Na bands which dominated in the spectra. Under these conditions, in the spectrum of sulfonate 12 a broad band was observed at ca 3480 cm-1 due to traces of water in KBr. That this band was due to water was checked by the presence of a band at ca 1650 cm-1 (deformation) and running some spectra in Nujol. The asymmetric stretching of S = O is in the region 1270–1180 cm-1 and the symmetric one is in the narrow region 1060–1015 cm-1 and they are split depending on the compound. Thus, for the anhydrous sulfonate 12 the asymmetric stretching is split (1265, 1222 and 1190 cm-1) while the symmetric one is not split (1060 cm-1). In the spectra of the anhydrous 19 and 27 [see Fig. 3(A2)] they were split in different ways probably indicating involvement of the hydroxyl groups in hydrogen bonding. Among the monohydrated sulfonates, 14 revealed a split pattern in the 3650–3350 cm-1 region and, because the CH vibrations were not affected, it can be concluded that the water molecule was coordinated to the -SO3Na group giving sharp bands at 1220, 1190 and 1066 cm-1. The hydrated hydroxyl-containing sulfonates 16 and 22, however, showed a very strong and broad band at ca 3440 cm-1 and somewhat broad asymmetric but sharp symmetric stretching bands at 1190 and at ca 1050 cm-1, respectively. In all sulfonates examined the S-O stretching varied in position in the region 800–600 cm-1 and it was sharp. The IR (neat) of the not perfectly dried acid from 14 showed a very strong and broad band 3750–2100 cm-1, and broad bands for asymmetric (ca 1160 cm-1) and symmetric (1040 cm-1) S = O stretching and broad S-O stretching (606 cm-1).
The 1H NMR (D2O) spectra of all sulfonates prepared showed, as expected, very complex splitting patterns, see for example Fig. 2 and Fig. 3. However, whenever was possible these complex patterns were analyzed, and J values have been measured.
In the reaction of 17 with Na2SO3 a mixture of two isomers (18 and 19) was obtained, but only isomer 18 was possible to be isolated. However, knowing the 1H NMR absorbances of 18 the absorbances of the non-isolated isomer 19 were identified, Fig. 2.
It was noted that the AB splitting of RCH2OH and of RCH2SO3Na, in e.g. 18 and 19 (Fig. 2), differed being somewhat larger for RCH2OH.
Comparing the spectra of the sulfonate 14 and its free acid in D2O, we see that both resonated at the same positions, implying that the spectra recorded correspond to their anion due to 100% ionizations.
Conclusions
The Strecker reaction on alkyl halides [11 and 13], hydroxy halides [15, 20, 25 and 26] and epoxides [17, 21 and 24] gave either pure sulfonates (hydrated or anhydrous), mixture of isomers where only one isomer could be isolated, e.g. 19, only by-products, e.g. from 24, or no products, e.g. from 25. The precipitated sulfonates are usually hydrated. Anhydrous 12 was obtained by drying at ≥100°C. Drying in vacuo over P2O5 gave anhydrous sulfonates in cases of 19 and 27, while 14, 16 and 22 were obtained as (mono)hydrates. The yields were at about 50% except in the case of 27 which was obtained in 8–12% yield as a by-product. The sulfonates did not melt but gradually decomposed. Mechanisms for some reactions, Fig. 4, Fig. 6 and Fig. 7 have been proposed and the IR (KBr) and 1H NMR (D2O) spectra are discussed.
References
1.
BittmanR., Applications of novel synthetic lipids to biological problems, Chem Phys Lipids129 (2004), 111–131.
2.
BlaserB., Process of the preparation of sulfones, US patent 3,164,608 (1965) to Henkel and Cie, Germany.
3.
BonnerW.D., BonnerL.G. and GurneyF.J., Azeotropic hydrobromic acid solutions at pressures 100 Mm to 1200 Mm, J Am Chem Soc55 (1933), 1406–1409.
4.
EngelR., Phosphonates as analogues of natural phosphates, Chem Rev77 (1977), 349–367.
5.
E. Erlenmeyer und L. Darmstaedter, Z Chem (1868), 342; cited by Schenck and Kaizerman.
6.
GiggR., PenglisA.A.E. and ConantR., Synthesis of 3-O-(6-deoxy-6-sulfo-α-D-glucopyranosyl)-1,2-di-O-hexadecanoyl-L-glycerol, “sulfoquinovosyl diglyceride”, J Chem Soc (1980), 2490–2493.
7.
GoldingB.T., SlaichP.K. and WatsonW.P., Reaction of guanosine with glycidaldehyde, IARC Scientific Publication, No 70, (1986), 227–231.
8.
HägerM. and HolmbergK., A substitution reaction in an oil-in-water microemulsion catalyzed by a phase transfer catalyst, Tetrahedron Letters41 (2000), 1245–1248.
9.
HainesT.H., The chemistry of sulfolipids, Progr Chem Fats Other Lipids11 (1971), 297–345.
10.
HuangJ. and WidlanskiT.S., Facile synthesis of sulfonyl chlorides, Tetrahedron Letters33 (1992), 2657–2660.
11.
IoannouP.V., Arsonolipids, pseudo arsonolipids, arsinolipids and arsonoliposomes: Preparation, biophysical, biochemical and biological properties, Main Group Chem17 (2018), 111–132.
12.
IoannouP.V., VachliotisD.G. and KouliO., The reaction of propargyl chloride with As(III) and S(IV) nucleophiles, Main Group Chem18 (2019), 365–381.
13.
JamieC., OrtuñoR.M. and FontJ., Interpretation of conjugated oxiranes behavior towards nucleophiles, J Org Chem53 (1988), 139–141.
14.
