
Abstract
Radioimmunotherapy capitalizes on the radiosensitivity of non-Hodgkin lymphoma (NHL) and the targeted nature of monoclonal antibodies. In an attempt to reverse bone marrow infiltration with B-cells and optimize the biodistribution of Yttrium-90 (90Y)–ibritumomab tiuxetan, we conducted a phase I study combining a single course of 90Y-ibritumomab tiuxetan after a 4-weekly course of rituximab in relapsed or refractory low-grade or transformed CD20+ B-cell NHLs with <25% marrow involvement. The 0.4 mCi/kg dose was associated with 80% grade-4 cytopenias. Dose escalation was held, and 6 patients were enrolled at a 0.3 mCi/kg cohort. As the 0.3 mCi/kg dose was well tolerated, the 0.4 mCi/kg cohort was expanded to 6 additional patients. In the expansion cohort, grade-4 cytopenia developed in 33%. Further dose escalation was held, and the maximum tolerated dose was determined at 0.4 mCi/kg. With this regimen, marrow involvement decreased in all patients with complete clearance in 50%. The overall response rate was 82%. With a median follow-up of 31.7 months, the median progression-free survival and time to next treatment were 12.3 and 10.9 months, respectively. Although this regimen was associated with a high response rate, the hematologic toxicity was higher than with the standard 90Y-ibritumomab tiuxetan regimen.
Introduction
Non-Hodgkin lymphomas (NHLs) constitute a heterogeneous group of lymphoid malignancies, predominantly of the B-cell phenotype. 1,2 Their clinical course is variable ranging from aggressive, rapidly advancing malignancies requiring immediate intervention to more indolent tumors where treatment can be safely postponed until symptoms develop. Follicular lymphoma constitutes the most common indolent B-cell NHL, and although its prognosis may be more favorable compared to its aggressive counterparts, it is still incurable with the current treatment modalities. 2 Indolent NHLs tend to present at advanced stage, 1 and despite the high response rates and long-term remissions that can be achieved with conventional chemotherapy, their clinical course is characterized by relapses with shorter remission intervals with successive lines of therapy. Moreover, indolent B-cell lymphoproliferative disorders can undergo histologic transformation, that is, evolution into a high-grade malignancy with a rapidly progressive clinical course associated with a significant negative impact on overall prognosis. 3
An important advance in the therapeutics of indolent B-cell NHL has been the introduction and incorporation of targeted immunotherapy in the treatment regimens. The first in-class immunotherapy was rituximab, 4,5 a chimeric monoclonal antibody comprised of a murine variable region that recognizes the cluster-of-differentiation 20 (CD20) phosphoprotein expressed on the surface of B-cell lymphomas and a human IgG1-kappa constant region that binds the complement. In an attempt to improve the responses obtained with anti-CD20 immunotherapy, several approaches have been undertaken, 6 including the combination of rituximab with interleukin-2 7 or interleukin-12 8 to enhance antibody-dependent cell-mediated cytotoxicity, use of higher doses of rituximab, 9 development of novel antibodies that recognize different epitopes on CD20, and conjugation of anti-CD20 monoclonal antibodies with interferon-α. 10 A particularly attractive approach has been the development of radioimmunotherapy, that is, the conjugation of monoclonal antibodies with radionuclides, a strategy that capitalizes on the radiosensitivity of lymphoma cells and the inherently targeted nature of monoclonal antibodies. Unlike external beam radiotherapy where tumor irradiation is fractionated and intermittent, radiolabeled antibodies exert their tumoricidal effect by targeted emission of continuous, exponentially decreasing, low-dose-rate radiation. 11 Moreover, the targeted emission of penetrating radiation has an effect on antigen-negative cells of the tumor microenvironment and may obviate the problem of limited antibody access in bulky or poorly vascularized tumors.
