
Abstract
This research examined the effects of solvent polarity and temperature on the tautomerization of a carbonitrile molecule at CAM-B3LYP/6-311G (d,p) level of theory. The selected solvents were n-hexane, diethyl ether, pyridine, ethanol, methanol, and water. The solvent effects were examined by the self-consistent reaction field theory (SCRF) based on conductor-like polarizable continuum model (CPCM). The solvent effects were explored on the energy barrier, frontier orbitals energies, and HOMO-LUMO gap. Dependencies of thermodynamic parameters (ΔG and ΔH) on the dielectric constants of solvents were also tested. Specifically, the temperature dependencies of the thermodynamics parameters were studied within 100–1000 K range. The rate constant of the tautomerism reaction was computed from 300 to 1200 K, in the gas phase.
Introduction
Compounds containing pyrazoles [1] are known to display diverse pharmacological activities such as antibacterial, antifungal, anti-inflammatory, analgesic, and antipyretic [2]. A number of heterocyclic compounds fused with pyrazole are known for their varied biological and industrial applications [3]. The pyrazolotriazole are precursors for photosensitive materials (e.g. inks and toners) [4] and ingredients in cosmetics [5]. Preparation of 7-amino-1,3-dioxo-1,2,3,5- tetrahydropyrazolo [1,2-a][1,2,4]triazole has been reported using magnetic Fe3O4 nanoparticles coated with (3 aminopropyl)-triethoxysilane as catalyst for the formation of the 7 amino-1,3-dioxo-1,2,3,5- tetrahydropyrazolo[1,2-a][1,2,4]triazoles [6].
Many investigations have been reported about tautomerization reactions [7–17]. Also, solvent polarity importance on the structure and properties of the molecules have been studied using quantum mechanics tools [18–31].
In this study, we investigate Solvent and Temperature effects on the tautomerization of 7 amino-1,3-dioxo-2,5-diphenyl-2,3-dihydro-1H,5H-pyrazolo[1,2-a][1,2,4]triazole-6-carbonitrile at the CAM-B3LYP/6-311G(d,p) level of theory.
Computational methods
Optimization and vibrational analysis were done using Gaussian 09 software package [32]. The standard 6-311G(d,p) basis set [33–36] was considered for the elements. CAM-B3LYP functional was used for optimizing the geometries of the compound. This functional is Handy et al.’s long range corrected version of B3LYP using the Coulomb-attenuating method [37]. The identities of the optimized structures as an energy minimum were confirmed via vibrational analysis.
For the solvation impact study, we utilized a self-consistent reaction field (SCRF) approach, using conductor-like polarizable continuum model (CPCM) [38, 39].
The identity of the reactants, transition states, and products was confirmed through vibrational analysis. All the transition states (TS) were checked by the intrinsic reaction coordinate (IRC) analysis at the same level of theory [40–43].
Gpop program was used for computing the reaction rate parameters [44]. The temperature dependence of the rate constants was investigated within 300–1200 K using transition state theory (TST) based on statistical thermodynamics. Further, the tunneling effect was considered by assuming asymmetric Eckart potential [45] and Shavitt’s correction [46] the corresponding correction factor for corrections of the rate constants values.
The Eckart potential function is often used to estimate quantum mechanical tunneling corrections to theoretically determined chemical rate constants. Eckart’s potential has the following form:
The potential has the limiting value of zero when x→ - ∞, goes through a single maximum of height V1 as x increases, and has a limiting value of V1–V2 as x→ + ∞. F* is the second derivative of V at its maximum. Shavitt recommended a simple equation for the tunneling correction:
Energy aspects
Figure 1 presents the structures of 7-amino-1,3-dioxo-2,5-diphenyl-2,3-dihydro-1H,5H-pyrazolo[1,2-a][1,2,4]triazole-6-carbonitrile

The structures of 7-amino-1,3-dioxo-2,5-diphenyl-2,3-dihydro-1H,5H-pyrazolo[1,2-a][1,2,4]triazole-6-carbonitrile
Total energy (E, a.u) and dipol moment values (μ, Debye) of tautomersof 7-amino-1,3-dioxo-2,5-diphenyl-2,3-dihydro-1H,5H-pyrazolo[1,2-a][1,2,4]triazole-6-carbonitrile and transition state between them (a.u) in the gas and solution phases.
The transition state geometry suggested for the studied tautomerization reaction is depicted in Fig. 1. TS has a four-membered ring structure. According to the frequency analysis calculations, all TSs have distinctive imaginary frequency. These values are outlined in Table 1.
The changes in the total energy upon alteration of the Ca-Ha and Na-Ha bond distances along the IRC path are shown in Fig. 2 in the gas phase. It can be observed that the Na–Ha bond rapidly lengthens from the reactant side. On the other hand, the Ca–Ha bond rapidly shortens from the reactant side. There are identical variations in the presence of other solvents.
