
Abstract
Density functional theory (DFT) calculations were performed to investigate electronic and structural properties of barbituric acid (BA) and sixtheen of its derivatives to show impacts of structural functionalization on the features of parent BA. The models were optimized and the minimum energy structures were confirmed by frequency calculations. Molecular and atomic descriptors were evaluated for the optimized models, in which the results of formation binding strength and molecular orbital features indicated significance of such functionalization processes on the observed properties. The highest occupied and the lowest unoccupied molecular orbitals (HOMO and LUMO) and their related parameters all indicated magnitudes of changes from one molecule to another one. Furthermore, atomic scale quadrupole coupling constants (Cq) were evaluated for the nitrogen and oxygen atoms of BA compounds showing significance of structural functionalization impacts on the atomic properties of parent BA. As a consequence, such structural analyses of BA compounds could show their characteristic features for further developments especially for their efficient pharmaceutical applications.
Introduction
Structural features play important roles for arising corresponding activities for the chemical compounds as focused in structure-activity relationship (SAR) concept [1]. Evaluating molecular and atomic descriptors could help to find the mysteries inside the chemical compounds for providing their further applications [2]. In this regard, performing quantum chemical calculations could very well help to achieve the purpose of structural features evaluations [3]. Molecular and atomic scale properties could be very well recognized by quantum based descriptors especially those of molecular orbital features [4]. Therefore, it is an advantage to perform such calculations for structural features evaluations of chemical compounds especially those of pharmaceutically related ones [5–7]. Barbituric acid (BA) or 6-hydroxyuracil is one of those pharmaceutically related compounds with important roles in living systems, which is indeed the starting compound of synthesizing barbiturate drugs [8]. Barbiturates have been dealing with the central nervous system as anxiolytics, hypnotics, or anticonvulsants [9]. Additionally, further functions of anti-oxidant, anti-cancer, and anti-diabetic agents have been also reported for barbiturates [10–12]. Earlier experimental works reported synthesis of BA derivatives for evaluating new functions for such important pharmaceutical related compounds [13]. Liao et al. [14] reported the synthesis of BA derivatives with expected antiproliferative and antimigratory effects, in which they found that new candidates of BA could work very well for the purpose. Interestingly, supramolecular structures of BA combinations with other substances were studied in the work of Okuno et al. [15] to show the advantage of BA participation for generating new molecular architecture features. Further efforts on developing new features and functions for BA derivatives have been still under investigations to characterize BA employing experimental methodologies [16, 17]. Hereby, it is important to perform computational structural characterization for BA compounds to find insightful information about their characteristic features for assisting the development of this area [5–7].
In this work, BA and sixteen of its related derivatives were investigated by performing quantum chemical calculations. Earlier works indicated the important of evaluating such atomic and molecular descriptors for BA compounds [18]. Based on importance of BA compounds as starting materials for generating other pharmaceutical compounds, several experimental and computational works have been reported about the topic up to now [19–22]. But the problem has not been solved yet and further investigations could reveal more insights about BA and related compounds. To this aim, this work was performed to see the impact of functionalization process on the structural feature of parent BA. The heterocyclic ring of BA has one free carbon atomic site, in which it was functionalized by other molecular groups to provide related compounds. All the compounds were known for pharmaceutically related functions. Geometry optimizations were performed and the properties were evaluated for the optimized structures to investigate the features of BA and derivatives. This indeed an important question how the atomic scale properties of skeleton of parent BA would detect the effects of functionalization processes. To reach a possible solution for this question, atomic scale properties could help in addition to molecular scale parameters, in which quadrupole coupling constants (Cq) have been seen useful elements for this purpose [23]. Electric field gradient (EFG) tensors originated from the electronic sites of atoms could detect any perturbation to these sites to show the effects of structural functionalization and modification at the atomic scales [24]. Such physical properties have been categorized as solid-state nuclear magnetic resonance (NMR) techniques of material characterizations, in which the parameters could be reproduced very well by performing quantum-chemical calculations [25]. Using such advantages, properties at the atomic and molecular scales were evaluated for BA and derivative to see the impact of structural functionalization on the original properties of parent BA and what would be generated new.