Two radiolabeled murine anti-CD20 monoclonal antibodies, 131Iodine (131I)–tositumomab and 90Yttrium (90Y)–ibritumomab tiuxetan, are currently licensed by the U.S. Food and Drug Administration (FDA), and both have been shown to induce high response rates and durable remissions in relapsed or refractory low-grade, follicular, and transformed low-grade NHLs. 12 –16
90Y-ibritumomab tiuxetan is a radiolabeled monoclonal antibody composed of ibritumomab, the parent murine anti-CD20 monoclonal antibody engineered to form rituximab; and tiuxetan, a linker covalently attached to ibritumomab that allows for the chelation of Indium-111 (111In) or 90Y for dosimetric and therapeutic purposes, respectively. 90Y is a pure β- (electron) emitting radioisotope with a higher maximum energy and range (2.28 MeV and 12 mm, respectively) compared to 131I (0.61 MeV and 2 mm, respectively), but a shorter half-life (T1/2) (64 hours vs. 8 days for 90Y and 131I, respectively). 17 In clinical studies, rituximab (or unconjugated ibritumomab 18 ) was administered before each dose of ibritumomab tiuxetan to bind nonspecific CD20 antigenic sites and to deplete normal blood CD20+ lymphocytes, thereby optimizing the biodistribution of the radioimmunoconjugate. 6,13 The most significant adverse event associated with 90Y-ibritumomab tiuxetan is reversible myelosuppression, which has been shown to correlate with the degree of bone marrow involvement. 16
We hypothesized that a 4-weekly course of rituximab preceding radioimmunotherapy will reverse bone marrow infiltration with malignant cells and eliminate nonspecific CD20-binding sites, thereby optimizing the biodistribution of 90Y-ibritumomab tiuxetan and allowing higher doses of radioimmunotherapy to be administered. At the pharmacokinetic level, the clearance of bone marrow by lymphoma cells as well as other CD20+ cells will eliminate localization of the radioimmunoconjugate in the bone marrow, and in doing so, decrease the associated myelosuppression. The elimination of nonspecific CD20-binding sites should optimize the tumor dosimetry of 90Y-ibritumomab tiuxetan and hence enhance its efficacy. At the pharmacodynamic level, radioimmunotherapy will eliminate any residual disease after a 4-weekly course of rituximab, which may be particularly relevant in cases of bulky disease.
To test the above hypothesis, we undertook a phase I, multicenter, open-label, dose–escalation study combining a 4-weekly course of rituximab followed by a course of radioimmunotherapy with 90Y-ibritumomab tiuxetan. The primary objectives of the study were to characterize the toxicities and establish the maximum tolerated dose (MTD) of 90Y-ibritumomab tiuxetan after 4-weekly doses of rituximab in patients with relapsed or refractory low-grade or follicular or transformed CD20+ B-cell NHL. Secondary objectives of the study were to determine the frequency of reversal of malignant bone marrow involvement with rituximab and the antitumor efficacy of this investigational approach. This trial has been registered at
Methods
Patient selection and eligibility criteria
Eligible patients were required to have histologically confirmed, relapsed or refractory low-grade or follicular or transformed B-cell NHL with a demonstrable monoclonal CD20+ B-cell population in the lymph nodes or bone marrow. All patients required treatment as determined by increasing overall tumor size, development or presence of B symptoms, and/or development or presence of masses that cause clinical symptoms. Eligible patients had failed at least one prior chemotherapy regimen without restriction to the number of prior regimens. Patients previously treated with rituximab were eligible, but when the protocol was amended, they must have achieved an objective response to rituximab with a time to progression of more than 6 months. To meet the eligibility criteria, patients had to be at least 19 years old, not pregnant, nor lactating, and, regarding patients with reproductive potential, to follow accepted birth control methods. The baseline performance status had to be ≤2 according to the World Health Organization scale, and the expected survival had to be at least 6 months.
