
Abstract
This study investigated the interaction between Al12N12 nano-cluster and titanocene dichloride anticancer drug complex using B3P86 functional in gas and solution phases. Non-covalent interaction (NCI) analysis of this complex was employed for illustration of the Cl⋯Al weak non-covalent interaction. The self-consistent reaction field theory (SCRF) based on the Polarizable Continuum Model (PCM) was applied for testing the solvent effects. The solvent effect on the interaction energy, dipole moment, frontier orbital energy, and global reactivity parameters was examined as well. The changes in the dipole moment, polarizability and electronic spatial extent (ESE) with solvent polarity were analyzed by applying different solvent polarity parameters based on Lippert-Mataga, Bakhshiev and Bilot-Kawski models. In addition, temperature and pressure effects on the thermodynamic parameters of this interaction were illustrated.
Keywords
Introduction
The discovery of the anticancer activity of titanocene dichloride (namely bis(cyclopentadienly)titanocene dichloride, TiCl2Cp2, Cp =η 5–(C5H5)2) dates back to 1980s; since then, many experimental studies have been done on it [1–3]. While the outcomes of phase II clinical trials were not suitable because of the lack of activity against the investigated tumors, the reported significant investigations on titanium compounds, excited the attention in novel titanium compounds with anticancer properties [4–7].
The action mechanism of the TiCl2Cp2 is unknown; initial studies recommended that it might be connected with the purine bases of DNA [8–10]. Theoretical studies have been conducted on the hydrolysis chemistry of anticancer drug titanocene dichloride [11]. Later, more synthetic attempts have been done for boosting the cytotoxicity of titanocene dichloride derivatives [12–14]. A novel process starting from titanium dichloride and fulvenes [15, 16] paved the way for directly accessing highly substituted ansa-titanocenes [17–19].
Nano-structures such as fullerene hollow nano- clusters of elements other than carbon are favored due to their special electronic and optical properties [20–29]. Group III-nitrides and Group V-nitrides rank among the most favorable nano-clusters [30–32]. In this family, suitable applications have reported for AlN nano-clusters. These nano-clusters have shown thermodynamic stability, and high thermal conductivity and low electron affinity [33]. The investigation of Aluminum nitride nano-clusters (n = 2-41) showed that (AlN)12 nano-cluster is energetically the most stable one, and can be reflected as an appropriated nano-cluster [34]. Al12N12 nano-cluster was used successfully in gas sensing and hydrogen storage [35]. Many researchers have evaluated the ability of carbon nanotubes (CNT) and nano-clusters for a drug delivery system development [36–39]. Drug delivery is possible either through filling the inner space of the tubes with metals [40], porphyrins [41] and biomolecules [42] or by attaching proteins and compounds (via a specific adsorption or covalent bond) on their external surface. Other studies focused on loading a phenanthroline-based Platinum(II) complex onto the surface of a carbon nanotube via π–π Stacking [43]. A DFT investigation of the solvent effect, structures, properties, and topologies of the interaction between the C20 Cage and cis-PtCl2(NH3)2 has been reported [37].
Indeed, it is an advantage of preforming such theoretical works to investigate the characteristic features of matters especially those of nano-related complex systems, at the lowest possible molecular and atomic [44, 45].
This theoretical research studied the interaction between Al12N12 nano-cluster and titanocene dichloride in gas and solution phases. Differences in the frontier orbital energies were shown. It was tried to show that this interaction is dependent on the temperature and pressure.
Computational methods
Optimization and vibrational analysis were done with Gaussian 09 software package [46]. The standard 6-311G(d,p) basis set [47–50] was considered in the calculations. The B3P86 functional was employed for geometry optimization [51, 52]. B3P86 defines clearly the same functional with the non-local correlation provided by Perdew 86. The identities of the optimized structures as an energy minimum were confirmed by vibrational analysis. Non-covalent interaction (NCI) analysis was performed by evaluating the reduced density gradient and low-gradient isosurfaces [53–55]. NCI studies were performed on the optimized geometries with the used level of theory for optimization using Multiwfn 3.7 software package [56]. The polarizable continuum model (PCM) was used to include implicit solvation effects [57]. This paper uses the Lippert–Mataga polarity function (called orientation polarizability of the solvent) [58, 59], Bakhshiev polarity function [60] and Bilot and Kawski [61]. These functions have the equations of:
Lippert–Mataga polarity function:
ɛ and n are dielectric constants and refractive index of the solvent.
