
Abstract
Background:
Altered glutamatergic neurotransmission after traumatic brain injury (TBI) contributes to excitotoxic cell damage and death. Prevention or suppression of such changes is a desirable goal for treatment of TBI. Memantine (3,5-dimethyl-1-adamantanamine), an uncompetitive NMDA receptor antagonist with voltage-dependent open channel blocking kinetics, was reported to be neuroprotective in preclinical models of excitotoxicity, brain ischemia, and in TBI when administered prophylactically, immediately, or within minutes after injury.
Methods:
The current study examined effects of memantine administered by single intraperitoneal injection to adult male rats at a more clinically relevant delay of one hour after moderate-severe controlled cortical impact (CCI) injury or sham surgery. Histopathology was assessed on days 1, 7, 21, and 90, vestibulomotor function (beam balance and beam walk) was assessed on days 1–5 and 71–75, and spatial memory (Morris water maze test, MWM) was assessed on days 14–21 and 83–90 after CCI injury or sham surgery.
Results:
When administered at 10 mg/kg, but not 2.5 or 5 mg/kg, memantine preserved cortical tissue and reduced neuronal degeneration 1 day after injury, and attenuated loss of synaptophysin immunoreactivity in the hippocampus 7 days after injury. No effects of 10 mg/kg memantine were observed on histopathology at 21 and 90 days after CCI injury or sham surgery, or on vestibulomotor function and spatial memory acquisition assessed during any of the testing periods. However, 10 mg/kg memantine resulted in trends for improved search strategy in the MWM memory retention probe trial.
Conclusions:
Administration of memantine at a clinically-relevant delay after moderate-severe CCI injury has beneficial effects on acute outcomes, while more significant improvement on subacute and chronic outcomes may require repeated drug administration or its combination with another therapy.
Introduction
Traumatic brain injury (TBI) is a leading cause of death and disability among working age individuals in the developed world and constitutes a major health care cost burden in the United States (Faul & Coronado, 2015; Maas, Stocchetti, & Bullock, 2008). A pathological consequence of TBI is rapid accumulation and sustained action of the excitatory neurotransmitter glutamate due to its increased release from synaptic terminals and damaged cells (Bullock et al., 1995; Bullock et al., 1998; Di, Harpold, Watson, & Bullock, 1996; Faden, Demediuk, Panter, & Vink, 1989) and/or decreased cellular re-uptake in the brain (Rao, Baskaya, Dogan, Rothstein, & Dempsey, 1998). Compared to levels reported in uninjured brain, glutamate concentration rises within three hours after injury in human brain, remains elevated for hours to days (Bullock et al., 1995; Bullock et al., 1998), and correlates with long term neurological deficits (Koura et al., 1998). A similar rapid rise in glutamate concentration is observed in animal models of TBI such as fluid percussion and controlled cortical impact (CCI) injury (Faden, Demediuk, Panter, & Vink, 1989; Palmer et al., 1993) with the extent and length of elevation influenced by injury severity (Faden, Demediuk, Panter, & Vink, 1989; Nilsson, Hillered, Ponten, & Ungerstedt, 1990; Palmer et al., 1993; Rose et al., 2002). Pharmacological interventions using competitive glutamate antagonists as neuroprotective agents were promising in experimental TBI models but failed and/or were discontinued in clinical trials [see (Muir, 2006) for review] due to disruption of physiological glutamate transmission (Ikonomidou et al., 1999; Pohl et al., 1999) and detrimental side effects (Ikonomidou & Turski, 2002; Lipton, 2004a, 2004b; Muir, 2006). An alternative agent for countering glutamate-induced toxicity is memantine (3,5-dimethyl-1-adamantanamine), a pharmacologically well-characterized uncompetitive antagonist of NMDA glutamate receptors (Chen & Lipton, 1997; Chen et al., 1992; Parsons, Gruner, Rozental, Millar, & Lodge, 1993). Memantine blocks NMDA receptors only in the presence of non-physiological glutamate concentrations (i.e., supra-physiological levels and/or sustained presence in the synaptic cleft) and without affecting physiological NMDA receptor-mediated neurotransmission [see (Lipton, 2004a) for review]. Unlike other glutamate antagonists, memantine administration is not complicated by adverse side effects in humans, and is currently approved for use in moderate to severe Alzheimer’s disease (AD) to ameliorate clinical symptoms (Reisberg et al., 2003; Tariot et al., 2004; Winblad & Poritis, 1999). AD is a chronic neurodegenerative disease that involves neuronal cell death believed to be due in part to excitotoxicity related to accumulation of amyloid-β peptides (Hynd, Scott, & Dodd, 2004; Smith-Swintosky & Mattson, 1994), and for which severe TBI is a risk factor (Barnes et al., 2018; Ikonomovic, Mi, & Abrahamson, 2017; Lye & Shores, 2000) – thus memantine therapy in brain injury might have the additional benefit of reducing this risk (Miguel-Hidalgo, Alvarez, Cacabelos, & Quack, 2002).
The therapeutic potential of memantine is high in the acute time period (i.e., hours to days) after a TBI. The role of excitotoxicity in neuron death in the injured brain is well-established, for example in ischemia models (Rothman & Olney, 1986) and after TBI, wherein brain concentrations of glutamate reach supra-physiological levels within an hour (Faden, Demediuk, Panter, & Vink, 1989) and are sustained (Koura et al., 1998). A recent study in patients with moderate TBI (initial Glasgow Coma Score, GCS, between 9–12) reported that memantine therapy reduced plasma levels of neuron-specific enolase (a marker of neuronal damage) and improved GCS score assessed 3 days after TBI (Mokhtari et al., 2018). Despite limitations of this study, including lack of details of time and route of memantine administration, these findings are in agreement with reports of neuroprotective effects of memantine in experimental animal models of neuronal injury including neuroinflammation (Cho, Lee, Ju, Kim, & Kim, 2013; Rosi et al., 2009; Rosi et al., 2006), excitotoxicity (Chen, Lin, Liu, & Lin-Shiau, 2008; Misztal, Frankiewicz, Parsons, & Danysz, 1996; Pellegrini & Lipton, 1993), subarachnoid hemorrhage (Huang, Wang, Shan, Pan, & Tsai, 2015), ischemia/reperfusion (Kilic, Yilmaz, Reiter, Yuksel, & Kilic, 2013), and hypoxia/ischemia [see (Parsons, Danysz, & Quack, 1999) for review]. Several experimental TBI studies reported neuroprotective effects acutely (within hours to days) when memantine was administered immediately or within minutes after injury. Attenuated apoptotic DNA fragmentation 1 day after cold injury-induced TBI in mice (Kelestemur et al., 2016), reduced lipid peroxidation 1 day after closed-head trauma in rats (Ozsuer, Gorgulu, Kiris, & Cobanoglu, 2005), and attenuated hippocampal neuron loss 7 days after moderate CCI injury in rats (Rao, Dogan, Todd, Bowen, & Dempsey, 2001) have been reported. In addition, memantine administered by intraperitoneal injection immediately after TBI and then once every 12 hours (1 mg/kg dose) reduced apoptosis, infarct volume, gliosis, and motor impairment 3 days after fluid percussion injury in rats (Wang, Wee, Hu, Chio, & Kuo, 2018). Memantine was also protective after repetitive axonal injury in vitro (Effgen & Morrison, 2017). When given 1 hour after the last injury exposure in a repetitive mild TBI model in mice, memantine attenuated microgliosis and tau hyperphosphorylation when these variables were assessed 3 days later but resulted in no behavioral improvement at 14 days (Mei et al., 2018). Collectively, these studies provide support and impetus for continued preclinical evaluation of memantine in TBI with the caveat that the immediate intervention paradigm (i.e., administration of therapy within minutes of injury) is difficult to apply to clinical situations in humans which typically involve a significant delay to getting medical care. In the current study, we administered memantine at a more clinically-relevant delay (one hour) after moderate-severe CCI injury in adult, male Sprague Dawley rats and examined effects on acute (1 and 7 days after injury), subacute (21 days after injury), and chronic (90 days after injury) neuropathological and neurological outcomes.
Materials and methods
Animals and husbandry
The University of Pittsburgh Institutional Animal Care and Use Committee approved all investigative procedures. The study included 170 adult, male rats (Sprague Dawley, Harlan, Indianapolis, IN; 250–275 g at start of experiment). Female rats were not included in this study to avoid potentially confounding effects of greater sensitivity to adverse effects of glutamate antagonists in female compared to male rats [see (Creeley, Wozniak, Labruyere, Taylor, & Olney, 2006)]. Rats were maintained on a 12-hour light/12-hour dark cycle with free access to food and water throughout the experiment.
Experimental paradigm
Details of experimental groups, group size, intervention, survival intervals, and endpoint analyses
Details of experimental groups, group size, intervention, survival intervals, and endpoint analyses
LC-MS/MS: groups consisted of 3 rats per time point (18 rats total); Exp #1: experimental groups consisted of 5–8 rats, and the naïve group consisted of 3 rats; Exp #2: all groups consisted of 10 rats except the behavioral testing at 21 d (20 rats per group, see below); *All rats underwent beam balance and beam walking on post injury days 1–5, and MWM testing on post injury days 14–21. After completion of testing on day 21, ten rats in each group were randomly selected and sacrificed for histological analysis while the remaining ten rats underwent a second round of testing starting on post injury day 71 (beam balance and walk) and post injury day 83 (MWM). These remaining ten rats per group were sacrificed after the final day of the second round of MWM testing (day 90). Abbreviations: CCI, controlled cortical impact; d, days; hr, hours; LC-MS/MS, liquid chromatography with tandem mass spectrometry; veh, vehicle (saline).