Within 6 weeks before initial treatment and on day 29 of the treatment, a bilateral bone marrow aspirate and biopsy were required. Within 4 weeks before initial treatment, a medical evaluation, including assessment of the performance status, was conducted. Within 2 weeks before initial treatment, the following were required: peripheral blood flow cytometry, absolute neutrophil count ≥1500/mm3, platelet count ≥100,000/mm3, hemoglobin ≥9 g/dL, bilirubin ≤1.5 mg/dL, and creatinine ≤1.5 mg/dL. Patients must not have received granulocyte colony-stimulating factor or granulocyte–macrophage-colony stimulating factor within 3 weeks preceding initial treatment. In addition, all prior chemotherapy was required to have been completed at least 3 weeks (6 weeks regarding rituximab, nitrosourea, fludarabine, or mitomycin C) before initial treatment.
Patients with bone marrow involvement by lymphoma ≥25% were excluded for safety reasons. Also, patients with impaired bone marrow reserve, as indicated by prior myeloablative therapies with stem cell rescue, hypocellular bone marrow (≤15% cellularity expected for the age of the patient), history of failed stem cell collection or external beam radiation therapy to >25% of active bone marrow, and marked reduction in bone marrow precursors of one or more cell lines, were excluded. Patients with central nervous system lymphoma, chronic lymphocytic leukemia, human immunodeficiency virus or acquired immunodeficiency syndrome-related lymphoma, serious nonmalignant conditions that, in the opinion of the investigators would compromise the protocol objectives, were excluded. Prior radioimmunotherapy and presence of anti-murine or anti-chimeric antibodies, as assessed in a central laboratory, were also exclusion criteria.
There was no limitation on bulky disease or the number of prior disease relapses. The study was approved by the institutional review boards of the participating sites, and written informed consent was obtained from all patients. The full protocol for the study is available in the Supplementary Appendix at
Study design and objectives
This was a multicenter (UAB, Stanford, and Mayo Clinic), dose–escalation, open-label study to determine MTD, safety, and efficacy of a single course of 90Y-ibritumomab tiuxetan after 4-weekly doses of rituximab in relapsed or refractory low-grade or follicular or transformed CD20+ B-cell NHL. A course of therapy included 4-weekly infusions of rituximab (first dose 250 mg/m2 plus 5 mCi of 111In-ibritumomab tiuxetan followed by 3-weekly doses of rituximab at 375 mg/m2) followed 2 weeks later (day 36) by rituximab 250 mg/m2 plus 5 mCi of 111In-ibritumomab tiuxetan (Fig. 1). The dose estimates to normal organs were determined by a gamma-camera on days 1 and 36 with images obtained at 0 and 4–6 hours and on days 1, 3, and 6 after infusion of 111In-ibritumomab tiuxetan. If dose estimates were in the safe, defined limits, the patients received on day 43 250 mg/m2 of rituximab plus the programmed dose of 90Y-ibritumomab tiuxetan for treatment. The latter was determined based on the results of bilateral bone marrow biopsies conducted on day 29. If there was <25% marrow involvement by lymphoma, and the marrow cellularity was >50% of the normal expected for the patient's age, and the platelet count was ≥150,000 per microliter, the patient would receive the dose of 90Y-ibritumomab tiuxetan assigned in one of the dose–escalation cohorts (0.4, 0.5, 0.6, and 0.7 mCi/kg; maximum dose 32, 40, 48, and 56 mCi, respectively). If the biopsies were clear or consistent with the NHL infiltration (<25%), and the platelet count was between 100,000 and 150,000 per microliter, the patient would receive 0.3 mCi/kg of 90Y-ibritumomab tiuxetan (Fig. 1).
