
Abstract
Formations of bimolecular barbituric acid (BA) complexes through hydrogen-bonding (HB) interactions were investigated in this work. BA has been known as a starting compound of pharmaceutical compounds developments, in which the molecular and atomic features of parent BA in homo-paring with another BA molecule were investigated here. The models were optimized to reach the stabilized structures and their properties were evaluated at the molecular and atomic scales. Density functional theory (DFT) calculations were performed to provide required information for achieving the goal of this work. Six dimer models were obtained finally according to examining all possible starting dimers configurations for involving in optimization calculations. N-H . . . O and C-H . . . O interactions were also involved in dimers formations besides participation of the X-center of parent BA in interaction. Molecular and atomic scales features were evaluated for characterizing the dimers formations. As a consequence, several configurations of BA dimers were obtained showing the importance of performing such structural analyses for developing further compounds from BA.
Introduction
Discovery of nucleic acids structures have leaded the researchers to focus on exploring new functions for each of pyrimidine and purine based structures for further applications in living systems, especially for pharmaceutical purposes [1–3]. In this regard, considerable efforts have been dedicated to characterize novel analogous of already known pyrimidine and purine structures through modifying the parent structures by additional atomic and molecular group [4–6]. Indeed, developments of materials for applications in biological media have been always an important issue for researchers to investigate their features for innovating new applications [7–9]. To this aim, several devices such as sensors have been investigated based on occurrence of interactions between the specified biological and inorganic materials to reach the purpose [10–12]. Barbituric acid (BA) is one of those pyrimidine derivatives, also known as 6-hydroxyuracil, with important functions in biological systems [13–15]. Uracil is a pyrimidine base, in which BA has been driven from structural modification of uracil at the carbon atomic site number six [16–18]. BA itself is not a real pharmaceutical compound, but it is indeed a parent material of synthesizing other potent compounds for pharmaceutical applications, generally called barbiturate drugs [19–21]. The originated compounds from BA parent are mainly medicated for patients with the central nervous system (CNS) related problems, in which phenobarbital has been a remarkable drug of this category [22]. Therefore, the parent BA structure has been seen always very important to be investigated employing several methods of computations and experiments [23–25]. One of the major important issues about such heterocyclic pyrimidine or purine based structures is their participation in intermolecular interactions, especially hydrogen bonds (HB) [26]. In this case, several efforts have been carried out to recognize proper protocols for recognizing HB interactions in the molecular systems, in which computer-based works have shown success for this purpose [27]. Earlier works indicated that the correct HB interacting systems could be very well recognized by evaluating molecular and atomic descriptors for singular and complex systems [28–31]. To this aim, examining the existence of dimers of HB interacting systems could very well describe the atomic sites features of molecules for involving in the interacting systems [32–34]. It is worth to note that employing quantum chemical approaches could help very much to reach the purpose at the reasonable accuracy and reliability of the obtained results [35–37].
Within this work, formations of dimers of BA were investigated employing density functional theory (DFT) calculations (Figs. 1 2). To this aim, singular and bimolecular models of BA were examined properly to reach the point of clarification of HB interacting systems with the importance of atomic sites features of parent BA. The required molecular and atomic scales descriptors were evaluated for discussing the problem of this work as listed in Tables 1 2 and Figs. 1–3. To carefully characterize the atomic features, the advantage of computational generation of solid-state nuclear magnetic resonance (NMR) descriptors were employed to describe HB interactions in the dimers of BA. As described extensively by the earlier works [38–40], quadrupole coupling constants (CQ) could be generated as the useful properties of solid-state NMR by performing quantum chemical calculations. Indeed, such technique was introduced among the most versatile techniques for characterizing materials at the atomic scale [41–43]. Therefore, such advantage was employed for recognition of HB interacting dimers of BA in this work to show the corresponding atomic and molecular features. Not only energy values, but also other atomic and molecular features could be used for detection of type and strength of interactions [44–46]. In addition to conventional HB interactions by locating one H atom between two electronegative atoms, other types of HB interactions have been also introduced [47–49]. As a consequence, investigating interactions inside the chemical systems has been always a complicated and important task to be considered carefully for recognizing possible applications of related materials.