Experiment #2 examined the effects of a single IP injection of 10 mg/kg memantine, the dose determined by mass spectrometry to result in therapeutic concentrations of memantine in plasma (see Mass spectrometry) and in reducing histological damage in Experiment #1, on vestibulomotor function (beam balance and beam walk) assessed on days 1–5 and 71–75, spatial memory (Morris water maze test, MWM) assessed on days 14–21 and 83–90, and histopathology assessed on day 21 and day 90 after CCI injury or sham surgery (Table 1). In this experimental paradigm, each group started with 20 rats. Following completion of the MWM test on day 21, 10 rats from each group were sacrificed for histopathological analysis, while the 10 remaining rats in each group were assessed a second time for vestibulomotor function (days 71–75) and spatial memory (days 83–90) after which they were sacrificed for histopathological analysis. Investigators were blinded to the treatment groups during drug or vehicle administration and histological and behavioral analyses. Blinding codes were broken only after completion of all analyses.
TBI was induced using the CCI injury model (Dixon, Clifton, Lighthall, Yaghmai, & Hayes, 1991). Rats were anesthetized with 2–4% isoflurane in N2O and O2 (2 : 1), intubated, mechanically ventilated with 2% isoflurane in 2 : 1 N2O/O2, and positioned in a stereotaxic frame. Body temperature was monitored using a rectal probe and maintained at 37°C using a heating pad. A midline incision was made in the scalp, tissue was reflected to expose the skull, and a 7 mm parasagittal craniotomy was performed over the right parietal cortex midway between bregma and lambda and centered 5 mm lateral to the sagittal suture. Rats were injured using a pneumatic CCI device (Pittsburgh Precision Instruments, Pittsburgh, PA) consisting of a vertically-directed 19.75 mm bore reciprocating double-acting pneumatic piston with a 5 cm stroke driving a 6 mm diameter flat-tip impounder. Prior to injury induction, the impounder tip was lowered to the dural surface, retracted, and then extended to achieve 2.7 mm deep cortical deformation. Impact velocity was 4.0 m/s and duration of impact (dwell time) was 150 ms. Following surgery, the scalp incision was sutured, anesthesia was discontinued, and rats were removed from the stereotaxic frame and monitored until they displayed righting reflex. Rats were returned to their home cages 30 minutes after cessation of anesthesia. Sham-operated rats were subjected to all aspects of the protocol (surgery, anesthesia, craniotomy) except for CCI. Age-matched naïve (no surgical intervention) rats were included in each experimental group (Table 1).
Mass spectrometry
Eighteen rats were injected with 10 mg/kg memantine (IP). At 1, 2, 6, 12, 24, and 48 hours after injection (n = 3 rats per time point) rats were deeply anesthetized with pentobarbital, blood was collected by transcardial puncture, and brain was cleared of blood by transcardial perfusion with 0.1 M sodium phosphate buffer (PB, pH 7.4). Blood plasma and brain tissue concentrations of memantine from each rat (3 rats per time point) were extracted by solid phase extraction using Waters Oasis HLB columns followed by liquid chromatography triple quadrupole mass spectrometry (LC-MS/MS) detection. The standard curve ranged from 5 to 2000 ng/ml (28 to 11,170 nM) and quality control samples (QC) were assessed at 40, 450, and 1250 ng/ml. 12.5 μl of the internal standard, 10 ng/μl d6-memantine, was added to all samples, QC, and standards. Solid-phase Oasis HLB columns were prepared by the addition of 1 ml acetonitrile followed by 1 ml LC/MS grade water prior to sample, standard, or QC solution addition. Internal standard spiked plasma (50 μl) or 3 ml of brain homogenate (1 : 3 tissue to PBS), QC, or standard solution were added to prepared solid phase columns. After sample addition the columns were washed 3 times with 1 ml of 5% acetonitrile and water. Columns were then eluted with 2 ml of 65 : 35 methanol:0.1% formic acid. Aliquots of the 2 ml solutions were added to injection vials for subsequent LC/MS/MS analysis.
LC-MS/MS was conducted using a Waters Acquity UPLC connected to a Thermo Quantum Ultra Triple Quadrupole Mass Spectrometer. Chromatographic separation was achieved using a BEH C18 1.7 μm, 2.1×100 mm column (Thermo, Pittsburgh, PA). The mobile phase consisted of an isocratic 65% formic acid (solution A) and 35% acetonitrile (solution B) at a flow rate of 0.3 ml/min with over a 3-minute run time. Memantine eluted from the column at a 1.2-minute retention time. Mass spectrometric detection of memantine was at an ion transition of m/z 180.1⟶163.1 and d6-memantine m/z 186.1⟶169.1 using a TSQ Quantum Ultra MS/MS with an electrospray ionization source (Thermo, Waltham, MA) under positive mode with a collision gas pressure of 1.5 mTorr, a spray voltage of 4000 kV; collision energy of 18 V; vaporizer and capillary temperature of 355°C and 350°C respectively; and sheath and auxiliary gas pressures of 50 PSI and 30 PSI, respectively. The XCalibur 2.0 software was used for data acquisition and analysis.
Histology
At time points listed in Table 1, rats were deeply anesthetized with pentobarbital and transcardial perfusion was performed with 50 ml PB followed by 300 ml of cold 4% paraformaldehyde fixative made in PB. Brains were removed and cryoprotected in graded sucrose solutions made in PB. After final infiltration with 30% sucrose, 40 μm coronal sections were obtained on a freezing sledge microtome (American Optical Corp., model 860, Buffalo, NY). FJ-C histochemistry was performed as described previously (Schmued, Stowers, Scallet, & Xu, 2005). Quantification of FJ-C labeling in the hippocampus was performed on three coronal sections spaced equally through its anterior-posterior extent, using Image J (ImageJ, U. S. National Institutes of Health, Bethesda, Maryland). Total number of pixels occupied by FJ-C labeled cells and processes were calculated separately in hippocampal regions CA1, CA3, and the dentate gyrus (DG), and values for the three sections were averaged for each rat. Cortical lesion size was determined using cresyl violet (Sigma, #C5042) histology to determine cytoarchitecture on three tissue sections through the lesion, approximately 1.3, 2.3 and 3.3 μm caudal to Bregma. Using Image J, cortical gray matter ipsilateral and contralateral to CCI injury was measured and percent of cortical tissue area spared in the injured hemisphere was calculated relative to the non-injured hemisphere.
Immunohistochemistry (IHC)
Sections were processed using the avidin-biotin method as described previously (Ciallella et al., 2002), with a mouse monoclonal antibody directed against synaptophysin (Sigma, #S5768, clone SVP-38, RRID:AB_477523; lots 118K4828, 127K4819, and 027K4834), an affinity-purified, biotinylated goat anti-mouse secondary antibody (Jackson ImmunoReseach Lab., West Grove, PA, #115-065-166; RRID:AB_2338569), the Vector Elite avidin biotin kit (Vector Labs, Burlingame, CA) and nickel-enhanced diaminobenzidine (NiDAB) as the chromogen. Sections were washed with imidazole acetate buffer before and after incubation in NiDAB. Hippocampal synaptophysin-immunoreactivity (-ir) was analyzed on coronal sections spaced equally through the extent of hippocampus by calculating the optical density (OD) of immunoreactive puncta in CA1 stratum radiatum (CA1sr), CA3 stratum lucidum (CA3sl), and DG stratum moleculare (DGsm) using Image J.
Beam balance and walk tasks
Gross vestibulomotor function as well as its finer components and coordination were assessed by the beam balance task and beam walk task, respectively, on days 1–5 and days 71–75 after CCI injury using procedures described previously (Dixon et al., 2003).
Morris water maze (MWM)
MWM testing was performed on days 14–21 and days 83–90 after CCI injury (Dixon et al., 2003). One-day prior to the test, rats were pretested to assess their ability to swim normally for 1 minute. The MWM test was performed in a blue-colored plastic circular pool (180 cm diameter; 60 cm height) filled with 26°C water to a height of 28 cm and positioned in a 3 meter×3 meter room with visual cues (visible to rats while in the pool) placed on the walls at each of the primary intercardinal directions (NW, NE, SW, SE), dividing the pool into four quadrants. A clear acrylic escape platform (10 cm diameter) was positioned 2 cm below the surface of the water (invisible to rats in the pool) and 26 cm away from the pool wall in the target (SW) quadrant and held constant for all rats. Memory acquisition of each rat was assessed in four trials per day on each of days 14–18 and days 83–87 after CCI injury or sham surgery. The first trial of each block consisted of placing the rat into the water facing the wall of the pool at a randomly chosen quadrant. For each of the subsequent three trials the rat was placed into the water in a randomly chosen quadrant not yet used for entry in previous trials in the block. During each trial, rats were allowed to explore the pool for 120 s and trials were stopped after rats found and climbed onto the platform or after 120 s elapsed. Rats that failed to find and climb onto the platform after 120 s were manually guided to the platform. In either scenario, the rat remained on the platform for 30 s after which the rat was removed from the pool and placed into a warmed incubator for four minutes between each trial, and before being returned to the home cage after the fourth trial. On days 19 and 20 and days 88 and 89 after CCI injury and sham surgery, the escape platform surface was raised to a position 2 cm above the surface of the water (visible to the rat) and each rat underwent four trials each day as described above. On days 21 and 90 after CCI injury or sham surgery, the escape platform was removed from the pool and each rat was placed in the pool for a 120 s probe trial. Swim speed, swim length, swim pattern, and time spent in the target quadrant during the probe trial were evaluated using a video tracking system (Chromatrack, San Diego Instruments, San Diego, CA). Additionally, the path taken by each rat during the probe trial was evaluated and categorized into one of three types: focal searching, circling, or thigmotaxis [thigmotaxis: tendency to explore while remaining close to maze walls; categories were adapted from (Tuma, Kolinko, Vozeh, & Cendelin, 2015)].