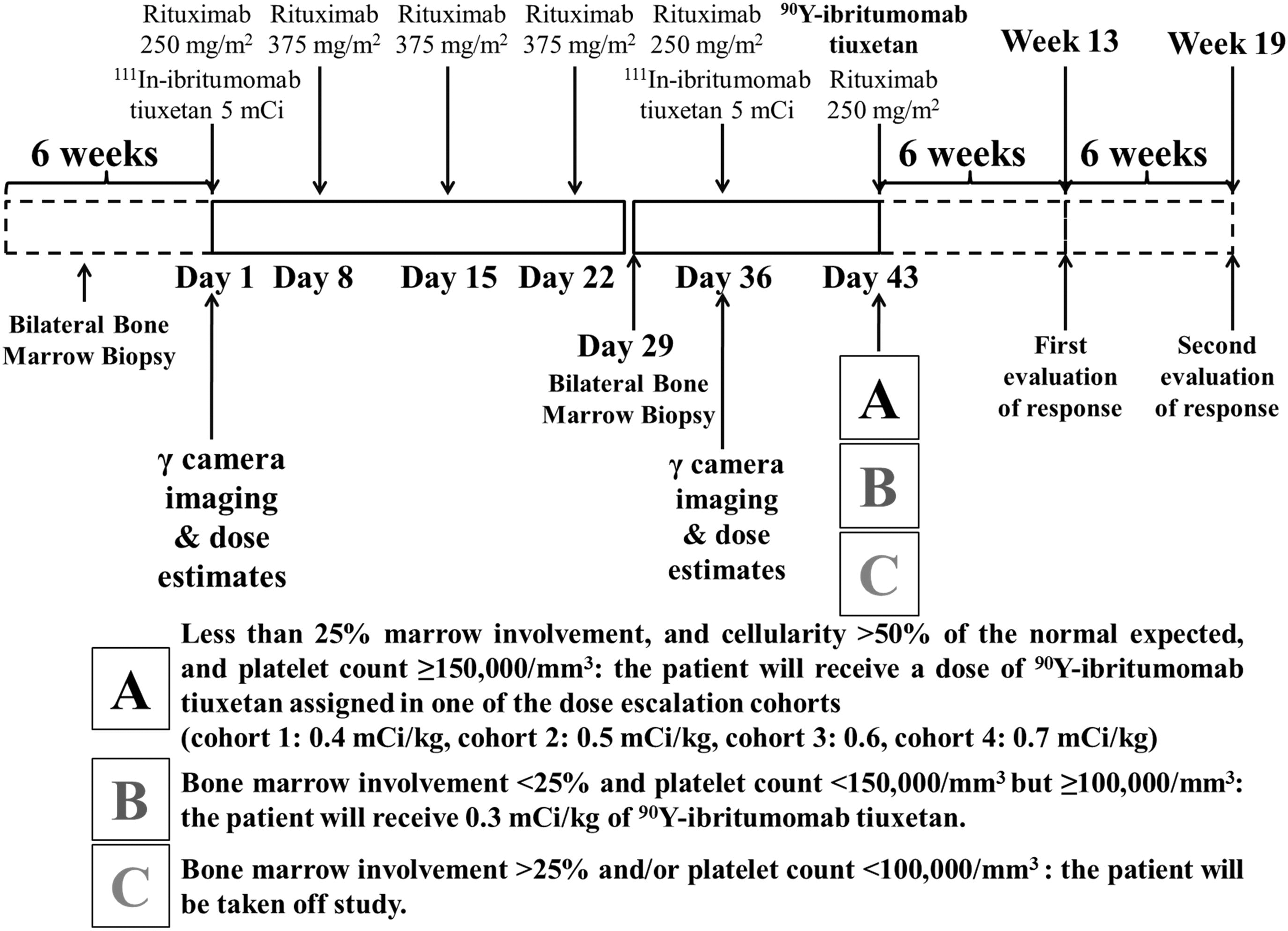
Study schema and dose determination. A course of therapy included 4-weekly infusions of rituximab, followed 2 weeks later by rituximab 250 mg/m2 plus 5 mCi of 111In-ibritumomab tiuxetan. Gamma-camera imaging for dose estimates was conducted on days 1 and 36. If dose estimates were in the safe defined limits, the patients received on day 43 250 mg/m2 of rituximab plus a dose of 90Y-ibritumomab tiuxetan, determined by the results of bilateral bone marrow biopsies conducted on day 29.
