
Abstract
Alveolar capillary dysplasia (ACD) is a rare neonatal lung disease characterized anatomically by a defective and hypoplastic development of pulmonary alveoli leading to persistent pulmonary hypertension (PPHN) and finally lethal respiratory failure. It is often associated with congenital left heart obstruction. Given the fatal prognosis an early diagnosis is important. However, due to the fast onset of PPHN in neonates and lack of pathognomonic signs for its cause, safe and fast detection of ACD is challenging. Therefore, following the exclusion of cardiac and common pulmonary causes, lung biopsy becomes essential for diagnosis.
We hereby report a case of ACD with atrial septal defect type one and hypoplastic aortic arch with an ante-mortem diagnosis and discuss the current state of medicine in relation to ACD.
Keywords
Case
We report a neonate delivered after 41 weeks of uncomplicated gestation in an external birth center. The child had a healthy family history, normal weight and Apgar, usual adaptation and was discharged home after four hours.
Eight hours later the newborn presented with severe hypoxemia (oxygen-saturation 55%) and dyspnea, suffering from both metabolic and respiratory acidosis (lactate of 12 mmol/l). X-ray revealed enhanced vascular markings similar to respiratory distress syndrome (Fig. 1). Echocardiography showed atrial septal defect type one (ASD 1), aortic coarctation with hypoplastic arch, open ductus arteriosus with right to left shunt and consecutive volume depleted left ventricle (Fig. 2). Blood samples did not show signs of infection.
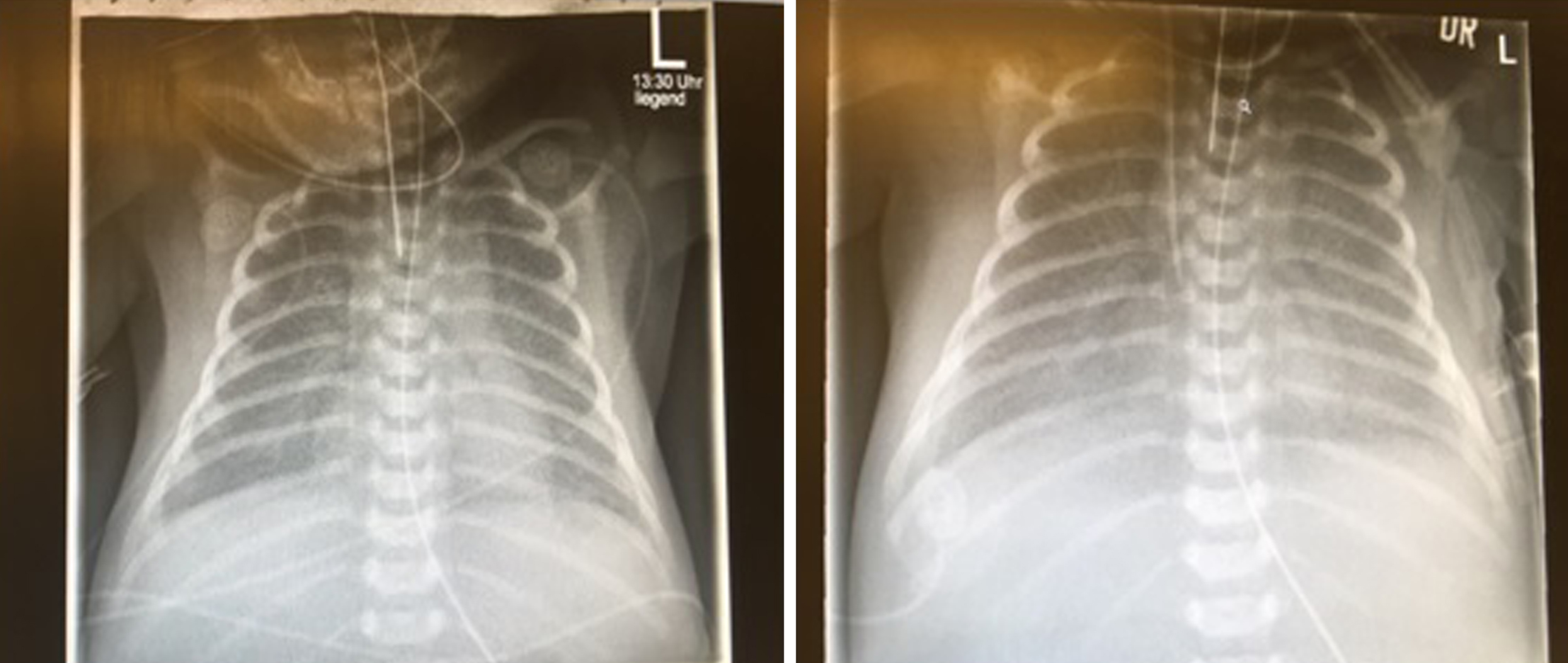
X-Ray of patient on day one (left) and day two (right). Decrease of transparency similar to respiratory distress syndrome, reticular condensation.