Statistical analyses
Experimental groups were compared by experimental measures using a one-way ANOVA with Bonferroni post hoc testing, Student t-test, or Fisher exact test where applicable. Behavioral outcome measures (beam balance, beam walk, and MWM) were assessed using a two-way repeated measures ANOVA and Bonferroni post-hoc testing. Statistical analyses were performed using GraphPad Prism 7.02 software (GraphPad Software, Inc., La Jolla, CA). To achieve greater than 80% power for detecting an effect size of 0.33 between groups, 0.39 for time and 0.35 for the interaction effect of groups and time, a minimum of ten rats per group were used in the behavioral studies. To achieve greater than 80% power for detecting an effect size of 0.45, a minimum of five rats per group were used in histological studies.
Results
Memantine dose-response analysis after CCI: acute histopathological outcomes
Histological outcomes of acute, subacute, and chronic experiments
Histological outcomes of acute, subacute, and chronic experiments
1Tissue spared = percent of cortical tissue ipsilateral to controlled cortical impact (CCI) injury compared to the contralateral (non-injured) side. 2FluoroJade (FJ-C) = measure of hippocampal cell degeneration assayed by calculating FJ-C positive particles (pixels) in high resolution microscopic composites of three equally spaced coronal sections through the dorso-rostral hippocampus. 3Synaptophysin = optical density (OD) of synaptophysin immunoreactive puncta in coronal sections equally spaced through the dorso-rostral extent of hippocampal subregions, expressed as percent ipsilateral compared to contralateral region relative to CCI injury. Abbreviations: CA, Cornu Ammonis; d, days; DG, dentate gyrus; n.s., not significant by ANOVA; sl, stratum lacunosum; sm, stratum moleculare; sr, stratum radiatum; veh, vehicle (saline).
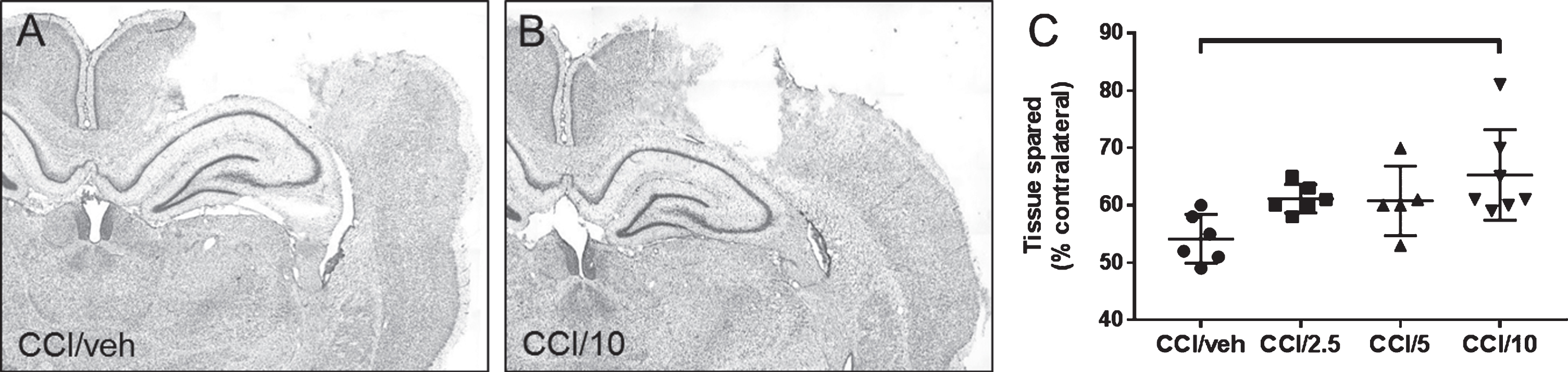
Photomicrographs of coronal brain sections (40 μm thickness) processed with cresyl violet to delineate tissue cytoarchitecture, taken at approximately 2.75 mm caudal to bregma, from representative rats receiving CCI and either vehicle (A, CCI/veh) or 10 mg/kg memantine (B, CCI/10) and sacrificed 1 day later. The scatter graph (C) shows the amount of tissue spared in CCI/veh compared to treatment with memantine doses of 2.5 mg/kg (CCI/2.5), 5 mg/kg (CCI/5), or 10 mg/kg. The 10 mg/kg memantine dose significantly reduced injury-induced tissue loss relative to the CCI/veh group (C). See Table 2 for the statistical differences between groups indicated in the graph by a horizontal bracket.
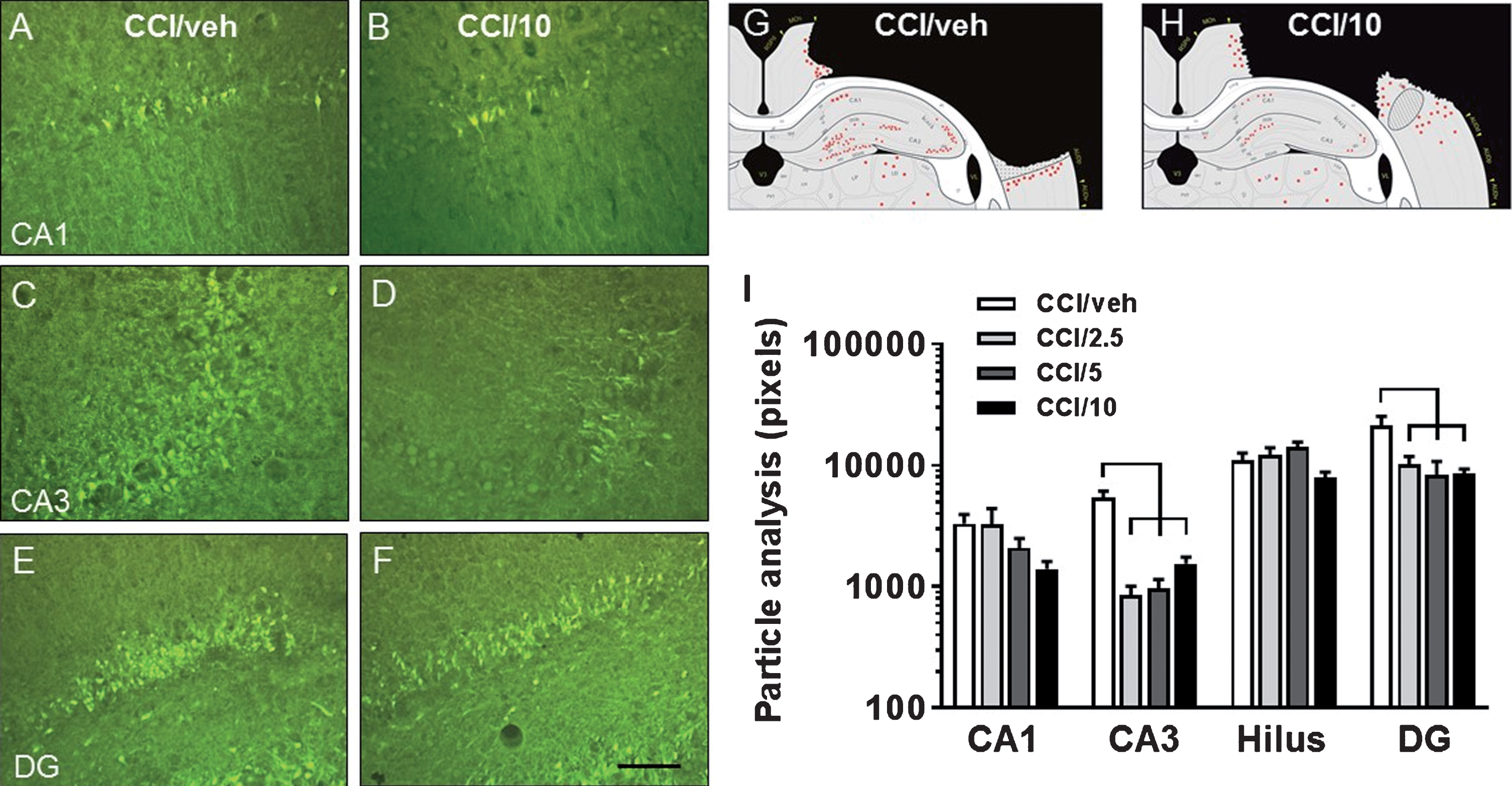
A–F: Fluorescent photomicrographs illustrating FluoroJade-C (FJ-C) labeled cells in hippocampus CA1 (A, B), CA3 (C, D), and DG (E, F) from rats receiving CCI and either vehicle (A, C, E; CCI/veh) or 10 mg/kg memantine (B, D, F; CCI/10) and sacrificed 1 day after CCI. G-H: Schematics illustrate representative distributions of FJ-C-labeled cells surrounding the midpoint of the rostrocaudal extent of the lesion in CCI/veh (G) and CCI/10 (H) groups. I: Compared to the CCI/veh group, all three memantine doses resulted in significantly decreased numbers of injury-induced FJ-C labeling of cells in the DG and CA3 region, while there were strong trends for beneficial effects of the 10 mg/kg dose in CA1 and in the hilus. See Table 2 for the statistical differences between groups, as indicated in the graphs by horizontal brackets. CA1, CA3 = cornu ammonis fields 1 and 3; DG = dentate gyrus. Scale bar = 75 μm.