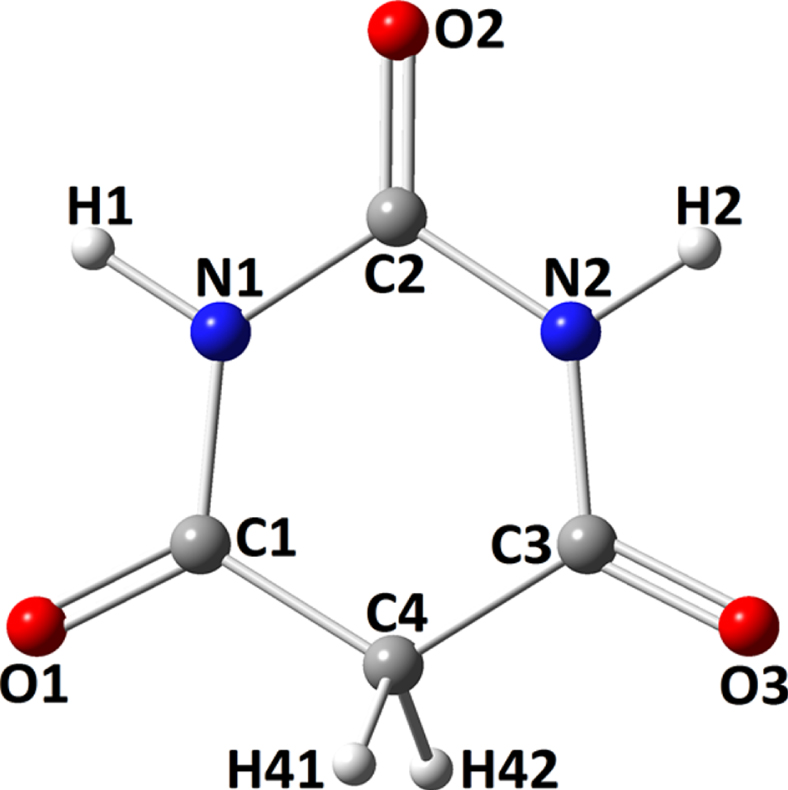
Barbituric acid.
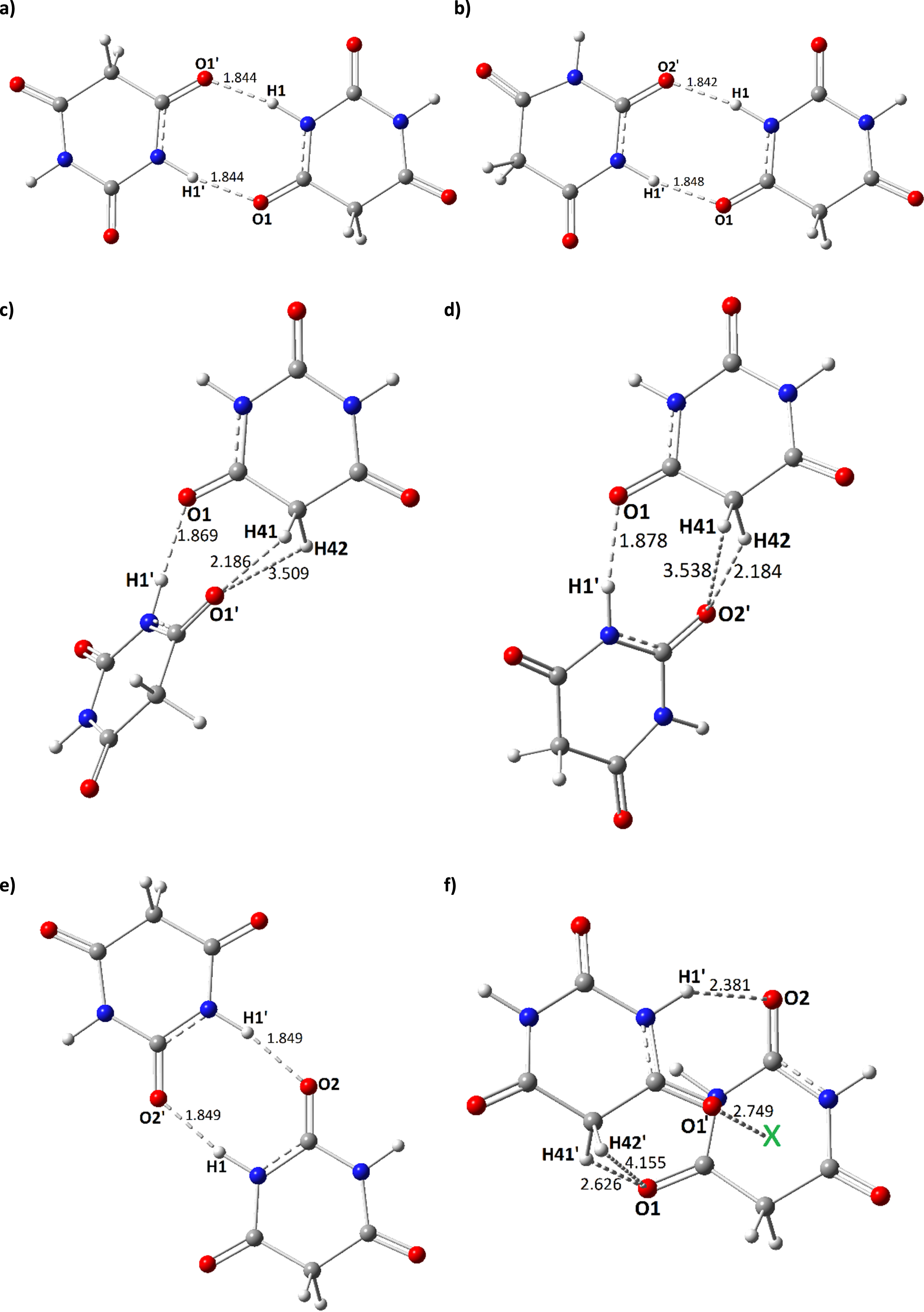
Hydrogen-bonded dimers of barbituric acid. Dashed-line distances are in Angstrom. In each dimer model, the prime signs of atoms show those of second molecule (left side) versus the parent molecule (right side).
Optimized molecular descriptors*
*The models were described in Figures 2. All energies are in eV, DM is in Debye, and V is in cm3.mol–1.
Quadrupole coupling constant (CQ MHz) atomic descriptors*
*The models were described in Figures 2. In each column, the parameters of second molecule of dimers are shown in brackets.

Earlier works revealed that dimerization could be possible for parent BA even by other substances to produce homo or hetero-bimolecular systems [50–52]. As described above, the parent BA is an important material for pharmaceutical compounds productions; therefore, synthesis of BA relatives has been always an important research subject for introducing novel drugs for medications [53–56]. To this point, characterization of the original features and functions of parent BA is an important task regarding the innovation of other relatives for such substance. Hereby, this work was performed to show the feature of homo-bimolecular formation of BA dimers through participating in HB interactions, as the most important intermolecular communicators in living systems.
Formations of HB interacting dimers of BA (Figs. 1 2) were investigated in this work by performing quantum chemical DFT calculations using the WB97XD/6-31++G** computational level as implemented in the Gaussian program [57–59]. Bimolecular configurations of BA were examined for this purpose by formations of dimers with considering all