Echocardiography with ASD type 1 (left), hypoplasic aortic arch (middle), open ductus arteriosus (right).
Despite immediate shock treatment by ventilation, inotropes, diuretics and prostaglandin E1, low cardiac output syndrome and pulmonary hypertension persisted. Implantation of venoarterial extracorporeal membrane oxygenation (ECMO) was therefore necessary. Persistent pulmonary hypertension (PPHN) was addressed with inhaled nitric oxide, high frequency oscillation and surfactant admission. Nevertheless, neither lung function nor adequate systemic circulation could be established without ECMO. Having excluded other left heart obstruction causing pathologies and reasons for PPHN, we performed an open lung biopsy on ECMO. Histology revealed immature lobules, thickened alveolar septae, medial hypertrophy of small pulmonary arteries and muscularization of distal arterioles – typical of alveolar capillary dysplasia (ACD). ECMO was explanted, therapy was switched to comfort care and the patient passed away in their parents’ arms at the age of twelve days.
Alveolar capillary dysplasia
Alveolar capillary dysplasia (ACD) is a rare neonatal lung disease often associated with the pathologies of other organ systems (gastrointestinal, urogenital and cardiovascular) in 50–80% of cases [1]. Congenital heart defects (CHD) are present in 10–13% of patients and among this group those with left heart obstruction are most common [2, 3]. Prevalence of ACD is still unknown but more than 200 cases are described in the literature [3].
ACD is congenital and is characterized by defective and hypoplastic pulmonary alveoli [1], suggesting a severe retardation in alveolar development. There is a relative disproportion between pathologically dilated alveoli with thickened walls, the interalveolar mesenchyme and its vascularity [4].
Neonates generally present within the first 48 hours of life due to severe breathing distress that rapidly leads to respiratory failure, despite high-end treatment strategies, including ECMO support. Clinical examination may show a muted auscultation over the chest and reduced breath sounds, similar to total atelectasis or asthma. Patients are in very poor condition. Extent may vary, involving both lungs uniformly or only a portion of a single lobe. A small group with only minimal lesion and late manifestation may survive. This small group of late manifestation cases may survive a honeymoon episode that can last from days to months [5]. However, despite maximum support, therapy is frustrated and mortality is 100% to date. The only option for curing the disease is lung or combined lung and heart transplantation. A first report in the literature describes a patient with onset of ACD without cardiac abnormalities at the age of two months who was bridged to transplant with a paracorporeal lung assist device [6].
Unfortunately, most patients are diagnosed post-mortem. Only 10% of cases are recognized ante-mortem, which prohibits therapy for patients, families and care providers [1].
Histology
In our case, histology revealed a lung with dystelectasis and interstitial thickened alveolar septal tissue. Lung capillaries were centrally located and lung veins were in para-arterial position. In addition, exudate showed swollen alveolar macrophages and prominent interstitial and peri-adventitial lung fibrosis (Fig. 3).