Seven days after CCI injury (Tables 1 and 2, Experiment #1), no gross damage to the hippocampus was detected in any CCI group. Cortical lesion size in the three CCI/memantine groups was not significantly different from the CCI/vehicle group (Table 2). In the CCI/vehicle group, synaptophysin-ir was reduced on average by ∼25–30% in CA1sr, CA3sl, and DGsm in the injured hemisphere compared to the non-injured hemisphere (Table 2; Fig. 3A, B, E). Loss of synaptophysin-ir in the DGsm was attenuated significantly in the CCI/10 mg/kg memantine group (Table 2; Fig. 3C–E). Sham rats had similar synaptophysin-ir optical densities in the hippocampus from the craniotomized hemisphere compared to the opposite hemisphere and compared to naïve rats (not shown). Overall, the studies in Experiment #1 identified the memantine dose of 10 mg/kg as most protective against acute neuronal degeneration and synaptic loss in the hippocampus after CCI injury, therefore this dose was used in the subacute and chronic studies in Experiment #2.
Photomicrographs of synaptophysin immunoreactivity in the dentate gyrus stratum moleculare (DGsm) 7 days after CCI in rats treated with either vehicle (A, B) or 10 mg/kg memantine (C, D). Changes in synaptophysin immunoreactivity were assessed by calculating the optical density of DGsm from the injured hemisphere (A and C) as a percent of the value obtained in the mirror area from the uninjured hemisphere (B and D, respectively) to injury. E–G are scatter plots of optical density measured in DGsm (E), CA1 stratum radiatum, CAsr (F), and CA3 stratum lucidum, CA3sl (G). Scale bar = 50 μm.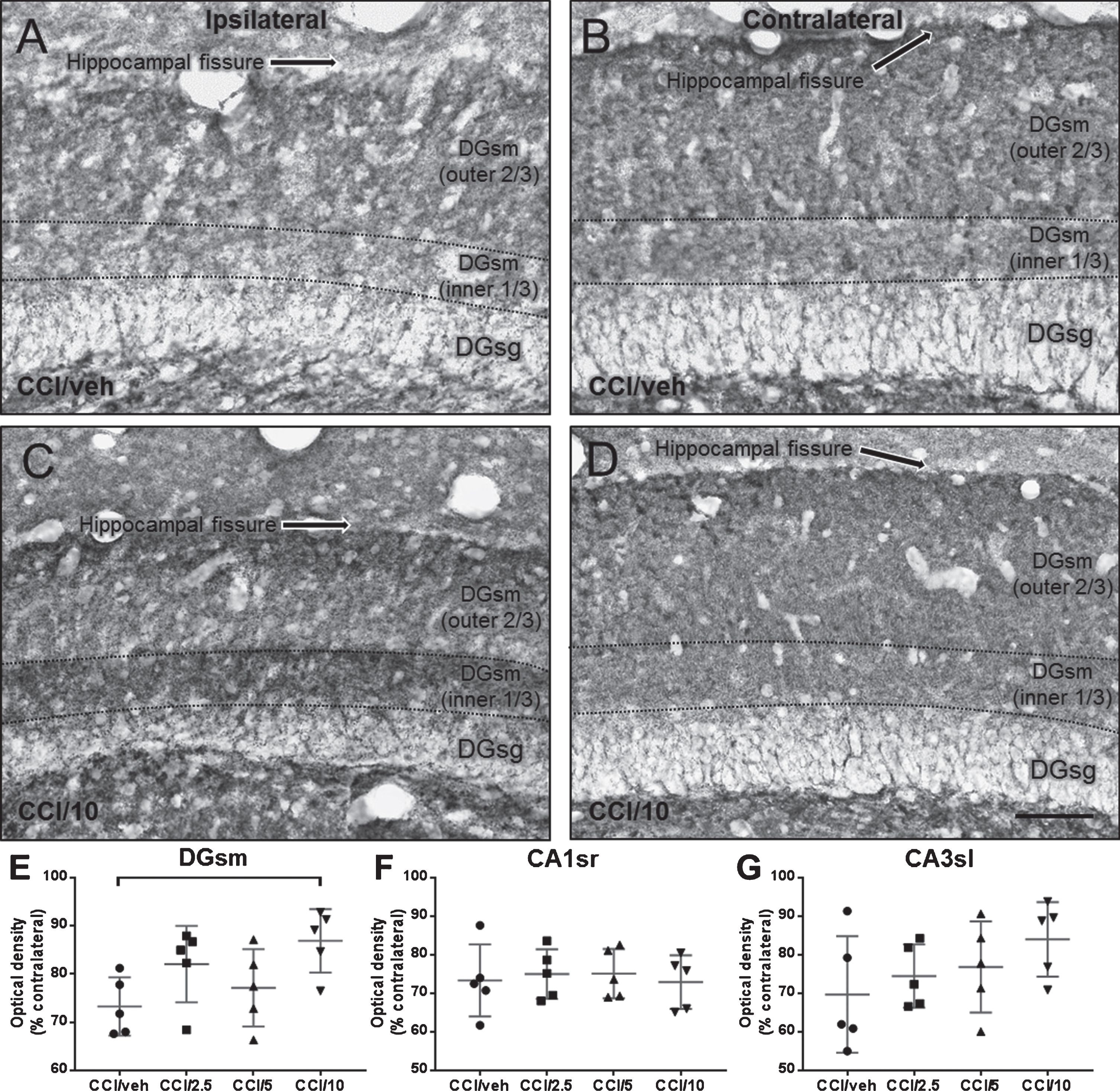
Sample LC-MS/MS chromatograms of memantine and d6-memantine are illustrated in Fig. 4. Standard curve linearity from 20 to 2000 ng/ml memantine concentrations is illustrated in (Fig. 4 (inset)). Based on the standard curve, intra-day assay reproducibility was evaluated with QC concentrations of 50, 400, and 900 ng/ml and the percent relative standard errors were 11.7, 2.93, and 2.21% respectively (n = 12 per QC concentration, not shown). For measurements of memantine concentrations in plasma and brain (cerebral cortex), rats that received one IP injection of 10 mg/kg of memantine were sacrificed after intervals of 1, 2, 6, 12, 24, or 48 hours. Plasma concentrations were highest 1 hour after injection (∼2,800 nM), reached therapeutic levels (∼1,000 nM) 2 hours after injection, and declined to negligible concentrations (∼30 nM; assay detection limit = 28 nM) after 12 hours (Fig. 4C). Plasma concentrations between 1 and 2 hours after injection were near the concentration range reported in other studies (More et al., 2008). Brain memantine concentrations in the cerebral cortex were approximately 15 times greater than plasma concentrations 1 and 2 hours after injury (Fig. 4D), in agreement with previous studies reporting up to 20 times higher memantine concentration in brain compared to plasma after IP injections in this dose range (Wesemann, Schollmeyer, & Sturm, 1982). Although we were not able to determine brain interstitial fluid (ISF) memantine levels in the current study, previous studies report that after injection in the 1–20 mg/kg range, memantine concentration in ISF is similar to its concentration in plasma even though total memantine concentration in brain is up to 20 times higher (Hesselink, De Boer, Breimer, & Danysz, 1999).
