
Abstract
To demonstrate the role of Bax in death receptor-induced apoptosis in the human colon cancer HCT116 cells. We treated HCT116 cells and HCT116 with p53−/− (KO) by 0.1 μg/mL TRAIL for 24 hours, which indicated that HCT116 parental cells are sensitive to p53-independent death receptor-induced apoptosis. Although the p53 signaling pathway is totally intact in this system, the down-regulation of Bax in HCT116 cells is dramatically resistant to TRAIL and failed to undergo apoptosis. However, the over-expression of Bax can rescue the sensitivity of apoptosis induced by the death receptor. Our study has revealed an essential role for Bax in death receptor-induced apoptosis in the human colon cancer HCT116 cells. It may aid in a molecular understanding of possible defects in signal transduction and a regulation of the death receptor-induced apoptotic process.
Introduction
Apoptosis is executed by caspases, which, based on their roles in apoptotic pathways, can be divided into two general groups. The intrinsic pathway mediates apoptotic responses to various stress signals such as DNA damage, hypoxia, and growth factor deprivation. It is generally thought that these signals eventually lead to the activation of pro-apoptotic members of the Bcl-2 family (e.g., Bax and Bak). 1 –3 The extrinsic pathway is initiated by the interaction of death ligands with their corresponding death receptors, including Fas, tumor necrosis factor receptor-1, TRAIL R1, and TRAIL R2. In certain cell types, death receptor-induced apoptosis requires the amplification of death signals via a mitochondrial pathway that is controlled by Bcl-2 family proteins. 4 –7 Within this pathway, caspase-8 cleaves Bid, a member of the proapoptotic Bcl-2 family of proteins. The resulting truncated Bid translocates from the cytoplasm to mitochondria, where it promotes the release of cytochrome c and other apoptotic proteins. 8,9
Colon cancer is a common malignancy in the gastrointestinal tract. Some scientists have reported that Bax deficiency causes cancer cells to be very resistant to Apo2L/TRAIL. 10 –12 Here, we tested the requirement of either p53 or BAX in death receptor-TRAIL-induced apoptosis of colon cancer cells. We found that the loss of Bax but not p53 is resistant to TRAIL-induced apoptosis. It may aid in a molecular understanding of possible defects in signal transduction and also a regulation of the death receptor-induced apoptotic process.
Materials and Methods
Cell culture
Human colon cancer cell lines HCT116 and various cell lines derived from HCT116, including HCT116 with Bax−/− (KO), HCT116 with p53−/− (KO) from Bert Vogelstein's Lab, were grown in McCoy's 5A supplemented with 10% fetal bovine serum (FBS) and 1% Penicillin-Streptomycin. The growth media, FBS, and antibiotics were obtained from Invitrogen. Both cells were cultured at 37°C in a 5% CO2 humidified incubator.
Retroviral production and infection
Retrovirus-mediated gene transfer was performed as previously described. 13,14 Briefly, 48 hours after transfection, the retrovirus-containing medium was filtered through a 0.45 μm filter (Millipore) and supplemented with 4 μg/mL Polybrene (Sigma). For retroviral infections, cells were seeded in a 10-cm dish and incubated overnight. The cultured medium was then replaced by the retrovirus-containing medium. After 48 hours, the viral supernatant was removed, cells were cultured in the growth medium containing either 1.0 μg/mL puromycin or 100 μg/mL hygromycin B for 3 days, and drug-resistant cells were pooled. The percentage of retrovirus-infected cells ranged between 80% and 90%, as estimated in parallel infections using the retrovirus-expressing enhanced green fluorescent protein. Analysis over-expression of Bax was verified by immunoblotting.
Immunoblotting
Cells were suspended in a standard sodium dodecyl sulfate sample buffer. Protein concentrations were determined with a Bio-Rad protein assay kit, using bovine serum albumin as a reference. 50 μg of protein was separated on 10% sodium dodecyl sulfatepolyacrylamide gels, transferred to nitrocellulose membranes, and probed with monoclonal antibodies against either Bax (B3428; Sigma), α-tubulin (B-5-1-2; Sigma), or polyclonal rabbit anti-p53 (FL-393; Santa Cruz). Horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit IgG (ICN) was used as the secondary antibody. Proteins were visualized with a SuperSignal West Pico chemiluminescence kit (Pierce).
Apoptosis induction and analysis
Exponentially growing cells at 70%–80% confluence were either untreated or treated with 0.1 μg/mL of TRAIL (Calbiochem). After treatment, adherent and floating cells were pooled, collected by centrifugation, and washed once with ice-cold phosphate-buffered saline (PBS). Apoptotic cell death was determined by staining of the collected cells with Annexin-V and 7-aminoactinomycin D, using a Guava cytometer according to the manufacturer's protocol (Guava).