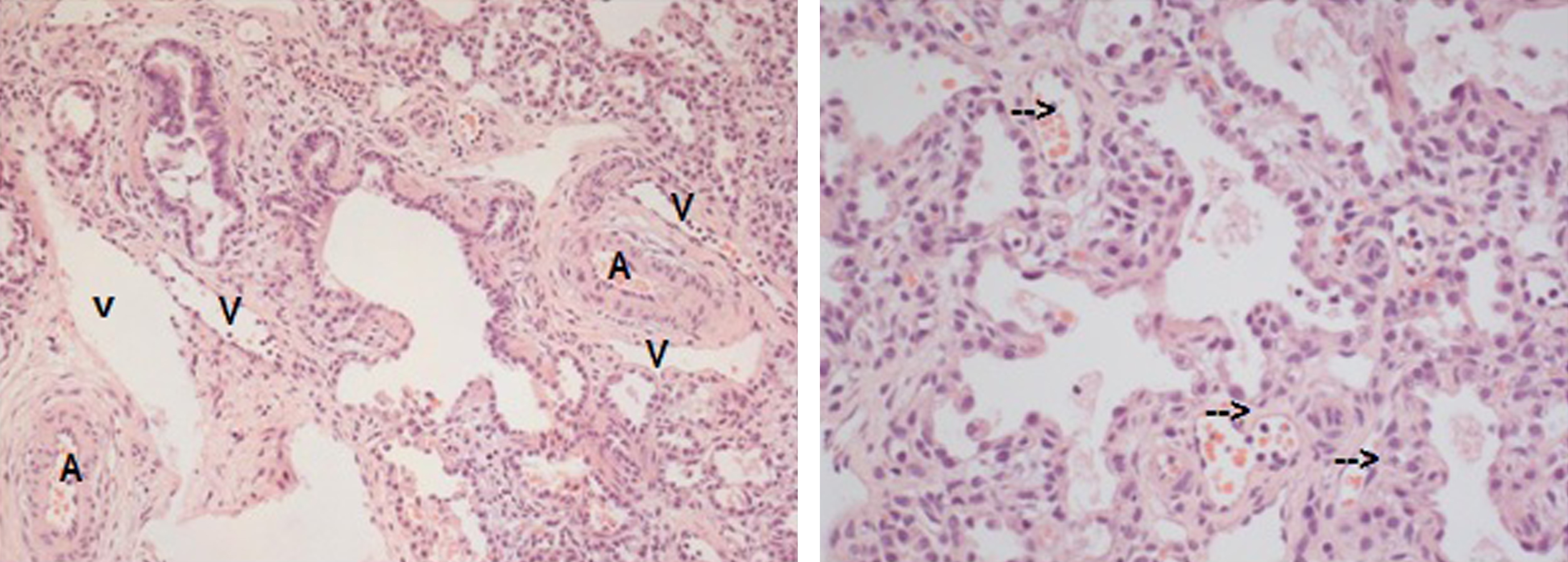
Patient’s lung biopsy reveals typical histological signs of ACD: Left: Atypical localization of distinct veins (V) besides parabronchial pulmonary arteries (A); Right: Irregularly distributed vessels (arrows) in thickened alveolar septa with often central localization and decreased contact with alveolar surface.
To summarize this and other described cases, the most striking change involves the alveolar spaces and their walls. There are too few alveoli and far too much interstitial mesenchymal stroma tissue.
There is no demonstrable basement membrane limiting the walls and much of the inner surface appears to be bare of epithelium. The walls are many times thicker than normal and this accounts for an almost complete reversal in the ratio of air space to stroma (Fig. 3). A second but less constant and less conspicuous finding in lungs showing congenital alveolar dysplasia is an exaggerated demarcation of the lobules by abnormally wide interlobular septal spaces [4].
The exact pathway from gene mutation to respiratory failure of the patient is still unknown. Nevertheless, during the last 70 years different concepts of pathogenesis have been hypothesized. In particular, it is unknown whether in patients suffering from ACD with CHD it is the heart or lung that is the “primary” organ system affected and that subsequently causes further morbidity.
“Congenital alveolar dysplasia of the lungs” was first mentioned in 1948 by MacMahon [4], who suggested that the lung arrests in the saccular stage of development. Thus, this primary lung hypoplasia causes incomplete alveolarization. MacMahon hypothesized that ACD has its origin in early embryonic development, probably dating back as far as the tenth or twelfth week of intrauterine life, since it was found in identical twins and has been encountered with and without developmental anomalies in other organs [7].
Later Cullinane and colleagues suggested that an unknown teratogen causes primary vascular abnormality and then secondarily development of CHD due to lower blood supply to the left ventricle [7]. Sirking et al. suggested an antenatal insult of the lung to be the reason for defect development of alveoli and capillaries [8]. Cater et al. described thickening of small pulmonary arteries as the primary pathology with consecutive deficient alveolar capillary development [9].
On the other hand, different variants of obstruction of the left part of the heart are suggested to cause defective lung development. As a result, an existing left heart obstruction causes inadequate blood flow through the ascending aorta. Thus, vasoconstriction in the lung maintains cerebral perfusion but also leads to retarded development of alveolar capillaries and pulmonary veins [10].
Genetics
In patients with ACD and CHD different transcription factors seem to be relevant. A microdeletion of the fork head box transcription factor gene cluster on 16q.24 can result in deletions and point mutations in genes FOXF1, FOXC2 and FOXL1. After examining several patients with ACD, with and without cardiac pathologies and other organ abnormalities, FOXF1 gene mutations seem to be responsible for pulmonary and cardiac development. FOXC2 and FOXL1 may be responsible for associated gastrointestinal and urogenital pathologies, as well as for the type of cardiac pathologies [3].