Materials and methods
3D structures of BA and sixteen pharmaceutical derivatives (Table 1) were obtained from ChemSpider data bank [26] for investigating their features in this work. Geometries of all structures were optimized using the B3LYP/6-31 + G** level of density functional theory (DFT) as implemented in the Gaussian program [27]. Molecular descriptors including binding energies (E), ionization potential (I), electron affinity (A), energy gap (Eg), chemical potential (μ), chemical hardness (η), chemical softness (σ), electrophilicity index (ω), dipole moment (Dm) and volume (V) were evaluated for the optimized structures as summarized in Table 2. These properties are very much important regarding the stability/reactivity of chemical structures at the molecular scale. Visual representations of distribution patterns for the highest occupied and the lowest unoccupied molecular orbitals (HOMO and LUMO) and electrostatic potential (ESP) surfaces were exhibited in Fig. 1. Further calculations were performed for the optimized structures to evaluate the atomic scale Cq properties as advantageous elements for analyzing the structures at the atomic scale. The investigated models and obtained results of this work were all prepared in Tables 1–3 and Fig. 1 for discussing about reaching a possible solution for the problem of this work. The optimized geometries were all summarized in a supplementary file. Indeed, this work was originated by the advantage of employing computer-based facilities for investigating matters at the smallest molecular and atomic scales [28–32].
Models description
Models description
Evaluated molecular descriptors
Units of Eb, I, A, Eg, μ, η and ω are in eV and that of σ is in 1/eV. Units of Dm and V are Debye and cm3/mol. Eb = ([Σ aiei] –Et)/Σai; ai is number of ith atom, ei is energy of ith atom, Et is total energy of molecule. I = –HOMO. A = –LUMO. Eg = LUMO –HOMO. μ= (HOMO + LUMO)/2. η= (LUMO –HOMO)/2. σ= 1/η. ω=μ2/2σ.
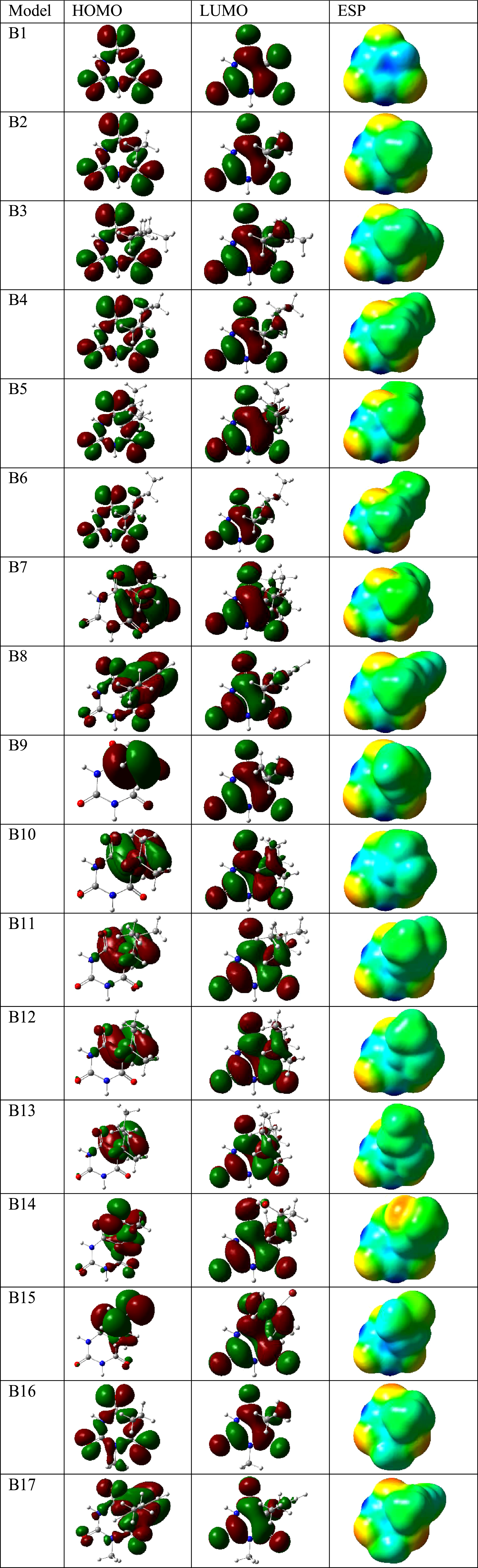
HOMO and LUMO distribution patterns and ESP surfaces.
Evaluated atomic descriptors
Within this work, structural and electronic features were investigated for BA and sixteen related compounds to show the impact of functionalization on the properties of parent BA. Descriptions of the model structures were all exhibited in Table 1 based on functionalization of additional groups to reach of R1, R2 and R3 atomic sites. With the exception of methyl group in R1 of B16 and B17, all other structures had hydrogen atom in this place. However, variations of molecular groups of R2 and R3 were significant among all B1-B17 structures. The parent BA was shown by B1 with hydrogen atoms in all of R places. For a quick analysis of the size of molecules, values of MW were all summarized in Table 1. It could be emphasized here that all the investigated structures were pharmaceutical compounds, which were analyzed in this work regarding the structural and electronic features. The structures were optimized to yield minimized energy geometries, in which they were confirmed by frequency calculations avoiding existence of any imaginary frequency.