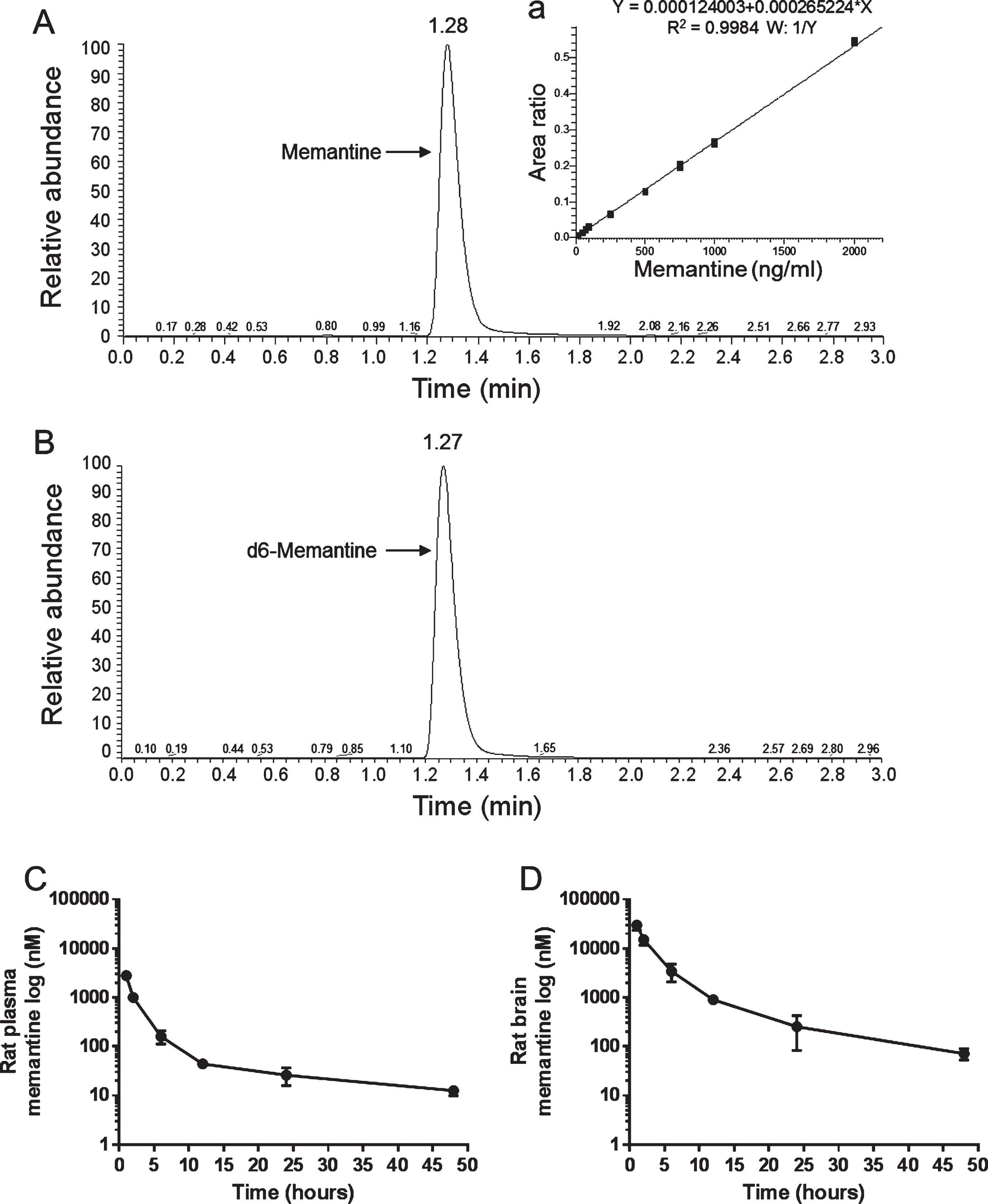
Example chromatograms of UPLC triple quadrupole mass spectrometry measurements of memantine in rat plasma spiked with memantine or d6-memantine illustrating peaks specific for (A) memantine (1.28 minutes) and (B) d6-memantine (1.27 minutes). Measurements performed on rat plasma spiked with increasing concentrations of memantine resulted in a standard curve with good linearity over concentrations ranging from 20 to 1000 ng/ml (inset a). In C and D, rats received intraperitoneal injection of 10 mg/kg memantine and were sacrificed after intervals of 1, 2, 6, 12, 24, and 48 hours (3 rats per timepoint). Plasma was collected via cardiac puncture and cerebral cortex was dissected immediately after pentobarbital overdose. Washout of memantine from rat plasma is illustrated in C and from brain in D.
Rats were sacrificed for histological assessment after behavioral testing 21 days and 90 days after CCI injury or sham surgery (n = 20 and n = 10 per group, respectively, (Tables 1 and 2), Experiment #2). At 21 days after injury, cortical lesion size in the CCI/vehicle and CCI/10 mg/kg memantine groups was similar to that observed after 7-days survival (i.e., there was no obvious expansion of the cortical lesion, (Table 2), and there was no gross tissue damage to the hippocampus due to CCI). However, in the CCI/vehicle group, hippocampal synaptophysin-ir in the injured hemisphere (relative to the non-injured hemisphere) was reduced on average by ∼8% in CA1sr, ∼13% in CA3sl, and ∼9% in DGsm (Table 2). Treatment with 10 mg/kg memantine did not affect synaptophysin-ir significantly, though there was a trend for attenuation of injury-induced synaptophysin-ir reduction in the DGsm (p = 0.064; Bonferroni post hoc test, not shown).
When assessed 90 days after CCI injury, cortical lesion size was slightly larger than after 21-days survival but there was no significant difference between the CCI/vehicle and CCI/10 mg/kg memantine groups (Table 2). Although there was no gross damage to the hippocampus from the CCI, the CCI/vehicle group had synaptophysin-ir reduced on average by ∼8–10% in the CA1sr, CA3sl, and DGsm in the injured hemisphere relative to the non-injured hemisphere. CCI injury-associated reductions in synaptophysin-ir were not attenuated by 10 mg/kg memantine when assessed at this chronic survival time point (Table 2).
Behavioral outcomes after CCI and memantine treatment
For the behavioral experiments, each group consisted of 20 rats except the CCI/vehicle group (19 rats). One rat in the CCI/vehicle group was eliminated after the animal failed to meet the MWM pre-assessment criteria (see Methods).
Beam balance latency
There were no differences among the groups during the pre-surgery assessment trials. Over days 1–5 after surgery, there was a significant overall group difference [F(4, 94) = 40.04, p < 0.0001]: the non-CCI groups, as expected, performed better than the CCI groups (p < 0.01). No differences were detected when comparing the CCI groups or the non-CCI groups. At 71–75 days after CCI, there were no significant overall group differences [F(4, 45) = 2.21, p = 0.08].
Beam walk latency
There were no differences among the groups prior to surgery (Fig. 5A). Over days 1–5 after surgery, there were significant overall group differences [F(4, 94) = 91.5, p < 0.0001]: the non-CCI groups performed better than both CCI groups (p < 0.01) but the latter two groups did not differ from each other. Within the non-CCI groups, a slight but non-significant negative effect on beam walk latency was detected over days 1–3 in the sham/vehicle group but not in the sham/10 mg/kg memantine group. At 71–75 days after surgery, no significant overall group differences were observed [F(4, 45) = 2.25, p = 0.08]. Similar CCI and sham-surgery effects were noted when beam walk scores were calculated (data not shown).
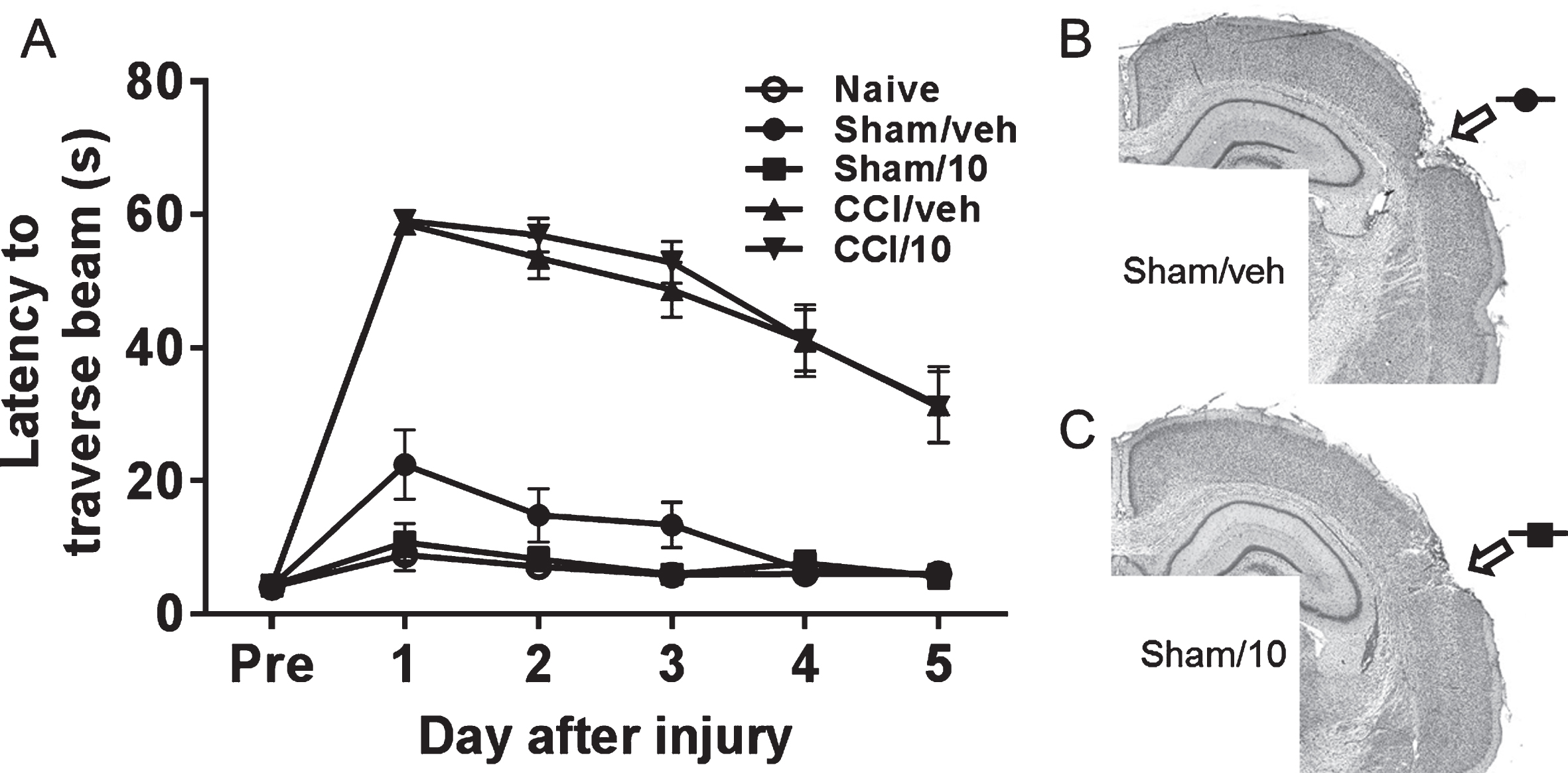
Beam walk latencies (A) of naïve rats and rats after CCI injury or sham surgery and treatment with either vehicle (sham/veh, CCI/veh) or 10 mg/kg memantine (sham/10, CCI/10). Tests were performed on days 1–5 after pretesting (Pre) and CCI injury or sham surgery. Minor cortical damage is evident in sham operated rats receiving vehicle (B) or 10 mg/kg memantine (C).