Statistical analysis
Quantitative data are expressed as the mean±SD. The two-tailed Student's t-test was performed for paired samples using the data analysis tools provided by the software. p<0.05 was considered statistically significant.
Results
Death receptor sensitizes HCT116 cells to apoptosis
As shown in Figure 1B and C, HCT116 cells were highly sensitive to death receptor TRAIL, and more than half of the cell populations lost their viability within 24 hours of treatment. The apoptotic cell death was confirmed by an Annexin-V binding assay in which Annexin-V binds to externalized phosphatidylserine on the surface of apoptotic cells. The parental control cells were markedly sensitive to TRAIL-induced apoptosis. Most of the cells exhibited morphological changes that are characteristic of apoptosis, such as cell shrinkage within 24 hours of treatment with 0.1 μg/mL TRAIL (data not shown).
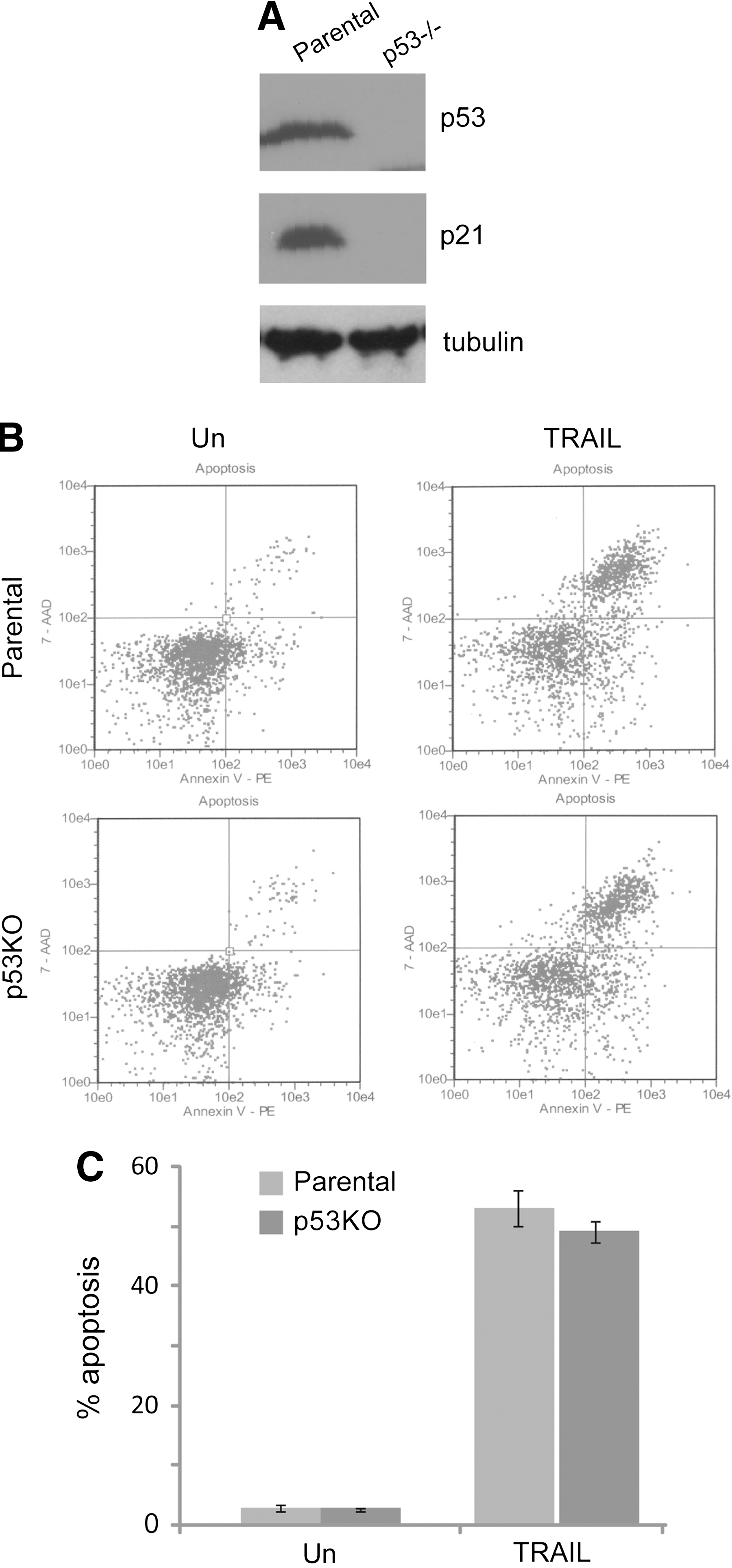
p53 is not required for death receptor-mediated apoptosis in HCT116 cells.