Bakhshiev polarity function:
Bilot and Kawski function:
Also, the studied systems were optimized at the B3LYP-D3/6-311G(d,p) and PBE1PBE1/level of theory for illustration of effect of method in the results. B3LYP-D3 model keeps the advantageousness of B3LYP method in one hand, and on the other hand, it corrects simulates the weak interactions well using Grimme term D3 [62]. PBE1PBE functional uses 25% exchange and 75% correlation weighting [63]. Single point calculations at the B3P86/6-311++G(d,p) [64–66] level of theory were done on the optimized molecules at the B3P86/6-311G(d,p) level of theory for studying of basis set effect.
Results and discussion
Solvation energies
The structure of Al12N12 nano-cluster (front view), TiCl2Cp2 anticancer drug and Al12N12 ⋯ TiCl2Cp2 complex are presented in Fig. 1. The total energy values of the Al12N12 ⋯ TiCl2Cp2 complex in the gas and solution phases are listed in Table 1. Selected solvents are tetrahydrofuran (THF), dichloromethane (CH2Cl2), Cyclohexanone (CH), Propanonitrile (PN), acetonitrile (MeCN) in this study. These values show that stability of the studied complex is greater in solution phase than gas phase. The stabilization energy values by solvents (solvation energy, Esolv) were calculated as:
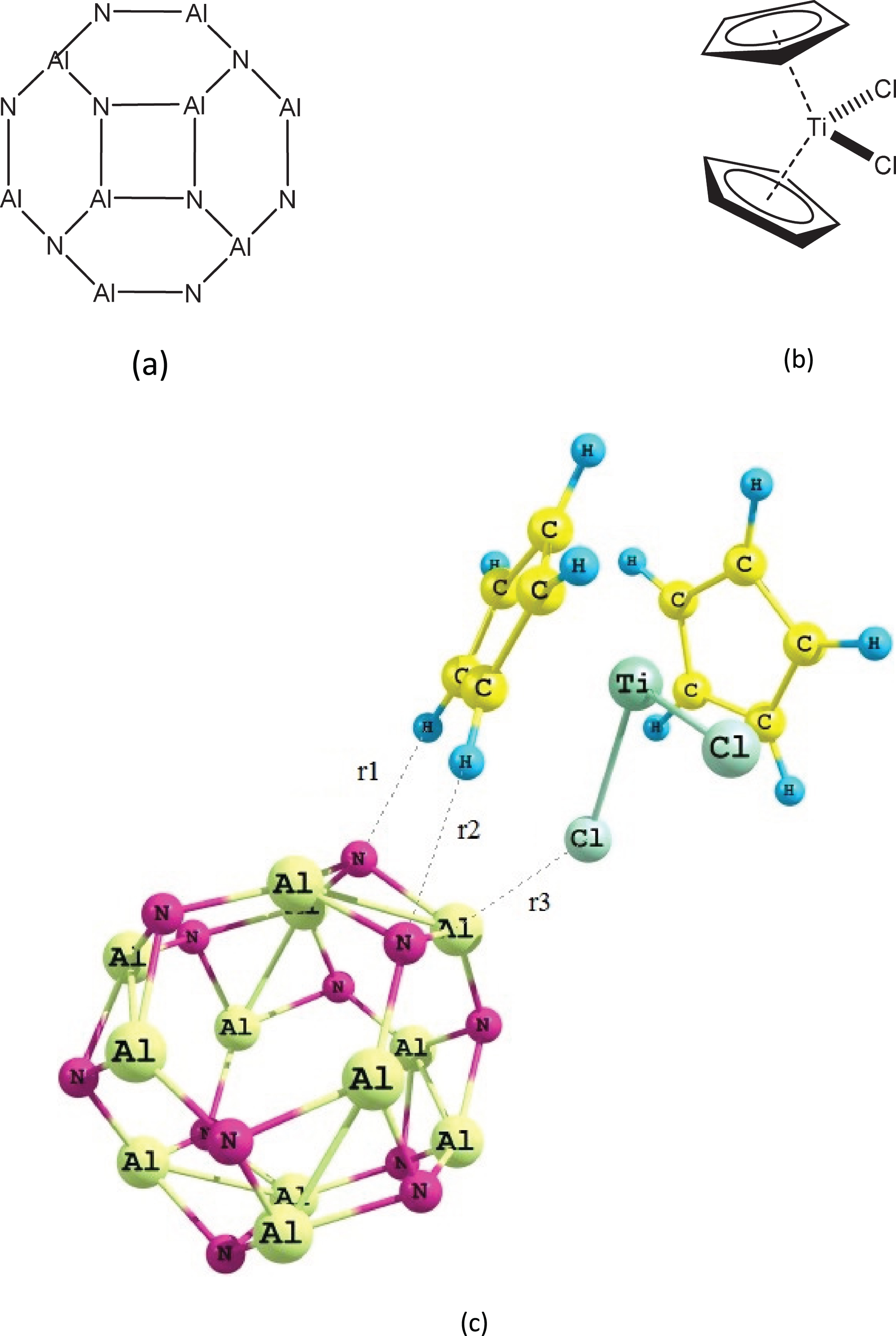
(a) Front view of the Al12N12nano-cluster, (b)TiCl2Cp2 anticancer dug, and (c) Al12N12 ... TiCl2Cp2 complex.
Total energy (E, au), solvation energy (Esolv, kcal/mol), and interaction energy (ΔEint, kcal/mol) values of the Al12N12 ... TiCp2Cl2 complex in gas and solution phases. ɛ and nD are dielectric constants and refractive index of the solvent
aB3P86/6-311G(d,p) level of theory. bB3LYP-D3/6-311G(d,p) level of theory. cPBE1PBE/6-311G(d,p) level of theory. dB3P86/6-311G(d,p) //B3P86/6-311++G(d,p) level of theory.