The primary objectives of the study were to characterize the toxicities and establish the MTD of escalating doses of 90Y-ibritumomab tiuxetan after 4-weekly doses of rituximab in patients with relapsed or refractory low-grade or follicular or transformed CD20+ B-cell NHL and <25% NHL marrow infiltration. The study also intended to establish the dosimetry and biodistribution of 111In-ibritumomab tiuxetan in the tumor sites, whole body, and normal organs before and after 4-weekly doses of rituximab. Secondary objectives of the study were to determine the frequency of reversal of bone marrow involvement with NHL after 4-weekly doses of rituximab and the antitumor efficacy of this investigational approach.
The treatment period included the time from the first rituximab infusion to 12 weeks after the 90Y-ibritumomab tiuxetan administration (totally, 19 weeks). History and physical examination were performed on days 29 and 43 and then every 6 weeks for 3 months, every 3 months for 2 years, and every 6 months for years 3 and 4, unless relapse occurred.
Dosimetry
Imaging and dosimetry were conducted at the clinical sites (methods published elsewhere). 19 Treatment with 90Y-ibritumomab tiuxetan was held if the predicted delivered dose of radiation exceeded 20 Gy to any nontumor organ or 3 Gy to the bone marrow.
Response assessment
Baseline imaging studies were conducted within 4 weeks before patient registration. The first and second evaluations of the response by means of computed tomography and/or magnetic resonance imaging were conducted 6 and 12 weeks after the administration of 90Y-ibritumomab tiuxetan, respectively.
Responses and efficacy endpoints are reported according to the Revised Response Criteria for Malignant Lymphoma (2007). 20 Complete responses need not be confirmed with a positron-emission tomography (PET), because when the protocol was written, the contemporary at that time International Workshop Response Criteria 21 were used, and the response rate was not the primary endpoint of the study.
Safety
Complete blood counts and differential, adverse events, and concomitant medications were documented weekly throughout the treatment period and thereafter on an ongoing basis. The use of prophylactic growth factors was not allowed, but therapeutic administration of growth factors and/or transfusions to treat cytopenias were permitted at the investigator's discretion. The anti-murine and anti-chimeric antibodies were assessed in a central laboratory before enrollment, 6 and 12 weeks after the administration of 90Y-ibritumomab tiuxetan. Adverse events were recorded using the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE), version 2.0. 22 The MTD was defined as the dose at which at least 50% of patients had grade 3 or greater, but no more than 50% of patients had grade 4 neutropenia and thrombocytopenia.
Statistical methods
This was a phase I study that followed a traditional dose–escalation design with a cohort expansion phase. Four cohorts with escalating doses of 90Y-ibritumomab tiuxetan were planned with at least 5 evaluable patients in each cohort. The protocol allowed for additional patients to be enrolled to account for those who could not receive the assigned dose of 90Y-ibritumomab tiuxetan secondary to bone marrow involvement by lymphoma on day 29. Descriptive statistics were used for the primary endpoint. Estimates of survival were determined using the Kaplan–Meier estimation method, and groups were compared using the log-rank test. SPSS statistical software, version 20., was used for all calculations. 23
Results
Patients
The study enrolled a total of 17 patients: 5 patients were enrolled to the first cohort (0.4 mCi/kg), 6 to cohort-1 (0.3 mCi/kg), and 6 to the 0.4 mCi/kg expansion cohort. Patient characteristics are shown in Table 1. All patients had a performance status of 0 or 1. The median age of the patients was 56 years (mean, 56.2; range 39–75). At the time of study, enrollment 6 patients (35%) had transformed follicular lymphoma, whereas 10 patients (59%) had grade I/II follicular lymphoma. Most patients (76%) had advanced-stage (III or IV) disease at enrollment, and of the 4 patients with stage II disease, 1 had grade III, and 2 had transformed follicular lymphoma. The median number of prior treatments was at least one (range, 1–7), and all patients had received rituximab before study enrollment, except one. An anthracycline-based regimen constituted the most frequent prior treatment with at least 13 patients having received cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) previously. The median time from original diagnosis to initiation of the investigational therapy was 41.5 months (mean, 49.2; range, 10.4–145.3). For 15 patients with the available date of last treatment, the median interval between completion of last treatment and initiation of the investigational therapy was 20.9 months (mean, 22.7; range, 1.2–93.1).