p53 signaling pathway is intact in HCT116 cells
We examined the functional status of p53 in HCT116 cells. Wild-type p53 protein has a short half life. 15,16 The response to DNA damage or other forms of genotoxic stress, p53 is stabilized, leading to a rapid increase in the protein level and its activity. Activated p53, in turn, induces the expression of its target genes, such as p21Waf1/Cip1 and MDM2. 17 –19 As shown in Figure 1A HCT116 expressed wild-type p53 and p21 (Fig. 1A); the result indicated that the p53 signaling pathway is functional in HCT116 cells.
p53 is not required for death receptor-mediated apoptosis in HCT116 cells
HCT116 cells have functional p53 pathways and provide a suitable system for examining the effect of p53 on death receptor-triggered apoptosis. Given that p53 is functional in HCT116 cells and the well-known role that p53 plays in regulating apoptosis, we examined the contribution of p53 to death receptor-induced apoptosis in HCT116 cells by abrogating endogenous p53 activity (Fig. 1A right lane). HCT116 with p53−/− (KO) still displayed remarkable increase in their sensitivity to TRAIL (Fig. 1B right bottom panel). p53 knockout had no effect on the sensitivity of HCT116 cells to the death receptor TRAIL (Fig. 1B, C). Those results showed that TRAIL induces apoptosis in HCT116 cells through a p53-independent pathway.
Bax is essential for death receptor-mediated apoptosis in HCT116 cells
Next, we defined the signaling pathway through which TRAIL sensitizes HCT116 cells to death receptor-induced apoptosis. Compared with HCT116 parental cells, Bax knockout cells were dramatically resistant to TRAIL and failed to undergo apoptosis after TRAIL treatment (Fig. 2B, C), Consistent with their TRAIL-sensitive phenotype, HCT116 parental cells showed higher levels of Bax (Fig. 2A). On the basis of these findings, we concluded that the function of Bax is essential for death receptor-induced apoptosis in HCT116 cells.

Bax is essential for death receptor-mediated apoptosis in HCT116 cells.
Rescue Bax can induce HCT116 death receptor-mediated apoptosis
To further investigate the role of Bax in TRAIL-induced apoptosis in HCT116 cells, retrovirus-mediated gene transfer was used to introduce Bax into Bax knockout cells. Compared with the parental HCT116 cells, a high level of Bax was observed in Bax knockout cells with an over-expression of Bax (Fig. 3A). As expected, Bax−/− (KO) cells with over-expression of Bax can rescue the sensitivity to TRAIL-induced apoptosis (Fig. 3B right column), which is equal to the sensitivity of the HCT116 parental cells with a similar level of Bax. However, HCT116 with Bax−/− (KO) cells stilled remained resistant to TRAIL (Fig. 3B).
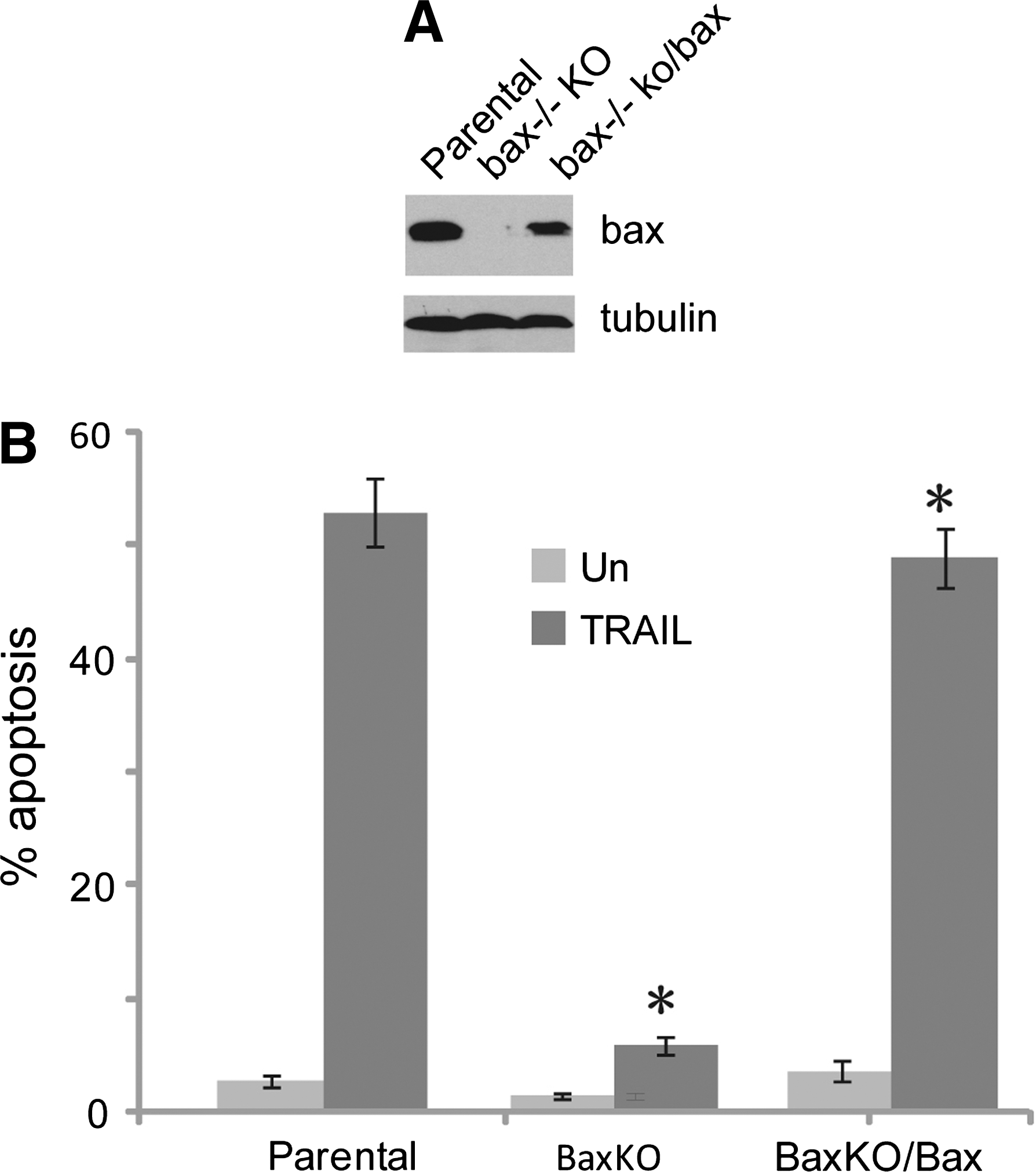
Rescue Bax-induced HCT116 death receptor-mediated apoptosis.