On days 14–18 after surgery there were significant overall group differences [F(4, 94) = 18.4, p < 0.0001] in latencies to find the submerged platform in the target quadrant (MWM acquisition test, platform not visible to rats): the CCI groups exhibited longer latencies compared to the non-CCI groups (post hoc: p < 0.01) (Fig. 6a). There were no differences in latencies between the two CCI groups or the three non-CCI groups. Similar results were observed when groups were compared by average overall latency (Fig. 6b). All groups exhibited shorter latencies on day 18 compared to day 14 (Fig. 6C) however this difference was less marked in the CCI/vehicle group (p = 0.03) compared to all other groups (all p < 0.0001). Comparing CCI/vehicle and CCI/10 mg/kg memantine groups by MWM acquisition on day 18 revealed a trend for better performance after memantine treatment (p = 0.11). On day 21 after CCI or sham surgery, there were significant overall group differences [F(4, 94) = 7.7, p < 0.0001] in percent time spent in the target quadrant (MWM probe trial, no platform in target quadrant): the CCI groups spent less time in the target quadrant compared to the non-CCI groups (Fig. 6D). The time spent in the target quadrant was not different between the two CCI groups and among the three non-CCI groups. Average swim speed and total swim length were similar among all groups by one-way ANOVA [F(4, 94) = 0.75, p = 0.56; F(4, 94) = 0.86, p = 0.49, respectively]. Analysis of search strategy used during the probe trial 21 days after surgery showed that in CCI groups, rats treated with 10 mg/kg memantine were less likely to exhibit thigmotaxis compared to those treated with vehicle (45% versus 68%; Fig. 7) and in sham groups, rats treated with 10 mg/kg memantine were more likely to exhibit a focal search pattern compared to those treated with vehicle (40% versus 25%; Fig. 7) though in both instances these trends did not reach statistical significance (Fisher exact test, p = 0.20 and 0.32, respectively).
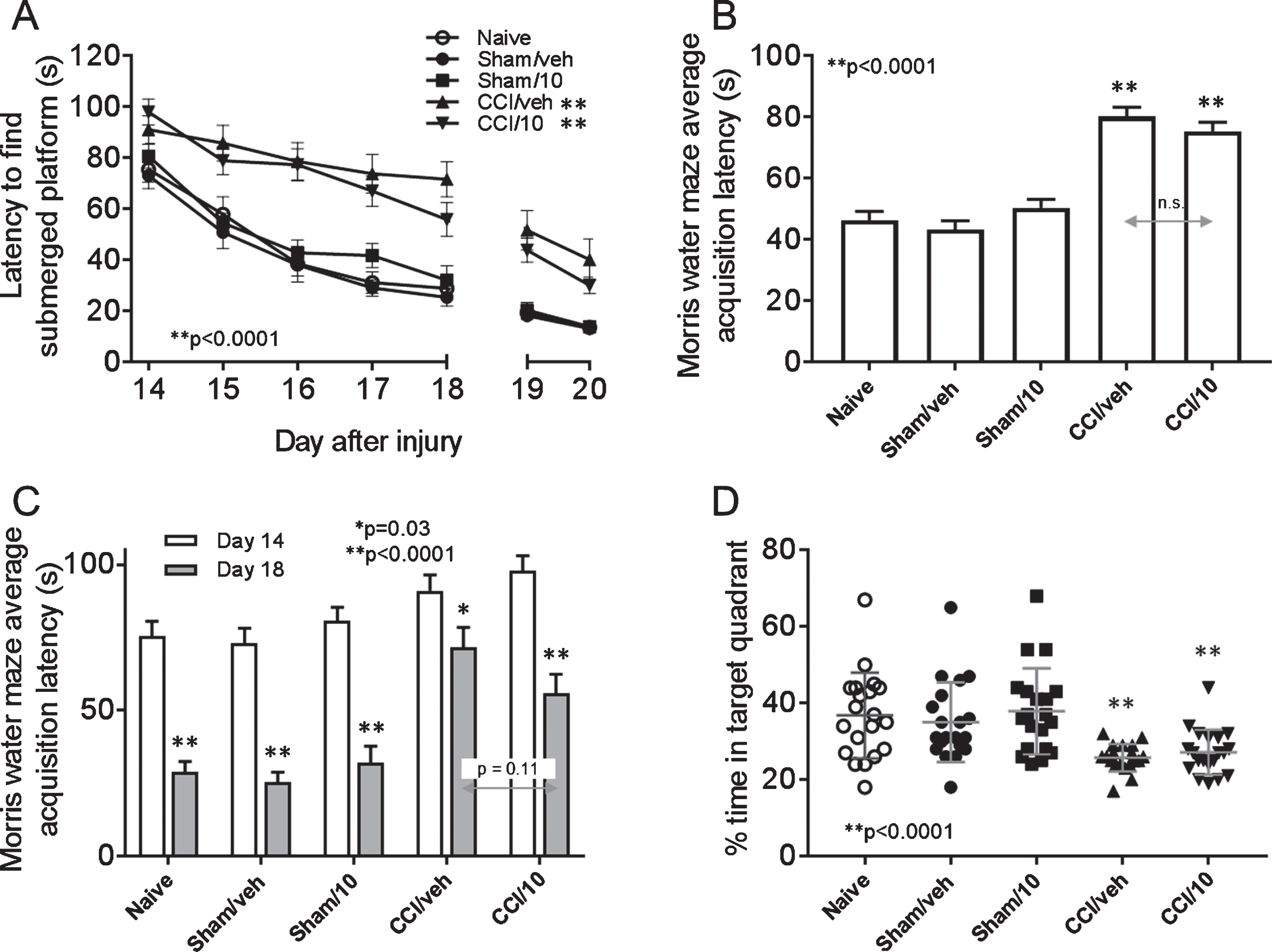
Morris water maze (MWM) test of spatial memory of naïve rats and rats receiving CCI injury or sham surgery and treatment with either vehicle (sham/veh, CCI/veh) or 10 mg/kg memantine (sham/10, CCI/10). Memory acquisition trials (time, in seconds, to find the submerged platform in the MWM) were run on days 14–18, and latency to visible platform was tested on days 19 and 20 (A). Overall average MWM acquisition in the five groups was compared in (B), and differences in MWM acquisition on day 14 was compared to day 18 in each group (C). Memory retention was tested on day 21 (D). See text for statistical results.