FLIPI (Follicular Lymphoma International Prognostic Index 31 ) was assessed at the time of study enrollment.
1 patient had cutaneous involvement.
Data for 2 patients were missing.
Primary objectives
Safety and toxicity
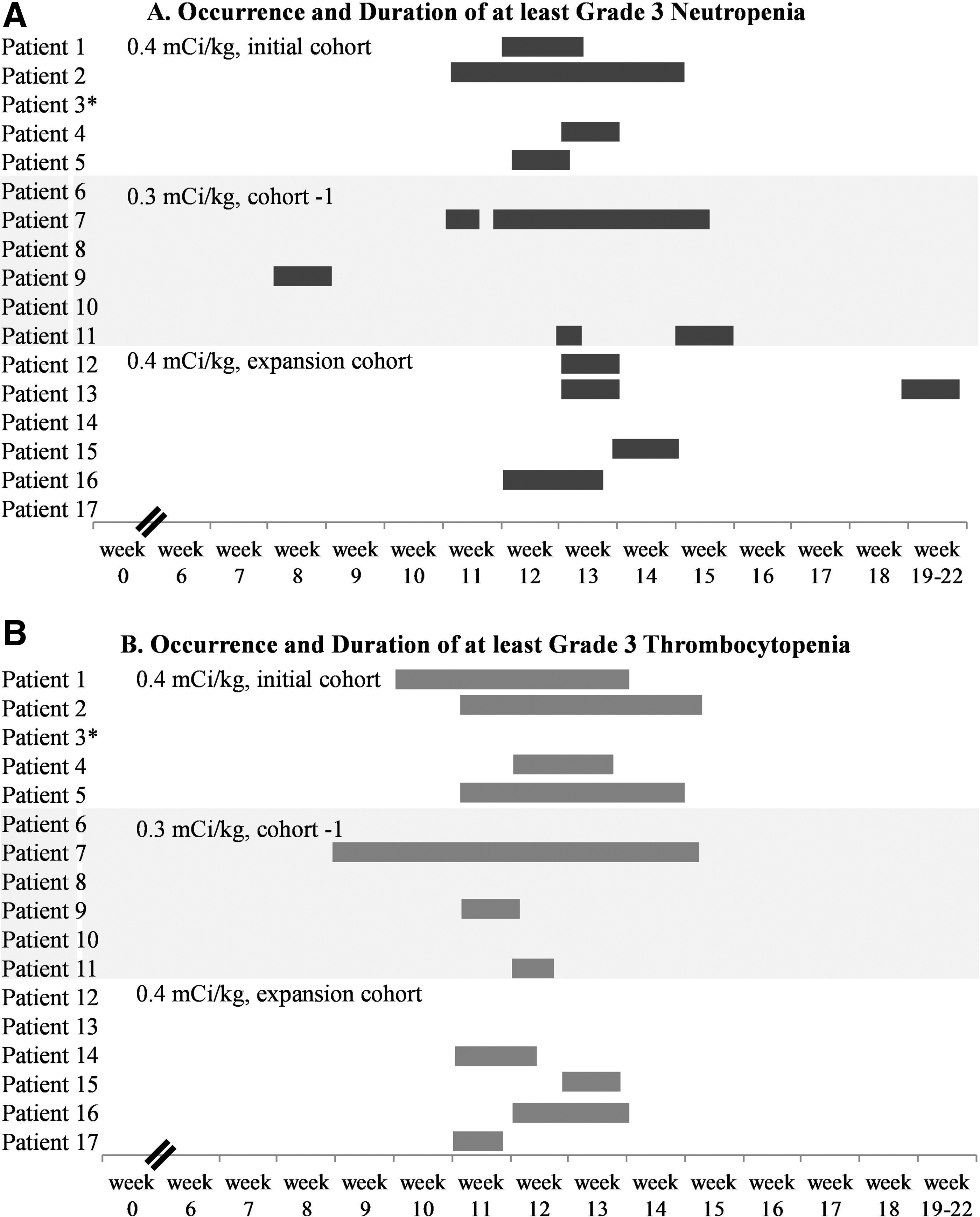
Cohort 1 (90Y-ibritumomab tiuxetan, 0.4 mCi/kg) enrolled 5 patients. Dose–escalation was held after the significant hematologic toxicities observed in those patients: 1 patient developed grade-4 and 3 developed grade-3 reversible thrombocytopenia; 4 patients developed grade-4 reversible leukopenia and neutropenia; and 1 patient developed grade-4 febrile neutropenia (Fig. 2). One patient in this cohort did not receive the study drug due to the toxicity observed in the previous patients, but was included in the dosimetry and antitumor efficacy studies; this patient experienced only reversible grade-2 neutropenia with rituximab infusions. No nonhematologic toxicities were observed, and no toxicities greater than grade 2 were observed with rituximab. All 4 patients who received the full course of radioimmunotherapy received growth factors at the nadir, and 2 patients received transfusions.
Dosimetry data from the first cohort showed no differences in the radiation-absorbed dose for the whole body, liver, and left kidney before and after a 4-weekly course of rituximab, except for the spleen, in which a reduction was observed in all patients (before rituximab: mean 55.2 rads/mCi, range 34–77; after rituximab: mean 40, range 29–47; p: 0.06, Wilcoxon signed-rank test). The radiation-absorbed dose to the bone marrow calculated from blood radioactivity, increased in 3 patients from 1.53 (mean; range, 1.2–1.9) to 5.37 rads/mCi (mean; range, 2.9–8.8), did not change in 1 patient with low tumor burden, and decreased in 1 patient with rituximab-refractory disease, high tumor burden, and rapid clinical progression. The dosimetry results for the entire study have been reported. 19