possibility of HB interactions. First, the singular molecule of BA was optimized to generate the stabilized structure. Next, dimers of BA were constructed using the optimized molecule for involving in further optimization calculations. To this point, all investigated structures were obtained for investigating further features at the molecular and atomic scales of bimolecular systems. The optimized geometries of models were shown in Table 1 including the interaction distances (DInt). Total energy (ET), interaction energy (EInt) and basis set supper position error (BSSE) were evaluated for the models to characterize the structures regarding molecular energy descriptors (Table 1). Molecular orbital based energies including energies of the highest occupied and the lowest unoccupied molecular orbitals (HOMO and LUMO) and their differences as energy gap (EG) were also obtained for the models in addition to their graphical representations of distribution patterns (Table 1 and Fig. 3). Moreover, electrostatic potential (ESP) surfaces were exhibited for the models in Fig. 3. Dipole moment (DM) and volume (V) are those two last descriptors for describing the investigated systems at the molecular scale (Table 1). For characterizing the models at the atomic scales, the absolute values of quadrupole coupling constant (CQ) were evaluated for each of oxygen, nitrogen and hydrogen atoms to show the effects of employed perturbation of HB interactions to the atomic sites features (Table 2). For doing such CQ evaluation, electric field gradient (EFG) tensors were first calculated for the mentioned atoms of optimized structures, and they were subsequently converted to the CQ parameters as atomic scale descriptors [60–62].