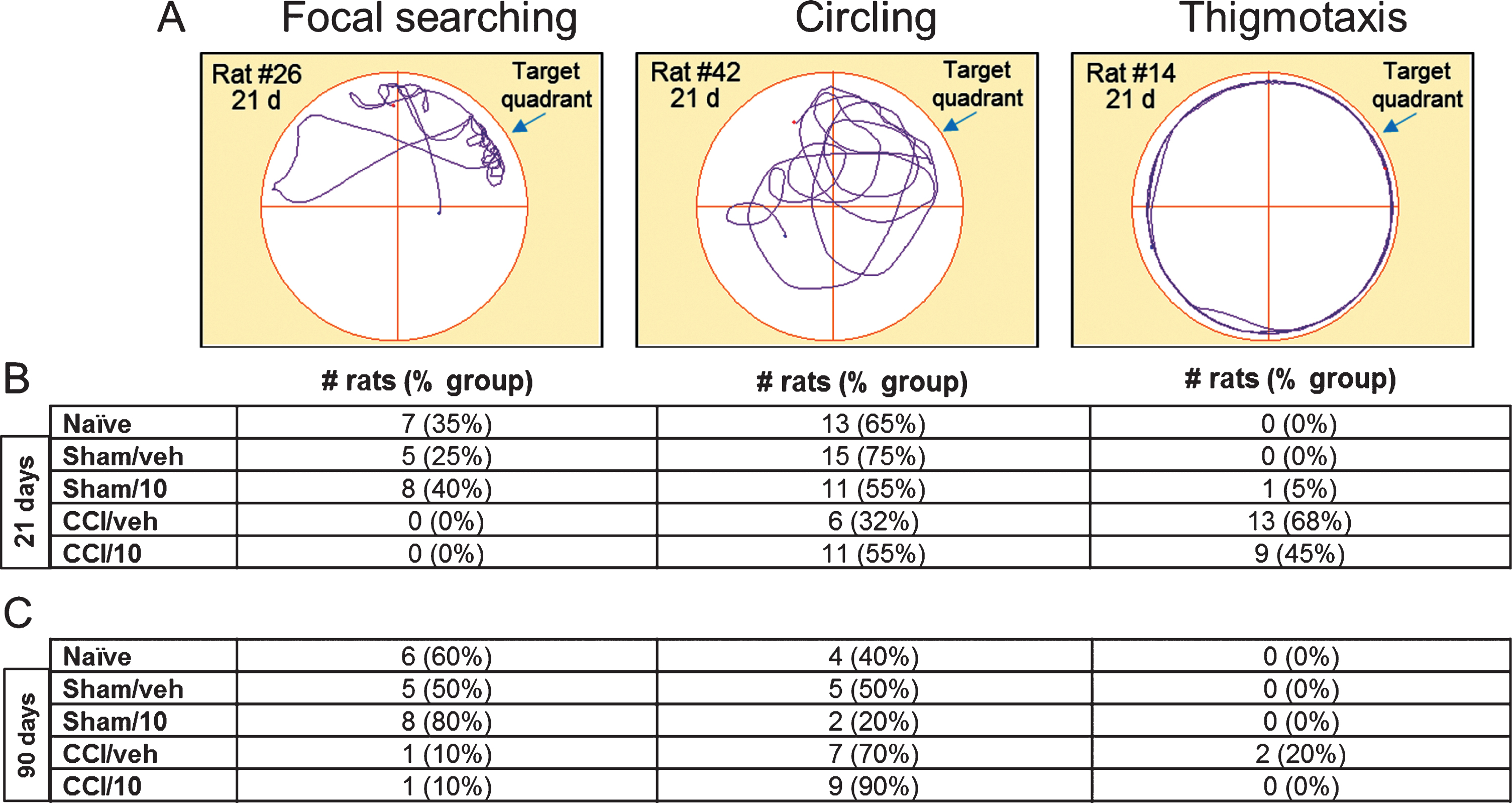
Example traces illustrating the three types of search strategies exhibited by rats in the Morris water maze probe trial (A) including focal searching, circling, and thigmotaxis. Numbers of rats in each experimental group exhibiting each search strategy are listed below the example traces, for rats tested at 21 d and 90 d after CCI injury or sham surgery.
After the MWM probe trial 21 days after surgery, half of the rats (10) in each group were sacrificed for histological assessment, while the other half survived for later testing over days 71–75 (vestibulomotor test, see Beam balance latency and Beam walk latency) and days 83–90 (MWM). When rats were assessed over days 83–87 after surgery, there were significant overall group differences [F(4, 45) = 17.82, p < 0.0001] in the MWM acquisition test: the two CCI groups continued to perform worse than the non-CCI groups. No differences were detected between the two CCI groups or the three non-CCI groups. On the MWM probe trial (90 days after CCI injury), both CCI groups spent less time in the target quadrant than the non-CCI groups [F(4, 45) = 9.29, p < 0.0001], but no differences were detected between the two CCI groups or among the three non-CCI groups. Average swim speed and total swim length were similar in all groups by one way ANOVA [F(4, 45) = 1.75, p = 0.16; F(4, 45) = 1.86, p = 0.13, respectively]. Analysis of search strategies used during the MWM spatial memory retention probe trial showed that two rats in the CCI/vehicle group (20%), but none in the CCI/10 mg/kg memantine group, engaged in thigmotaxis (Fig. 7). More rats in the sham/10 mg/kg memantine group exhibited a focal search pattern of behavior compared to sham/vehicle group (80% versus 50%; Fig. 7).
Previous studies reported improved acute outcomes (hours to days) after memantine treatment initiated immediately or within minutes after single brain injury (Kelestemur et al., 2016; Ozsuer, Gorgulu, Kiris, & Cobanoglu, 2005; Rao, Dogan, Todd, Bowen, & Dempsey, 2001; Wang, Wee, Hu, Chio, & Kuo, 2018). The current work investigates acute (1 day and 7 days), subacute (14–21 days), and chronic (71–75 and 83–90 days) effects of a single memantine injection delivered IP at a clinically relevant delay (1 hour) after moderate-severe CCI injury in adult, male rat. We demonstrate that compared to 2.5 mg/kg and 5 mg/kg doses, a 10 mg/kg memantine dose significantly reduced injury-induced neuronal degeneration and loss of synaptic protein synaptophysin in the hippocampus at acute time points after injury. However, beneficial effects of memantine did not persist out to five weeks or longer after injury. Interestingly, while the 10 mg/kg memantine dose did not attenuate memory acquisition and retention deficits (latencies) it appeared to improve search strategy.
The current intervention paradigm (1 hour after CCI injury) is clinically relevant in the context of severe TBI in humans, as it achieves therapeutically relevant plasma concentrations of drug within hours after injury, within the time period glutamate is elevated above control levels in experimental models and in human TBI patients (Bullock et al., 1998; Faden, Demediuk, Panter, & Vink, 1989; Palmer et al., 1993; Timofeev et al., 2011; Vespa et al., 1998; Yokobori et al., 2011). Using LC-MS/MS we observed that plasma concentration of memantine was 1 μM [considered therapeutically-relevant in experimental studies using rodents (Kornhuber, Weller, Schoppmeyer, & Riederer, 1994; Misztal, Frankiewicz, Parsons, & Danysz, 1996; More et al., 2008)] two hours after intraperitoneal injection of 10 mg/kg memantine. Brain memantine concentration at two hours after injection into naïve rat were ∼15-fold higher than observed in plasma, similar to what was reported in other studies following memantine administration to animals and humans (Danysz, Parsons, Kornhuber, Schmidt, & Quack, 1997; Hesselink, De Boer, Breimer, & Danysz, 1999; Misztal, Frankiewicz, Parsons, & Danysz, 1996; Wenk, Danysz, & Roice, 1996; Wesemann, Schollmeyer, & Sturm, 1982; Wesemann, Sturm, & Funfgeld, 1980). It should be noted that these data reflect total brain memantine concentration which was reported to be higher than free brain extracellular fluid concentration, the latter more closely reflecting plasma memantine concentration (Hesselink, De Boer, Breimer, & Danysz, 1999).
From a translational perspective, an acute dose study in humans reported that following a 20 mg/kg dose (per os), plasma memantine concentrations reached ∼11 ng/ml (∼50 nM) at 1 hour, 5–6 ng/ml at 2 hr, and <1 ng/ml at 6 hours after administration (Periclou, Ventura, Rao, & Abramowitz, 2006), and analysis of plasma of subjects administered 20 mg/kg/d over several weeks showed steady state levels of 70–95 ng/ml (∼0.4 to 0.5 μM), considered therapeutically-relevant (Periclou, Ventura, Rao, & Abramowitz, 2006) and close to the therapeutic range determined in rodent experimental studies. However, caution should be exercised extrapolating dosing paradigms used in the current study to species other than rodent as the elimination half-life of memantine in humans (60–80 hours) is approximately 20× that of rat and mouse (2–3 hours) (Beconi et al., 2011). While there is strong evidence that extracellular glutamate concentration increases immediately (within minutes) and peaks within hours after experimental TBI and is related to injury severity in experimental studies (Faden, Demediuk, Paner, & Vink, 1989; Nilsson, Hillered, Ponten, & Ungerstedt, 1990; Palmer et al., 1993; Rose et al., 2002), the peak extracellular glutamate concentration after a human TBI is not well-defined, due to variations in the injury to assay interval. However, in a human TBI microdialysis study Bullock (Bullock et al., 1998) reported peak glutamate concentrations at 3 hours after TBI (the earliest time point assay samples could be obtained) that slightly declined (but remained significantly above normal ISF glutamate concentrations) for at least 30+ hours to days after injury. In the same study, four patients exhibited “unexplained, massive delayed increases in glutamate 32 hours after injury, which was unassociated with ICP, CPP, or PaO2 changes” monitored in the same patients. These data, together with the reports of elevated ISF glutamate levels over the course of hours to days in experimental models and human ISF microdialysis studies (Bullock et al., 1998; Faden, Demediuk, Paner, & Vink, 1989; Palmer et al., 1993; Timofeev et al., 2011; Vespa et al., 1998; Yokobori et al., 2011) support the clinical relevance of our one-hour delay paradigm. In other words, the window of opportunity for therapies designed to counter excitotoxic levels of glutamate is variable and, in some instances, unpredictable, and is influenced by severity of TBI as well as TBI-related ischemic or hemorrhagic co-pathologies warranting systematic preclinical testing of memantine using our current 1-hour delayed, single injection paradigm. A recent study of severe TBI requiring craniectomy for management of ICP (patient n = 108) reported a median interval from injury to surgical craniectomy of approximately three hours, indicating that early intervention (within 1–2 hours) is feasible in at least some patients being treated for a TBI (Moskowitz et al., 2018). Technological advances facilitating rapid response and field assessment of injury state will likely continue to narrow this window. So, while our 1-hour delay intervention paradigm is unlikely to be applicable to all patients with severe TBI (no paradigm will be optimal for all patients being treated for a TBI), it is still clinically relevant and perhaps more so for some patients than the longer delay used in clinical trials.