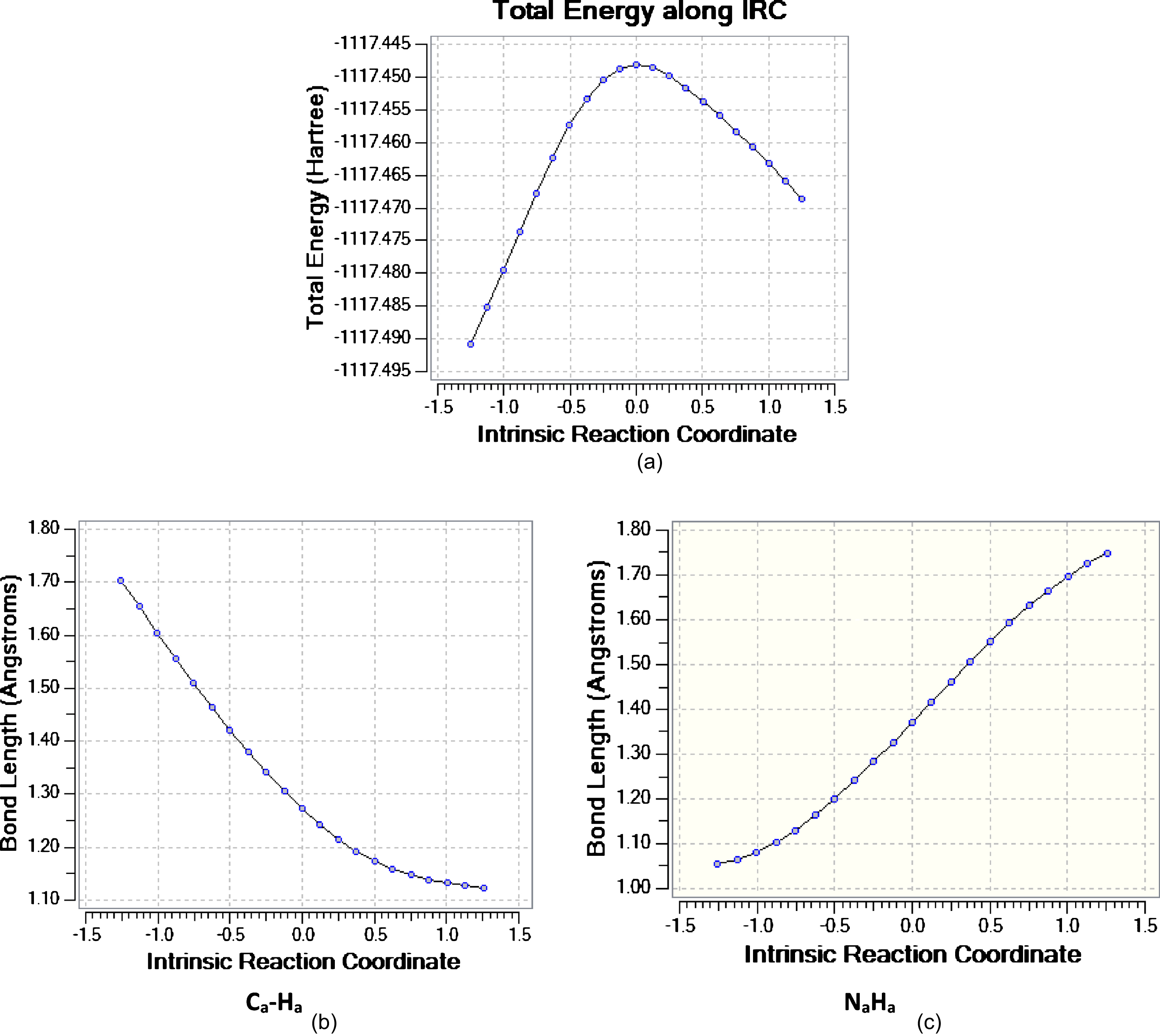
(a)Energy, (b) and (c) bond distances versus reaction coordinate (amu1/2. Bohr) in thetautomerization reaction.
The activation energy (
Solvent effects
The thermodynamic parameters of the studied tautomerization reaction are outlined in Table 2. The negative values of the reaction free energy (
Thermodynamic parameters of tautomerismof 7-amino-1,3-dioxo-2,5-diphenyl-2,3-dihydro-1H,5H-pyrazolo[1,2-a][1,2,4]triazole-6-carbonitrile in gas phase and various solvents
Thermodynamic parameters of tautomerismof 7-amino-1,3-dioxo-2,5-diphenyl-2,3-dihydro-1H,5H-pyrazolo[1,2-a][1,2,4]triazole-6-carbonitrile in gas phase and various solvents
In addition, as the results show, the activation free energy values (ΔG‡) are larger in the solution phase than in the gas phase. The more polar solvents have led to greater ΔG‡ values.