Calculated solvation energy values are shown in Table 1. As shown, the solvation energies depend on the size of the dielectric constant of solvents, and these values decrease with the increase of dielectric constants of solvents. Therefore, the stability of Al12N12 ⋯ TiCl2Cp2 complex increases in more polar solvents. This is because a dipole in the molecule will induce a dipole in the medium, and the electric field applied to the solute by the solvent (reaction) dipole will in turn interact with the molecular dipole to result in net stabilization. Hence, the stability of the Al12N12 ⋯ TiCl2Cp2 complex is greater in polar solvent than in the gas phase. The calculations at the B3LYP-D3/6-311G(d,p), PBE0/6-311G(d,p) and B3P86/6-311G(d,p)//B3P86/6-311++G(d,p) levels of theory predict a same trend.
Solvation energy values are fitted with quadratic equation with dielectric constants of solvents at the B3P86/6-311G(d,p) level of theory:
The computed interaction energy values (ΔEint) for the Al12N12 ⋯TiCp2Cl2 complex in gas phase and various solvents are shown in Table 1. The comparison of interaction energy value in gas phase and solution phase show weaker interaction between Al12N12 nano-cluster and TiCp2Cl2 anti-cancer drug in solution phase. As expected, the interaction between two fragments decreases in the presence of more polar solvents.
Very strong interactions are unfavorable for drug delivery, for, desorption process will be difficult. Therefore, Al12N12 cluster is a suitable carrier for drug delivery of TiCp2Cl2 complex in the presence of more polar solvents. The identical trend is observed in the similar systems [67–69].
Identical trend is observed at the B3LYP-D3/6-311G(d,p), PBE0/6-311G(d,p) and B3P86/6-311G(d,p)//B3P86/6-311++G(d,p) levels of theory.