Results and discussion
As mentioned above, BA is an important material for development of several other drug compounds with medical impacts on CNS. Therefore, the structural features of this parent compound should be very well characterized to provide more insightful information about it for developing further applications. In this case, participating in intermolecular interactions are those of important perturbating factors with serious effects on the original features of a compound. In addition to hetero-molecular interactions, each molecule could participate in interactions with other homo-molecules of its own structure. For BA (Fig. 1), formation of such homo-pairing is possible through occurrence of HB interactions within molecule of BA in the form of dimer formation. In this regard, such dimers formations of BA (Fig. 2) were carefully investigated in this work by means of performing DFT calculations. All possible configurations of BA molecules versus each other were investigated by performing several times of optimization calculations with different starting configurations. By the results, six models of dimers were obtained for formations of BA homo-pairing system as discussed below with each configuration. Molecular and atomic descriptors (Tables 1 2) were obtained for achieving the goal of this work for discussing about the formations of BA dimers. For formation of dimers, values of BSSE were also obtained showing almost negligible effects on the obtained values of energies of total structures of interaction energies.
The singular model of BA was shown in Fig. 1, in which the atoms were assigned by numbers to be followed in the interacting systems of formations of dimers models. The model was optimized to obtain minimum energy structure, in which its 2D representation could mean similarities of properties for O1/O3 and N1/N2 regarding their identical position in the molecule. Calculations of CQ parameters actually approved this similarity hypothesis with identical values for atoms of two sides of molecule. Indeed, this achievement was important for defining starting configurations of molecular dimers for occurrence of intermolecular interactions through performing optimization calculations. As a consequence, atoms with centers of O1, N1, and O2 were considered for defining the starting configurations of homo-paring of BA. Furthermore, C4 was also considered for examining its possible role for participating in intermolecular calculations. By such expectations and achievements, six dimer models were finalized for further analyses named by 2a-f as shown by corresponding panels of Fig. 2. Each panel showed one possible final configuration of BA dimer with the assistance assigned atomic centers.
Dimer 2a. This model was optimized by locating each of O1 and N1 atomic centers versus each other but in opposite direction. A clean model was obtained by the optimization process with interactions distances of 1.844 Å for both of occurred N-H . . . O HB interactions and EInt of –0.554 eV. This model was obtained in a planar configuration with the identical atomic centers paring. Obtained molecular descriptors indicated that the energy levels of HOMO and LUMO were changed in the obtained 2a model and their corresponding value of EG was also changed in comparison with the parent PA. Values of DM and V could mean that the model detected a more identical electric charge distribution in comparison with the parent BA but with a larger space occupation due to such planar configuration far from each other. The results of Fig. 3 showed very well that such identical electric charge distribution was obtained for 2a dimer with the exhibited distribution patterns of each of HOMO and LUMO in addition to the ESP surface. To examine the atomic scale CQ parameters, it could be mentioned that the properties of those atoms (O1 and N1) with direct contribution to interactions in 2a dimer were significantly changed in comparison with the parent BA. Moreover, those atoms (O2, O3, and N2) far from the interaction regions also detected the indirect effects of occurrence of such intermolecular interactions. The hydrogen atom could work as a bridge for formation of HB interactions between two atomic centers of N and O, in which H1 detected the effects HB interactions by playing the bridging role of N-H . . . O formations whereas the properties for H2, H41 and H42 were almost unchanged. For 2a model, O1/O1’ and N1/N1’ detected identical effects confirming the mentioned seminaries of this model by the obtained molecular descriptors. Based on both values of ET and EInt, 2a model was the most favorable dimer model among the obtained dimers of BA.