The enthalpy values of the reaction (
The results also indicate that the activation enthalpy values (ΔH‡) are larger in the solution phase than in the gas phase. The more polar solvents have led to greater ΔH‡ values.
A plot of the barrier heights (
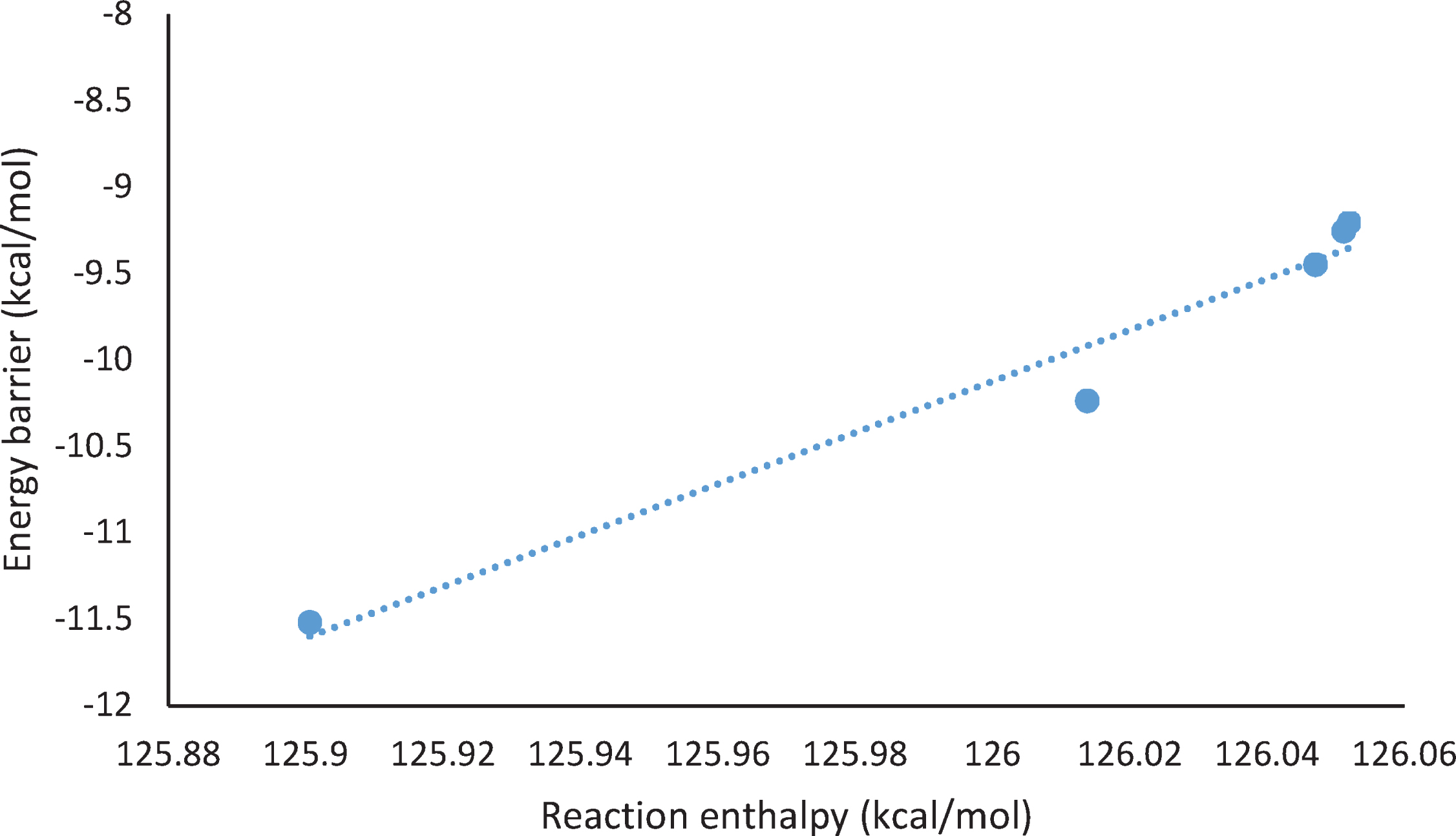
Linearcorrelation between barrier heights (
The thermodynamics parameters of the studied tautomerization reaction are listed in Table 3 within the 100–1000 K temperature range. The negative values of the reaction free energy (ΔGr) show that the studied reaction is spontaneous within the studied temperature range. It can be seen that this reaction is more appropriate at higher temperatures. There is a good linear correlation between ΔGr values with temperature (T):
Thermodynamic parameters of tautomerismof 7-amino-1,3-dioxo-2,5-diphenyl-2,3-dihydro-1H,5H-pyrazolo[1,2-a][1,2,4] triazole-6-carbonitrile in various temperatures
Thermodynamic parameters of tautomerismof 7-amino-1,3-dioxo-2,5-diphenyl-2,3-dihydro-1H,5H-pyrazolo[1,2-a][1,2,4] triazole-6-carbonitrile in various temperatures
In addition, as the results show, the activation free energy values (ΔG‡) are larger at higher temperatures. There is a good linear correlation between (ΔG‡) values with temperature (T):
The enthalpy values of the reaction (
On the other hand, these parameters are fitted with a quadratic equation:
In addition, as the results show, the activation enthalpy values (ΔH‡) are larger at lower temperatures. There is a good linear correlation between
On the other hand, these parameters are fitted with a quadratic equation:
As a larger dipole moment can increase the overall energy corresponding to a conformation, the population corresponding to the conformation with a larger dipole moment can be reduced compared with the conformation with a less dipole moment [47]. A greater dipole moment is associated with greater charge distribution (polarization). Thus, the conformations with greater dipole moments can be softer than those with smaller dipole moments.