Non-covalent interaction (NCI) analysis
The non-covalent interaction (NCI) method, which is also well-known as reduced density gradient (RDG) method, is a very common technique for illustrating weak interactions. NCI analysis of the Al12N12 ⋯ TiCp2Cl2 complex reveals the presence of Cl⋯Al weak non-covalent interaction (Fig. 2). The presence of a green surface accounts for the presence of a non-covalent interaction.
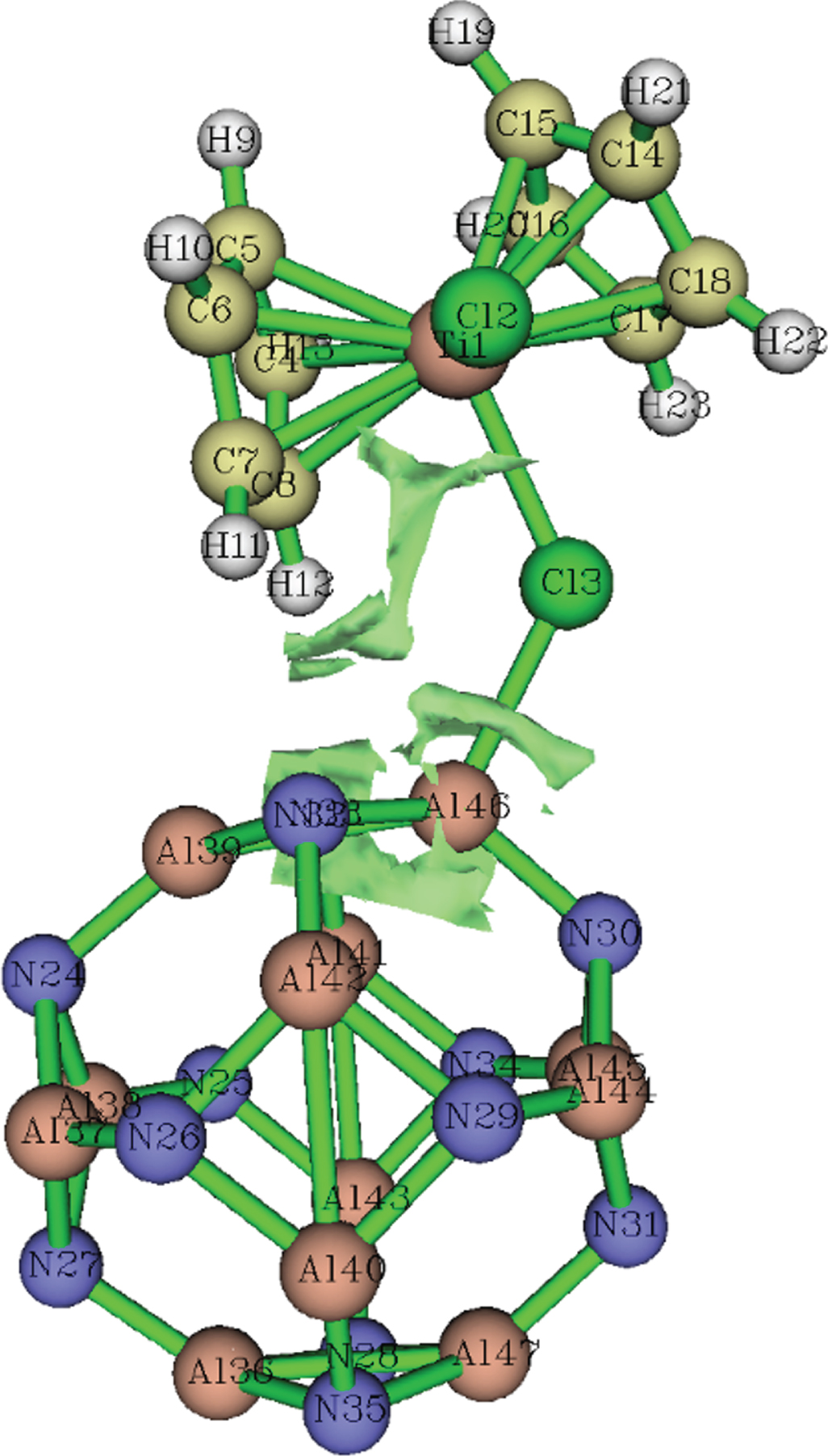
NCI gradient isosurfaces of Al12N12 ... TiCl2Cp2 complex.