Nevertheless, gene mutations can still only be detected in about 40% of patients [11]. Moreover, to date the exact pathological pathway of ACD is still unknown and diagnosis remains a challenge.
Diagnosis and differential diagnosis
Respiratory failure in neonates can be caused by many different pathologies and ACD does not show a specific pathognomonic sign. Because of this, diagnosis of ACD using routine diagnostic tools such as laboratory analysis, X-ray, echocardiography, CT, MRI or cardiac catheter is impossible, but these diagnostic tools allow other reasons for PPHN to be excluded.
Respiratory failure, PPHN and low cardiac output are most likely to be caused by left heart obstruction prohibiting regular blood flow from lung to body circulation. Using echo, we were able to rule out intra-atrial membrane, pulmonary vein stenosis and total anonymous pulmonary vein connection, but found ASD type one and hypoplastic aortic arch. MRI, CT or cardiac catheter may also be helpful in detecting such pathologies. In this case, the low cardiac output was not caused by the hypoplastic aortic arch. Open ductus arteriosus (via prostaglandin E1) guaranteed systemic blood flow as soon as blood supply was given.
After excluding cardiac reasons for the low cardiac output, we addressed lung pathologies. Common reasons for pulmonary failure causing PPHN are meconium aspiration syndrome, infections such as pneumonia or sepsis. Another cause could be congenital lung hypoplasia, appearing isolated or in combination with congenital diaphragmatic hernia. However, blood results, X-ray, echo or CT would reveal such pathologies quickly [12]. If these routine diagnostic features do not lead to the source of the disease and the neonate cannot be stabilized through ECMO, the next step is an open lung biopsy to evaluate rare pathologies like surfactant type B deficiency and alveolar capillary dysplasia, even if the patient is on ECMO.
Open lung biopsy on ECMO
Neonates requiring ECMO support due to respiratory failure can be subclassified into two groups. In one group, a reversible or a rectified process is responsible for respiratory failure, such as meconium aspiration syndrome, pneumonia, sepsis or left heart obstruction due to intra-atrial membrane, pulmonary vein stenosis or total anonymous pulmonary vein connection. In the other group, an irreversible process such as surfactant deficiency type B or alveolar capillary dysplasia causes insufficient pulmonary function. At time of implantation of ECMO exact differentiation of the cause is often not possible. As soon as the child is referred to ECMO further diagnostic steps may reveal the reason support is needed. If routine diagnostic features fail and ECMO weaning is not possible within 7 to 14 days, open lung biopsy on ECMO should be considered [13]. According to Houmes et al. open lung biopsy on ECMO is a safe procedure with a low risk of perioperative bleeding and mortality. In particular, biopsy results frequently (88%) lead to the reason for the child’s severe disease. In over half the cases, determination of further therapy is based on the results of the lung biopsy [14].
Until today lung biopsy remains the gold standard for diagnosis as soon as ACD is suspected. Prenatal screening for families at high risk is rare, but possible in some specialized centers [15].
In our case, after several unsuccessful attempts at ECMO weaning we performed open lung biopsy on day seven of ECMO. Biopsy was uncomplicated and three specimens were analyzed, revealing ACD.
Results and diagnosis were discussed within our team and with parents. Due to the lack of curative options for our patient end-of-life care was pursued. The patient died after twelve days of extensive treatment.
Conclusions
ACD is a rare and lethal lung disease associated with congenital heart defects and should be considered in cases of obstructive left heart lesions and PPHN. It is defined by pathologically designed lobules and alveoli, wrongly located lung vessels and abnormally shaped epithelia. We conclude that this may partly be causative for its associated CHD.
As soon as obstructions such as intra-atrial membrane, pulmonary venous stenosis, APVC etc., as well as meconium aspiration, infection, congenital diaphragm hernia and pulmonary hypoplasia, are excluded, a lung biopsy is essential in order to identify ACD.
As the sensitivity of the FOX gene cluster examination is too low, lung biopsy remains the gold standard. However, genetic diagnostics are useful in completing the diagnosis. In order to deal with the fatal prognosis as early as possible, ante-mortem diagnosis is essential and avoids futile interventions for patients, families and care providers. Whether transplantation is in fact an option for the patient requires individual interdisciplinary decision-making, including the family, and consideration of the ethical aspects.
Disclosure statements
There are no financial or human disclosures. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Footnotes
Acknowledgments
No funding was received for this work.