KhireU.R. and ChordiaM.D., Sulfonamide derivatives as MEK inhibitors and their preparation, pharmaceutical compositions and use in the treatment of diseases, WO patent 2011047055 (2011) to Allostem Therapeutics LLC.
15.
LacosteA.-M., DumoraC., AliB.R.S., NeuzilE. and DixonH.B.F., Utilization of 2-aminoethylarsonic acid in Pseudomonas aeruginosa, J Gen Microbiol138 (1992), 1283–1287.
16.
LalaM.A., TsivgoulisG.M. and IoannouP.V., Preparation of 2,3,4-trihydroxybutylarsonic acid: A starting compound for novel arsonolipids, Phosphorus, Sulfur and Silicon182 (2007), 2747–2760.
17.
ManzoE., FiorettoL., PaganoD., NuzzoG., GalloC., De PalmaR. and
FontanaA., Chemical synthesis of marine-derived sulfoglycolipids, a new class of molecular adjuvants, Mar Drugs15 (2017), 288–297.
18.
Moss IIIF.R.,
ShukenS.R.,
MercerJ.A.M.,
CohenC.M.,
WeissT.M.,
BoxerS.G. and
BurnsN.Z., Ladderane phospholipids from a densely packed membrane with normal hydrazine and anomalously low proton/hydroxide permeability, Proc Nat Acad Sci USA115 (2018), 9098–9103.
19.
MullerD.J. and KnepperW., Preparation sodium allyl and methallyl sulfonate, US patent 4171324A (1976) to Huels AG.
20.
NollerC.R. and GordonJ.J., The preparation of some higher aliphatic sulfonic acids, J Am Chem Soc55 (1933), 1090–1094.
21.
OehrleinR. and BaischG., Colored charged silsesquioxanes for electrophoretic displays, WO patent 2013150444 (2013) to BASF SE; BASF Schweiz AG; BASF Company Limited.
22.
PüschelF., und
KaiserC., 2-Hydroxy-n-alkan-sulfonsäuren-(1), Chem Ber97 (1964), 2903–2916.
23.
von RadA., Ueber Allylsulfonsäure und einige ihrer Salze, Ann Chem Pharmac161 (1872), 218–224.
24.
ReedR.M. and TartarH.V., The preparation of sodium alkyl sulfonates, J Am Chem Soc57 (1935), 570–571.
25.
RogersJ., YuB.-Z., ServesS.V., TsivgoulisG.M., SotiropoulosD.N., IoannouP.V. and JainM.K., Kinetic basis for the substrate specificity during hydrolysis of phospholipids by secreted phospholipase A2, Biochemistry35 (1996), 9375–9384.
26.
SchenckR.T.E. and KaizermanS., The reaction of bisulfite with epoxy compounds, J Am Chem Soc75 (1953), 1636–1641.
27.
ServesS.V., SotiropoulosD.N., IoannouP.V. and DixonH.B.F., On the mechanism of the Meyer reaction with epoxides and 2-haloalcohols as substrates, Phosphorus, Sulfur and Silicon90 (1994), 103–109.
28.
ServesS.V., TsivgoulisG.M., SotiropoulosD.N., IoannouP.V. and JainM.K., Synthesis of (R) and (S)-1,2-diacyloxypropyl-3-arsonic acids: Optically active arsonolipid, Phosphorus, Sulfur and Silicon71 (1992), 99–105.
29.
SmithG.M.T., BurtonP.M. and BrayC.D., Sultones and sultines via a Julia-Kocíenski reaction on epoxides, Angew Chem Int Ed54 (2015), 15236–15240.
30.
StewartJ.M. and CordtsH.P., Oxidative ring cleavage reactions of propylene sulfide, J Am Chem Soc74 (1952), 5880–5884.
31.
StreckerA., Ueber eine neue Bildungweise und die Constitution der sulfosäuren, Ann Chem Pharm148 (1868), 90–96.
32.
SudarmaI.M., YuanitaE. and SuanaI.W., Markovnikov addition of chlorosulfuric acid to eugenol isolated from clove oil, Indo J Chem13 (2013), 181–184.
33.
TianL., XuG.-Y., YeY. and LiuL.-Z., 1,3-Dipolar cycloaddition reactions of nitrile oxides to prop-1-ene-1,3-sultone, J Heterocyclic Chem40 (2003), 1071–1074.
34.
TsivgoulisG.M., SotiropoulosD.N. and IoannouP.V., 1,2-Dihydroxypropyl-3-arsonic acids: A key intermediate for arsonolipids, Phosphorus, Sulfur and Silicon57 (1991), 189–193.
35.
TsivgoulisG.M., LalaM.A. and IoannouP.V., Preparation of DL-2,3,4-trihydroxybutylarsonic acid and DL-2,3-dihydroxybutane-1,4-bis(arsonic acid): starting compounds for novel arsonolipids, Chem Phys Lipids148 (2007), 97–104.
36.
VivianD.L. and ReidE.E., The preparation of some of the lower alkyl sulfonic acids, J Am Chem Soc57 (1935), 2559–2560.
37.
XiaoD., ZhuL., ShixinS., LiangZ. and HuW., Preparation of N-arylamino(oxo)pyridinyl sulfonamides and N-arylamino(oxo)pyrazinyl sulfonamides as MEK inhibitors and useful in the treatment of inflammatory diseases, WO patent 2010145197 (2010) to Chemizon (Beijing) Ltd, Peop. Rep. China
38.
YonedaG.S., GriffinM.T. and CarlyleD.W., A kinetic study of the reaction between sulfite ion and propylene oxide, J Org Chem40 (1975), 375–377.
39.
ZuffantiS., The preparation of some branched chain aliphatic sulfonic acids, J Am Chem Soc62 (1940), 1044.