In consultation with the FDA, dose–escalation was held, and the protocol was amended to allow enrollment of patients at a lower dose level (0.3 mCi/kg) and exclude patients with rituximab-resistant disease (no response to rituximab or relapse within 6 months). Six patients were enrolled at a lower dose level (cohort −1, 0.3 mCi/kg): 1 patient experienced grade-4 neutropenia, whereas grade-3 leukopenia, neutropenia, and thrombocytopenia were seen in 2, 3, and 3 patients, respectively (Fig. 2). Four patients received growth factors at their nadir, and 1 patient received transfusions (of both platelets and red blood cells). Possibly related non-neutropenic cellulitis and sinusitis occurred in 1 patient each and resolved with antibiotics.
As the 0.3 mCi/kg dose was well tolerated, the 0.4 mCi/kg cohort was expanded to enroll 6 additional patients. In the expansion cohort, grade-4 leukopenia and neutropenia were seen in 1 and 2 patients, respectively. Grade-3 leukopenia, neutropenia, and thrombocytopenia were seen in 3, 4, and 4 patients, respectively, and only 1 patient received growth factors and a transfusion. No infectious complications were observed. Further escalations of the 90Y-ibritumomab tiuxetan dose were held, and the MTD was determined at 0.4 mCi/kg.
Overall, all hematologic toxicities were reversible, and no non-hematologic toxicities were seen with the exception of grade-1 fatigue in 2 patients and reversible infections. Among those who developed hematologic toxicities, the median duration of at least grade-3 leukopenia, neutropenia, and thrombocytopenia (first date in grade 3 to first date of improvement to grade 2 or better) was 12 (range, 7–37), 10 (range, 7–30), and 12 (range, 3–44) days, respectively. No significant differences were observed in the duration of cytopenias among the patients who received 0.4 and 0.3 mCi/kg of 90Y-ibritumomab tiuxetan: the median duration of at least grade-3 leukopenia, neutropenia, and thrombocytopenia was 12 (range, 7–37), 10 (range, 7–28), and 12 (range, 6–29) days, respectively, in the 0.4 mCi/kg cohorts, versus 7 (range, 7–7), 9 (range, 7–30), and 7 (range, 5–44) days, respectively, in the 0.3 mCi/kg cohort (Fig. 2).