Dimer 2b. This model was optimized by locating O1 and N1 atomic centers of parent molecule versus N1 and O2 of second molecule with the prime signs. H1 played the bridging role for formations of two N-H . . . O HB interactions for dimer formation. Two interacting distances of 1.842 and 1.848 Å were obtained for 2b dimer formation with the value of –0.553 eV for EInt. The energy value of HOMO and LUMO were changed to lower levels with a lower value of EG in comparison with the parent BA even dimer 2a. Significant effects of dimer formation of this model were recognized by HOMO and LUMO distribution patterns of Fig. 3, in which both molecular orbitals were mainly distributed at the second molecule and the parent molecule released almost all of them. This achievement was very much important to show insights about the role of locating configurations of molecules versus each other. The values of DM and V also indicated significant variations for this model with discrepancy from similar electric charges distribution. As mentioned above, O1 and N1 atomic centers of parent molecule and O2’ and N1’ atomic centers of second molecule were participated in the formation of HB interactions with H1/H1’ bridging atom. In this case, CQ parameters indicated insightful information for the dimers in complementary with obtained distribution patterns of HOMO and LUMO as localized at the second molecule for dimer 2b. The values of CQ were decreased for all atoms of parent molecule whereas such values were increased for those atoms of second molecule in both direct and indirect contributed atoms to HB interactions. This achievement could show the advantage of evaluations of molecular and atomic scales descriptors for characterizing the features of materials systems. Although the changes of CQ parameters for those atoms with direct contribution to HB interactions were detected significant changes of dimer formation, but the changes for other atoms of indirect contribution were almost notable. As a consequence, dimer 2b model was achieved as a typical model of dimer formation for BA.
Dimer 2c. In this dimer model formation, O1 and C4 atomic centers of parent BA were considered for participating in interactions with O1’ and N1’ of second molecule by the hypothesis of participation of H41 and H42 of C4 in intermolecular interactions. Indeed, possibility of formation of C-H . . . O semi-HB interaction was investigated by this model. The model was optimized and O1’ was relaxed versus H41 and H42 with reasonable distances of 2.186 and 3.509 Å and N1’ was relaxed versus O1 through H1’ bridging atom with H1’ . . . O1 distance of 1.869 Å. Yes, it seemed that the hypothesis of contributions of H41 and H42 to intermolecular interactions was almost confirmed by such optimized geometries with the value of –0.421 eV for EInt of dimer 2c formation. Indeed, the stability of 2c was lower than both of 2a and 2b, but the values of EInt was still meaningful for confirming the occurrence of intermolecular interactions in addition to favorable interacting distances. The energy values of HOMO, LUMO, and EG all detected variations for this dimer model formation, in which Fig. 3 showed the major distribution of HOMO at the second molecule but the distribution of LUMO at both molecular counterparts. The obtained values of DM and V were also in agreement with such achievements. Analyses of atomic scale CQ parameters indicated impacts of dimer formation for all atoms with direct and indirect contribution to interlobular interactions of dimer 2c formation. A very much interesting result was obtained for CQ of H41, in which its variation was meaningful for contributing to interaction with O1’. Accordingly, O1’ showed effects of occurrence of such interaction by variation of CQ parameter. As exhibited in Fig. 2, panel c, distance of H41 . . . O1’ was shorter than that of H42 . . . O1’; therefore, significant change of atomic feature was obtained for H41 in comparison of H42. This is indeed an advantage of performing computations to significantly detect the features of materials at the lowest molecular and atomic scales to provide insightful information for their further developments.