H⋯N and Al⋯Cl distances of the Al12N12⋯TiCp2Cl2 complex are listed in Table 2 both gas and values show that r2(N⋯H) and r3(Al⋯Cl) distances are shorter in solution phase than gas phase. In contrast, r1(N⋯H) distance is longer in solution phase than gas phase. It can be observed the most variation in the r1(N⋯H), r2(N⋯H) and r3(Al⋯Cl) distances are 0.228, 0.023, 0.024 Å, respectively. Therefore, the most significance variations is occurred for r1(N⋯H). As seen, There is shortest r1(N⋯H) distance in the gas phase. As a result, N ... H hydrogen bonding occurs in the gas phase. This result explains stronger interaction between Al12N12 and TiCp2Cl2 in gas phase than solution phase. Increasing attentions have been reported in so-called “weak” CH⋯A (where A = N or O) hydrogen bonds [70–72]. Their important roles in a wide range of chemistry from catalysis [73] to gelation [74] to polymer structure [75] have been proposed.
H ... N and Cl ... Al distances (in Å) of the Al12N12 ... TiCp2Cl2 complex in gas and solution phases
H ... N and Cl ... Al distances (in Å) of the Al12N12 ... TiCp2Cl2 complex in gas and solution phases
Dipole moment values of the Al12N12⋯TiCp2Cl2 complex are determined in gas and several solvents (Table 3). These values indicate that dipole moment values of the complex increase in solution phase in comparison with the gas phase. These values increase in the presence of more polar solvents. Increased dipole moment values can be ascribed to long range interactions solvent with the solute molecules. Linear correlation between dipole moment values and dielectric constant of solvent show the following equations:
Dipole moment (μ, Debye), Electronic spatial extent (ESE, in au), isotropic and anisotropic polarizability (α
iso, α
aniso, in Bohr3) of the Al12N12 ... TiCp2Cl2 complex in gas and solution phases
Dipole moment (μ, Debye), Electronic spatial extent (ESE, in au), isotropic and anisotropic polarizability (α iso, α aniso, in Bohr3) of the Al12N12 ... TiCp2Cl2 complex in gas and solution phases
As shown, these parameters are not linearly correlated.
It was tried to study the linear correlations between the dipole moment of this complex and solvent polarity function frequently covering both the refractive index [76] and the dielectric constant of the liquid medium.
The linear correlations between μ and these solvent polarity functions are:
As shown, a better correlation is observed between the dipole moment and Bakhshiev function in the investigated complex. Therefore, this model reveals a better linear correlation between dipole moment values and solvent polarity functions by considering the dielectric constant and refractive index.
Isotropic and anisotropic polarizability values of the Al12N12⋯TiCp2Cl2 complex are calculated in gas and several solvents (Table 3).
Isotropic polarizability
These values indicated that isotropic polarizability values of the complex increase in solution phase in comparison with gas phase. These values increase in the presence of more polar solvents. Linear correlation between isotropic polarizability values and dielectric constant of solvent show the following equations:
The linear correlations between α
iso and these solvent polarity functions are:
As shown, a better correlation is observed between the isotropic polarizabiliy and Bakhshiev function in the investigated complex. Therefore, this model shows a better linear correlation between isotropic polarizability values and solvent polarity functions by considering the dielectric constant and refractive index.
Anisotropic polarizability
These values show that anisotropic polarizability values of the complex decrease in solution phase in comparison with the gas phase. These values decrease in the presence of more polar solvents. Linear correlation between anisotropic polarizability values and dielectric constant of solvent show the following equations:
The linear correlations between α
aniso and these solvent polarity functions are:
As shown, a better correlation is observed between the anisotropic polarizability and Bakhshiev function in the investigated complex. Therefore, this model shows a better linear correlation between isotropic polarizability values and solvent polarity functions by considering the dielectric constant and refractive index.