In addition to reports of neuroprotective effects of memantine in preclinical studies of brain injury (Kelestemur et al., 2016; Mei et al., 2018; Ozsuer, Gorgulu, Kiris, & Cobanoglu, 2005; Rao, Dogan, Todd, Bowen, & Demspey, 2001; Wang, Wee, Hu, Chio, & Kuo, 2018), memantine was reported to be beneficial after ischemia [see (Seyedsaadat & Kallmes, 2018) for review] and subarachnoid hemorrhage (Huang, Wang, Shan, Pan, & Tsai, 2015), both of which may contribute to pathology after TBI. However, chronic effects of acute memantine therapy in the context of neuropathology and behavioral changes after moderate to severe TBI are not well understood. The spatial pattern of tissue loss and cell degeneration in the current report was consistent with other studies using CCI injury in rodents (Chen, Pickard, & Harris, 2003; Dixon, Clifton, Lighthall, Yaghmai, & Hayes, 1991; Hall et al., 2005). In our analyses of acute histopathology outcomes of CCI injury (cortical tissue spared, cell death, synaptophysin-containing synaptic terminal densities), the 10 mg/kg dose was more protective than the 2.5 mg/kg and 5 mg/kg doses, in agreement with previous studies delivering memantine minutes after a middle cerebral artery occlusion (Gorgulu et al., 2000) and immediately after CCI in rats (Rao, Dogan, Todd, Bowen, & Dempsey, 2001). The sparing of cortical tissue was greater in memantine-treated compared to vehicle-treated rats 1 day after CCI injury, but not after 7, 21, or 90 days survival. This contrasts reports of greater tissue sparing 7 days after CCI in rats treated immediately after injury with 10 or 20 mg/kg memantine compared to vehicle-treated rats (Rao, Dogan, Todd, Bowen, & Dempsey, 2001). Such discrepancy could be due to differences in the time of initiation of memantine therapy and perhaps indicates that the earlier memantine treatment is initiated, the more robust the histological protection. One limitation of the acute arm of current study is lack of insight into potential effects of memantine treatment on cerebral edema after CCI injury. Edema was detected within 2 hours, peaked at 24 hours, and largely resolved by 7 days after severe CCI injury in adult male rat (Kochanek et al., 1995), suggesting that if memantine were to influence edema, it would be most likely to achieve an effect if administered early in this time frame as in the current study. However, since non-NMDA receptors could more strongly influence glutamate-induced changes in intracellular calcium related to cytotoxic edema, as suggested in a middle cerebral artery occlusion study (Di, Alves, & Bullock, 2003) and due to the disruption of the blood brain barrier and consequent vasogenic edema, memantine’s effects on edematous changes could be limited, possibly reflecting the lack of differences in cortical cavitation at 7, 21, and 90 days after CCI injury in the current study.
At the chronic survival point after CCI injury (90 days), in addition to lack of effects on cortical tissue sparing, we no longer observed beneficial effects of memantine on synaptophysin-containing presynaptic terminal density observed at 7 days after CCI injury. These observations are difficult to interpret since they could be confounded by sprouting of inter-hippocampal axons or noradrenergic, cholinergic, and other hippocampal afferents. The contributions of acute synapse preservation and axonal sprouting to hippocampus-based behavioral tasks are therefore not straightforward and require more refined investigations of synapses in this region in the context of brain injury.
Despite the acute attenuation of histopathology, memantine had no significant effects on behavioral impairment assessed subacutely (14–21 days) and chronically (83–90 days) after CCI injury. Discordance between histological and functional outcomes is not uncommon, as observed in several instances in the study Operation Brain Trauma Therapy (Kochanek et al., 2018). In the current study, this discord could be explained by transient effects of the memantine single injection paradigm in the setting of rapidly-initiated but prolonged CCI-induced elevations of ISF glutamate known to occur in experimental and human TBI (Bullock et al., 1998; Faden, Demediuk, Panter, & Vink, 1989; Palmer et al., 1993; Timofeev et al., 2011; Vespa et al., 1998; Yokobori et al., 2011). Such a transient effect could be due to the rapid washout of the single IP memantine injection, supported by our MS/MS analyses which showed that a therapeutic plasma concentration of memantine achieved within 2 hours after a single dose of 10 mg/kg memantine administered IP to adult male rat decreased rapidly to negligible levels by 12 hours after administration. Thus, the short bioavailability of memantine warrants additional studies using repeated doses in long-term survival studies of experimental moderate-severe TBI. In the same CCI injury model, repeated daily treatment with amantadine, a weak glutamate antagonist with additional dopaminergic effects, improved MWM performance with no adverse behavioral or histological effects (Dixon et al., 1999), supporting the feasibility of a chronic, repeated administration strategy. An additional promising therapeutic strategy would be to combine memantine with other FDA-approved medications as has been proposed and explored for treatment of cognitive impairment in AD (Kishi et al., 2017) as well as in models of TBI in vitro and in vivo (Day, Carle, & Floyd, 2017; Kelestemur et al., 2016; Lamprecht & Morrison, 2015), ischemia (Babu & Ramanathan, 2009; Culmsee et al., 2004; Jung et al., 2009; Kilic, Yilmaz, Reiter, Yuksel, & Kilic, 2013; Landucci et al., 2018; Liu, Lin, Wu, & Qiu, 2009; Lorrio, Negredo, Roda, Garcia, & Lopez, 2009; Wang et al., 2015; Watanabe et al., 2010), and excitotoxicity (Chen, Lin, Liu, & Lin-Shiau, 2008). Additionally, a recent study in adult rats supported the use of memantine as a cognitive enhancer either alone or in combination with crocins, potentially nootropic chemicals derived from saffron (Pitsikas & Tarantilis, 2018). This is supported further by our observations that compared to vehicle, memantine treatment of sham controls resulted in more rats exhibiting a focal search pattern of behavior.
Subtle differences in search strategy that we observed between the CCI-injured groups during the MWM testing might be due to preservation of hippocampal synapses and neurons in memantine-treated rats, which could improve hippocampal function. These results are similar to those reported following treatment of rats with methamphetamine alone (thigmotaxis) or combined with IP injection of 5 mg/kg memantine (less thigmotaxis) (Camarasa, Rodrigo, Pubill, & Escubedo, 2010). Because less thigmotaxis that we observed in CCI-injured, memantine-treated rats could reflect memantine-induced reductions in injury-induced anxiety (Minkeviciene, Banerjee, & Tanila, 2008), future studies could use fear conditioning or open field assessments to evaluate anxiolytic effects of memantine as possible non-hippocampal mechanisms of action influencing performance on spatial memory tasks, such as alterations in amygdala function. In addition, more selective assessment of the function of cortical areas most severely affected by the CCI lesion may be required. In our CCI injury model, both acutely and chronically, the cortical lesion was most prominent in the somatosensory cortex, including the barrel fields. Thus, tests more sensitive for somatosensory function after CCI with or without memantine treatment are warranted.
Our observations of vestibulomotor function deficits in both memantine-and vehicle-treated CCI groups contrasts the findings of Lyeth and colleagues (Lyeth, Gong, Shields, Muizelaar, & Berman, 2001) who reported that deficits in beam walk latency after TBI were ameliorated by administration of the group 1 metabotropic glutamate antagonist (RS)-1-aminoindan-1,5-dicarboxylic acid. Differences in injury model (CCI versus fluid percussion), and drug mechanism of action as well as route of drug administration (IP versus parenchymal injection) might account for this discrepancy. Alternatively, lack of improvement on vestibulomotor function after memantine administration observed in the current study could be due to involvement of primary motor cortex in the cortical lesion (irreparable tissue loss due to mechanical destruction and acute neuronal degeneration occurring prior to drug administration). Notably, sham-operated rats showed subtle deficits on the beam walk test in the group treated with vehicle, but not in sham-operated rats treated with memantine despite similar levels of moderate tissue damage in the cortex due to effects of craniotomy.
A limitation of current and previous investigations of effects of memantine on outcome after experimental TBI is the lack of direct measurements of glutamate, its receptors, or glutamate-related second messenger pathways. Evaluation of glutamate-induced excitotoxicity requires investigations of complex interactions between excitatory (NMDA, AMPA) and inhibitory (GABA) receptor systems as well as the intracellular Ca2+ buffering system of the cells that could be probed in future mechanistic studies. Alternatively, [I-125]MK801 MRI and/or autoradiography could provide insight into the effects of memantine treatment on the functional status of the NMDA channel in relation to glutamate activity (Di, Alves, & Bullock, 2003; Di & Bullock, 1996).
In summary, this study reports that IP administration of a single 10 mg/kg dose of memantine 1 hour after severe TBI protects acutely against cell degeneration and synapse loss in the hippocampus but does not resolve subacute and chronic cognitive deficits assessed using the MWM test. Repeated doses of memantine over long-term recovery periods will be required to determine the full potential of this drug.
Footnotes
Acknowledgments
This work was supported by a Merit Review Award I01 RX000511 from the United States Department of Veterans Affairs, Rehabilitation Research and Development Service, and research grant NAM-MD-45 from Forest Research Institute. We gratefully acknowledge the contributions of Lan Shao, Michelle Ma, Sherman Culver, and Tricia M. Miller. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.