Discussion
Our findings demonstrated an essential role for Bax in death receptor-induced apoptosis in the human colon cancer HCT116 cells. Bax knockout cells were dramatically resistant to TRAIL and failed to undergo apoptosis. Over expression of Bax in Bax−/− knockout cells to rescue Bax expression level can immediately induce TRAIL-mediated apoptosis. The data showed that the function of Bax is required for death receptor-induced apoptosis in HCT116 cells.
Death receptors induce rapid apoptosis in HCT116 cells through p53-independent pathways, althoughHCT116 parental cells usually carry wild-type p53. A number of studies also revealed that the p53 signaling pathway is functional in various human colon cell lines. 20 –22 Knockout of p53 still showed a marked increase in their sensitivity to TRAIL-induced apoptosis in HCT116 cells. It means that p53 is not required in this apoptotic process. All these data suggested a possible role of mitochondria in mediating TRAIL-induced apoptosis in HCT116 cells.
Bax is required for apoptosis that is induced either by DNA damage drug or by growth factor deprivation. Bax plays a crucial role in cross-talk between apoptosis pathways across mitochondria and cell death receptors. In human colon cancer cells, Bax deficiency have provided a direct evidence that Bax plays a key role in mediating the apoptosis induced by certain anti-cancer agents. 23 Although Bax and Bcl-2 have in vivo competition, Bax can regulate apoptosis independently, which is not affected by Bcl-2. 24,25 In HBV or HBx transfection of a hepatoma cell line, apoptosis induced by TRAIL is related with Bax protein expression, 26 andBax has no effect on TRAIL-induced caspase-8 activation and subsequent cleavage of Bid. 27 However, it depends on the release of Smac/DIABLO to mediate the mitochondrial pathway to death receptor-mediated apoptosis. 28
TRAIL is a promising agent for cancer therapy because of its selective cytotoxicity to human cancer cell lines in vitro and to tumor xenografts in immunodeficient mice. 29 –31 However, the molecular basis for this differential sensitivity is poorly understood. It was initially suggested that the presence of TRAIL decoy receptors in normal, but not cancer, cells might account for the selectivity. 32 –34 Our findings may aid in a molecular understanding of possible defects in signal transduction and regulation of this apoptotic process.
Footnotes
Acknowledgments
The authors are grateful to Dr. Han-fei Ding for his invaluable support. This study was supported by the National Basic Research Program of China (No. 2012cb114600), and the National Natural Science Foundation of China (Nos. 31172268 and 31271462).
Disclosure Statement
The authors are not employed by any commercial companies that may influence the study performed here.