Electronic spatial extent
The electronic spatial extent (ESE) for each molecule is defined as the surface area covering a volume around the molecule and describes the gross receptivity of the molecule from an external electric field.
ESE values of the studied complex are reported in the gas and solution phases (Table 3). These values show that ESE values of the complex increase in solution phase in comparison with gas phase. These values decrease in the presence of more polar solvents. Linear correlation between ESE values and dielectric constant of solvent show the following equations:
As shown, these parameters are not linearly correlated.
The linear correlations between ESE values and these solvent polarity functions are:
As shown, a better correlation is observed between the ESE values and Bakhshiev function in the investigated complex. Therefore, this model reveals a better linear correlation between ESE values and solvent polarity functions by considering the dielectric constant and refractive index.
Molecular orbital analysis
Frontier orbital energy values of the studied complex are calculated in gas and various solvents (Table 4). As shown, the stability of the HOMO increases in solution phase; however, stability of LUMO decreases. The solvent, especially polar solvents, could really influence the geometry through solvent-solute interactions, thus stabilizing HOMO. In contrast, weak solvent-solute interactions destabilize LUMO.
Frontier orbital energy, HOMO-LUMO gap, hardness (η), chemical potential (μ), electrophilicity (ω) values (in eV) and electrophilicity-based charge transfer (ECT) of the Al12N12 ... TiCp2Cl2 complex in gas and solution phases
Frontier orbital energy, HOMO-LUMO gap, hardness (η), chemical potential (μ), electrophilicity (ω) values (in eV) and electrophilicity-based charge transfer (ECT) of the Al12N12 ... TiCp2Cl2 complex in gas and solution phases
The HOMO-LUMO energy gap characterizes the kinetic stability and chemical reactivity of the molecule. The low energy gap between the donor and acceptor shows the suitable transfer of energy between them. As shown, the HOMO-LUMO gap values of the complex increase in solution phase in comparison with the gas phase (Table 4). These values increase in the presence of more polar solvents. It is observed that in the polar solvents, the HOMO of the molecules is stabilized more than the LUMO via solvent-solute interactions thereby giving rise to a wider HOMO-LUMO gap as against the lower HOMO-LUMO gap in non-polar solvents.
Global reactivity descriptors
Some global reactivity descriptors such as hardness, chemical potential and electrophilicity index facilitate characterizing the nature and interaction of the molecule.
Electrophilicity-based charge transfer (ECT)
Electrophilicity-based charge transfer (ECT) is calculated defined as the difference between ΔNmax values of interacting molecules [77]:
In this equation ΔNmax is defined as:
ω and χ are electrophilicity and electronegativity of the system. They are defined as global reactivity descriptors [78–81] and determined on the basis of Koopman’s theorem [82]. These values for Al12N12 ... TiCl2Cp2 complex in the gas and solution phase are summarized in Table 4. The negative values of the ECT values reveal that charge transfer from TiCl2Cp2 to Al12N12. It can be observed, the variations of the ECT values with the dielectric constant of solvents are obeyed the following quadratic equation:
Thermodynamic parameters
Free energy, enthalpy and entropy changes values (ΔG, ΔH and ΔS, respectively) of Al12N12 ⋯TiCp2Cl2 complex formation are calculated according to the following reactions:
The calculated ΔG, ΔH and ΔS values of this reaction in the 298 K and 1atm pressure are –6.05 kcal/mol, –18.01 kcal/mol and –40.10 cal/mol.K, respectively. The negative ΔG and ΔH values of the Al12N12⋯TiCp2Cl2 complex formation shows that this reaction is spontaneous and exothermic, respectively. The negative ΔS values of these reactions seem logical since formation of one molecule after interaction between two molecules decreases the entropy of reaction. Formation constant values (K) of the Al12N12⋯TiCp2Cl2 complex is calculated by the following formula:
The calculated K value is 2.74×104.