Dimer 2d. In this dimer model formation, possibility of occurrence of C-H . . . O interaction was investigated again by obtaining a dimer model by relaxing O2’ of second molecule versus H41 and H42 of parent molecule and H1’-bridging assisted N1’ versus O1 of parent molecule. The obtained values of ET and EInt of dimer 2d model were almost similar to those of dimer 2c model with the difference of contribution H42 to shorter distance with O2’ than H41 in reverse with those distances of H41 and H42 with O1’ of dimer 2c model. The energy values of HOMO, LUMO, and EG all detected the effects of dimer formation, in which their distribution patterns showed almost similarity to those obtained for dime 2c model. Values of DM and V also showed the electric charge distribution features for the model. In addition to all observed direct and indirect effects of dimer formation on CQ parameters of BA, the variation of CQ parameter for H42 was dominant for approving its contribution to interaction with O2’. The obtained distance of H42 . . . O2’ was 2.184 Å and that of H41 . . . O2’ was 3.538 Å meaning that H42 was placed in a more suitable position for contributing to interaction with O2’ as indicated by values of CQ parameters for H41 and H42. As a consequence, the results of dimers 2c and 2d indicated the important role of C4 atomic center and its H41 and H42 bridging atoms for participating in intermolecular interactions in BA dimer formation.
Dimer 2e. In this dimer model, each of N1 and O2 of two BA molecules in opposite direction were relaxed versus each other to obtain the optimized structure in a planar configuration. The results of ET and EInt showed that the stability of dimer 2e model could be comparable with those of 2a and 2b models; however, the obtained interaction distance of N-H . . . O was obtained 1.849 Å slightly longer than those mentioned dimer models. As a consequence, a lower stability was obtained for dimer 2e in comparison with each of 2a and 2b models. Variation of energy values for HOMO, LUMO, and EG were obtained for 2e model whereas the distribution patterns exhibited their localization at both molecular counterparts. The obtained values of DM and V also indicated corresponding features similar to those 2a and 2b models. The atomic scale values of CQ parameters indicated significant impacts on features of N1/N1’ and O2/O2’ atoms of both molecules and their bridging H1/H1’ atoms. In this regard, the properties for other atoms at parallel positions were found almost similar. This model was the second dimer model after 2a model with homo-paring with parallel positions.
Dimer 2f. The last dimer model with the least stability and interacting energy among the models was 2f model, in which both of O1 and O2 atomic centers of parent BA were contributed to intermolecular interactions with the second molecule. O1 was contributed to interactions with H41’ and H42’ and O2 was contributed to H1’ of N1’ atomic center. N1 of parent molecule was almost kept safe from a direct interactions, in contrast, O1’ was in interaction with the center of BA molecule assigned by X in Fig. 2, panel f. In addition to variations of each of HOMO, LUMO, and EG, distribution patterns indicated localization of HOMO at the parent molecule whereas localization of LUMO at the second molecule. For this dimer with non-planar configuration, typical results were achieved in this case in which values of DM and V almost approve such typical mode. Additionally, the values of CQ parameters indicated that all atoms detected notable effects of such dimer 2f formation with more or less variations of electronic features for atomic sites of molecules. Moreover, such configuration showed that availability of non-conventional interactions could be expected for the molecular systems leading their expected functions in different ways.
Conclusion
In this work, formations of HB interacting dimers of BA were examined through evaluating molecular and atomic scale features by means of performing DFT calculations. The results indicated that the parent BA could have atomic sites with similar properties, in which it was leaded to participating one side of BA with other BA molecule for formation of dimer models in homo-paring system. Six models were obtained, in which there of them were in planar configuration and three of them were non-planar. The results indicated that 2a model was the most favorable dimer and 2f model was the least favorable dime among six dimer models indicated by 2a-f. In addition to occurrence of conventional N-H . . . O HB interactions, C-H . . . O interactions were observed for the formation of dimer models. Moreover, the X-center of parent BA was also suitable for providing interaction possibility with the second BA molecule. Analyses of HOMO and LUMO features in both of quantities and distribution patterns indicated significant impacts of dimer formation in addition to values of DM and V. Furthermore, the obtained values of CQ atomic descriptors showed the roles of atoms for contributing to intermolecular interactions by more or less significant impacts of dimer formations in comparison with the results of parent BA.