The results show that the dipole moments of the
On the other hand, these values show that dipole moment values of I and II-isomers increase in the solution phase as compared to the gas phase. These values increase in the presence of more polar solvents. The elevation of the dipole moment values is attributed to long range interactions solvent with the solute molecules.
Molecular orbital analysis
The energies of the frontier orbitals (HOMO, LUMO) and the corresponding HOMO–LUMO energy gaps values of the investigated molecules are given in Table 4.
Frontier orbital energy and HOMO-LUMO gap values (eV) of tautomers of 7-amino-1,3-dioxo-2,5-diphenyl-2,3-dihydro-1H,5H-pyrazolo[1,2-a][1,2,4]triazole-6-carbonitrile
Frontier orbital energy and HOMO-LUMO gap values (eV) of tautomers of 7-amino-1,3-dioxo-2,5-diphenyl-2,3-dihydro-1H,5H-pyrazolo[1,2-a][1,2,4]triazole-6-carbonitrile
As seen in Table 4, HOMO of

Plots of frontier orbitals of (a) (
As shown in Table 4, HOMO-LUMO gaps values of I-isomer are larger in the solution phase as compared to the gas phase. In contrast, HOMO-LUMO gaps values of II-isomer are smaller in the solution phase as compared to the gas phase.
No direct experimental investigation has reported the rate constant of the investigated tautomerization reaction at any temperature, to the best of our knowledge. The rate constant values of the studied isomerization reaction are summarized in Table 5. Furthermore, the tunneling factor is considered for calculations of the rate constant values. Asymmetric Eckart potential and Shavitt’s correction the corresponding correction factor of tunneling are assumed for corrections of the rate constants (Table 5). These values have been calculated in the gas phase and within the temperature range of 300–1200 K. Th equations fitted to the gas phase Arrhenius equation are collected in Table 6.
The gas phase calculated rate constants for studied reactions (s–1): (a) excluding tunneling factor, and corrected rate constants for tunneling by assuming (b) asymmetric Eckart potential, (c) Shavitt’s correction
The gas phase calculated rate constants for studied reactions (s–1): (a) excluding tunneling factor, and corrected rate constants for tunneling by assuming (b) asymmetric Eckart potential, (c) Shavitt’s correction
Fitted equations to the gas phase Arrhenius equation for studied reactions: (a) excluding tunneling factor, and corrected rate constants for tunneling by assuming (b) asymmetric Eckart potential, (c) Shavitt’s correction
Computational investigation of the effects of solvent polarity and temperature on the tautomerization of 7-amino-1,3-dioxo-2,5-diphenyl-2,3-dihydro-1H,5H-pyrazolo[1,2 a, 1,2 a][1,2,4]triazole-6-carbonitrile were studied at CAM-B3LYP/6-311G (d,p) level of theory reveal that: I-isomer was more stable than I-isomer in both gas and solution phases. ΔE‡ and ΔEr values were larger in the solution phase than in the gas phase. These values increased in more polar solvents. The negative values of ΔGr and ΔHr showed that the studied reaction was exothermic, respectively in both gas and solution phase. Dipole moment values of I and II-isomers increased in the solution phase when compared to the gas phase. These values increased in the presence of more polar solvents. HOMO-LUMO gaps values of I-isomer were larger in the solution phase than in the gas phase. In contrast, HOMO-LUMO gaps values of II-isomer were smaller in the solution phase as compared to the gas phase.