Solvent effect
Absolute free energy and enthalpy values of studied Al12N12⋯TiCl2Cp2 complex in the gas and various solvents are reported in Table 5. The solvation free energy and enthalpy values are computed via the following equation:
Solvation free energy (ΔGsolv, kcal/mol), solvation enthalpy (ΔHsolv, kcal/mol), and solvation entropy (ΔSsolv, cal/mol.K), values of the Al12N12 ... TiCp2Cl2 complex in various solvents
Solvation free energy (ΔGsolv, kcal/mol), solvation enthalpy (ΔHsolv, kcal/mol), and solvation entropy (ΔSsolv, cal/mol.K), values of the Al12N12 ... TiCp2Cl2 complex in various solvents
According to Table 5, the amount of ΔGsolv and ΔHsolv of Al12N12⋯TiCl2Cp2 complex decreases with increasing the dielectric constant. There is a good relationship between ΔGsolv and ΔHsolv with dielectric constant values:
As shown, these parameters are not linearly correlated. These calculated parameters are fitted with quadratic equations:
There is a good relationship between ΔGsolv and dipole moment values:
Therefore, in more polar solvents, the increase in dipole moment of Al12N12⋯TiCl2Cp2 complex affects its interaction with the solvent.
The effect of temperature on the thermodynamic properties of the interaction is explored. The temperature dependent behaviors of this interaction is investigated under various conditions (T = 100–1000 K) (Table 6).
Changes of the free energy (ΔG, kcal/mol), enthalpy (ΔH, kcal/mol) and entropy (ΔS, cal/mol.K) values of the Al12N12 ... TiCp2Cl2 complex in various temperature (in K)
Changes of the free energy (ΔG, kcal/mol), enthalpy (ΔH, kcal/mol) and entropy (ΔS, cal/mol.K) values of the Al12N12 ... TiCp2Cl2 complex in various temperature (in K)
During the interaction process, the enhancement of ΔG, ΔH and ΔS are observed distinctly along with the increasing of the temperature. Therefore, the interaction occurs easily under lower temperature. There are good linear correlations between of ΔG, ΔH and ΔS with temperature:
In addition, during the interaction process, the decreasing of K values is observed distinctly along with the increasing of the temperature. Therefore, the interaction occurs easily under lower temperature. There are good linear correlations between of log (K) values with temperature:
The effect of pressure on the thermodynamic properties of the interaction is explored. The pressure-dependent behaviors of this reaction is investigated under various conditions (P = 0.1–1000 atm) (Table 7).
Changes of the free energy (ΔG, kcal/mol), enthalpy (ΔH, kcal/mol) and entropy (ΔS, cal/mol.K) values of the Al12N12 ... TiCp2Cl2 complex in various pressure (in atm)
Changes of the free energy (ΔG, kcal/mol), enthalpy (ΔH, kcal/mol) and entropy (ΔS, cal/mol.K) values of the Al12N12 ... TiCp2Cl2 complex in various pressure (in atm)
During the interaction process, the decreasing of ΔG values is observed distinctly along with the increasing of the pressure. Therefore, the interaction occurs easily under higher pressure. There are good linear correlations between of ΔG with logarithm of pressure:
The variation of ΔS during the interaction increases distinctly along with the enhancement of pressure. There are good linear correlations between of ΔS with logarithm of pressure:
Computational investigation of the interaction behavior of TiCp2–Cl2– anticancer drug with Al12N12 cluster was explored in the gas and solution phases. The calculated interaction energy values revealed the weaker interactions in the presence solution phase rather than gas phase. NCI analysis of the Al12N12⋯TiCp2Cl2 revealed the presence of Cl⋯Al weak non-covalent bond. The changes in the dipole moment, polarizability and electronic spatial extent (ESE) with solvent polarity showed good correlation with Bakhshiev function models. The HOMO-LUMO gap values of the complex increased in solution phase in comparison with gas phase. These values increased in the presence of more polar solvents. Thermodynamics analysis showed easy interaction under lower temperature and higher pressure.