The evaluated molecular descriptors were summarized in Table 2 for all optimized structures of B1-B17 models. The exact value of Eb implies for the extracted energy of formation of each compound, in which a larger value could show more stability for comparison of the structures. B8 was the most stable structure among all seventeen models by the largest value of Eb (4.868 eV) whereas B6 was the least stable structures with the smallest value of Eb (4.451 eV). For further analysis of these two structures, R2 was similar for both of them but a phenyl group was attached instead of R3 in B8 versus a pentyl group of R3 in B6. As a consequence, it could be mentioned here that the addition of aromatic group to R3 increased stability of BA structure. Furthermore, increasing the length of alkyl group of R3 could decrease stability of structure.
The evaluated molecular orbital based properties including I, A, Eg, μ, η, σ and ω could show that the models detect variations of electronic features through functionalization processes. These properties were all obtained regarding the energy levels of HOMO and LUMO as two dominant levels of molecular orbitals for the models structures. I and A stand for ionization potential and electron affinity, which both describe the electron transfer possibilities for the molecular systems. Such property is also very much important for determining potency of a molecule for participating in interactions with other substances. In the case of this work, I and A are two important factors showing significant impacts of functionalized molecules on the properties of BA structures with different tendency of molecule for electron donating or accepting. Eg shows energy difference of HOMO –LUMO levels or I –A levels, in which lower value of Eg could imply for easier electron transfer of two molecular orbital levels. Eg is also a common parameter for comparing the molecular structures, in which such comparison could show different tendency of internal electron transferring properties for the investigated models. Each of μ, η and σ imply for tendency of a molecule for participating in chemical reactions as important factors for changes of structures from state to state. These parameters could show significant impact of functionalization on properties of BA showing their different tendency for participating in chemical reactions. In this regard, it could be mentioned that structural modification is a procedure of lead optimization for obtaining a structure with specified features and properties. To this point, such features were changed for BA derivatives from one structure to another one showing the importance of structural analysis to show the characteristic features for molecular structures. It is indeed important to show the molecular descriptors for analyzing chemical structures, in which their visual representations could also help to reach the purpose. In Fig. 1, distribution patterns for HOMO and LUMO and surfaces for ESP were shown for a graphical analysis of the structures. The results indicated that the models detected variations of molecular orbital representations and the related surfaces through functionalization process with the most significant changes of localization of HOMO in the model systems. These all show that the chemical structures are important regarding their own features, which could determine the next function for them. Furthermore, each of Dm and V could show electric charge distribution and occupied space for molecules.
Atomic Cq descriptors were evaluated for the optimized models to show the impact of functionalization process on the properties of atomic sites of parent BA molecule. N1, N2, O1, O2, and O3 where those atoms of BA ring structure with measurable values of Cq, in which such results were evaluated and summarized in Table 3. The electronic environments of atomic sites are very much important for determining their Cq values, in which changes of Cq parameters could show variations of such environments in the model systems. For the parent BA (B1), it was expected that the values of Cq for N1 and N2 atoms were identical, in which their localization were in similar electronic environments. The same results were obtained for O2 and O3 atoms with similar electronic environment but different from O1. This achievement could imply for the importance of electronic chemical environment for determining the values of Cq for the model systems. In the functionalized models, the effects of additional functional groups were very well detected by the evaluated Cq parameters of all N and O atoms, in which the magnitude of variations of parameters were a clue for showing the significance of impact of structural modification at the atomic scale.
Conclusion
In this work, impacts of structural functionalization on the structural and electronic properties of BA were investigated by performing DFT calculations. Molecular and atomic descriptors were evaluated for the optimized parent BA and sixtheen of its derivatives to show the significance of such functionalization process in describing the model systems. The obtained results indicated that the models were different in formation binding strength, in which functionalization of phenyl group increased such stability whereas functionalization of long alkyl groups decreased it. Further analyses based on molecular orbital systems indicated that different tendency of models to participate in electron transferring processes from one molecule to another molecule. Energy differences of HOMO and LUMO levels also indicated that the internal electron transferring mode was also detected the effects of such molecular functionalization process. Chemical reactivities were also examined for the models using extra molecular orbital based parameters, in which such feature was significantly changed in the models in addition to their localization for orbitals and also electric charge distributions. Atomic scale Cq properties also indicated the atomic sites of parent BA could detect the changes of electronic environment of structures though functionalization processes. As a consequence, analysis of this work indicated that the structural and electronic features of BA molecules could be very much varied in the functionalization process to be considered for developing further applications for such molecules especially in pharmaceutical applications.