In terms of hematologic adverse events, the major difference between the NCI CTCAE version 2.0 22 and version 4.03 24 is in the grading of thrombocytopenia (grade 4 equals platelet count <10,000/mm3 in version 2.0 vs. <25,000/mm3 in version 4.03). In 4 patients, grade-3 thrombocytopenia would be graded as grade 4 according to the most updated criteria; however, all 4 patients had also grade-4 neutropenia and/or leukopenia according to both versions.
Pharmacokinetic parameters
Pharmacokinetic data assessed in whole blood from 9 patients showed that after the administration of rituximab, the area under the curve (AUC) and maximum concentration (Cmax) of 111In-ibritumomab tiuxetan increased in 4 patients, remained stable (±15%) in 3, and decreased in 2. Whole-blood T1/2 changed variably and either remained unchanged (±10%) or increased. Both patients in whom Cmax and AUC decreased had bulky disease with a sum product of diameters of 34.6 and 69.5 cm, respectively. The patients in whom T1/2, Cmax, and AUC remained stable or increased had both bulky and nonbulky disease, but as a group had lower tumor burden (sum product of diameters, mean 21.8 cm, range 3–32.7). The whole-blood clearance of 111In-ibritumomab tiuxetan before and after a 4-weekly course of rituximab was determined in the first cohort (n=5) and followed a pattern of changes opposite to that of the T1/2, Cmax, and AUC. The whole-blood clearance decreased in 3 patients from 2.1 (range, 1.18–3.23) to 0.8 mL/hour/kg (range, 0.39–1.36), remained unchanged in 1 patient with low tumor burden (0.54 vs. 0.63 mL/hour/kg before and after rituximab, respectively), and increased in 1 patient with rituximab-refractory disease, high tumor burden, and rapid clinical progression (1.09 vs. 1.83 mL/hour/kg before and after rituximab, respectively). Overall, there were no significant differences in the pharmacokinetic parameters of 111In-ibritumomab tiuxetan before and after a 4-weekly course of rituximab (Fig. 3).

Pharmacokinetic data as assessed in whole blood of 111In-ibritumomab tiuxetan before and after a 4-weekly course of rituximab.
Secondary objectives
Reversal of bone marrow involvement
Six patients had bone marrow involvement by NHL at registration ranging from <5% to 20%. After 4-weekly doses of rituximab, marrow involvement decreased in all patients with complete clearance in 3 of them. Among the 3 patients with persistent NHL in the marrow, the level of involvement decreased further on subsequent biopsies after the administration of 90Y-ibritumomab tiuxetan with complete clearance in 1 patient. Nonetheless, dosimetric studies in the patients with marrow involvement at registration showed that the radiation dose to bone marrow from 90Y-ibritumomab tiuxetan in blood remained unchanged or even increased after a 4-weekly course of rituximab, and they all experienced at least grade-3 neutropenia and/or thrombocytopenia except one.
Antitumor response
In an intention-to-treat analysis, complete remission (unconfirmed by PET), partial remission, and stable and progressive disease were achieved in 6 (35%), 8 (47%), 2 (12%), and 1(6%) patients, respectively (Fig. 4A). The patient with progressive disease had rituximab-refractory transformed follicular lymphoma, with high tumor burden (sum product of diameters, 69.5 cm). The patient experienced a transient clinical improvement, but at the time of first evaluation of response, her disease progressed. The median interval from the administration of 90Y-ibritumomab tiuxetan to best response (per-protocol analysis) was 8 weeks (range, 5.1–24.9). Six patients (4 with partial and 2 with complete remission) achieved their best response beyond 12 weeks after the administration of 90Y-ibritumomab tiuxetan.
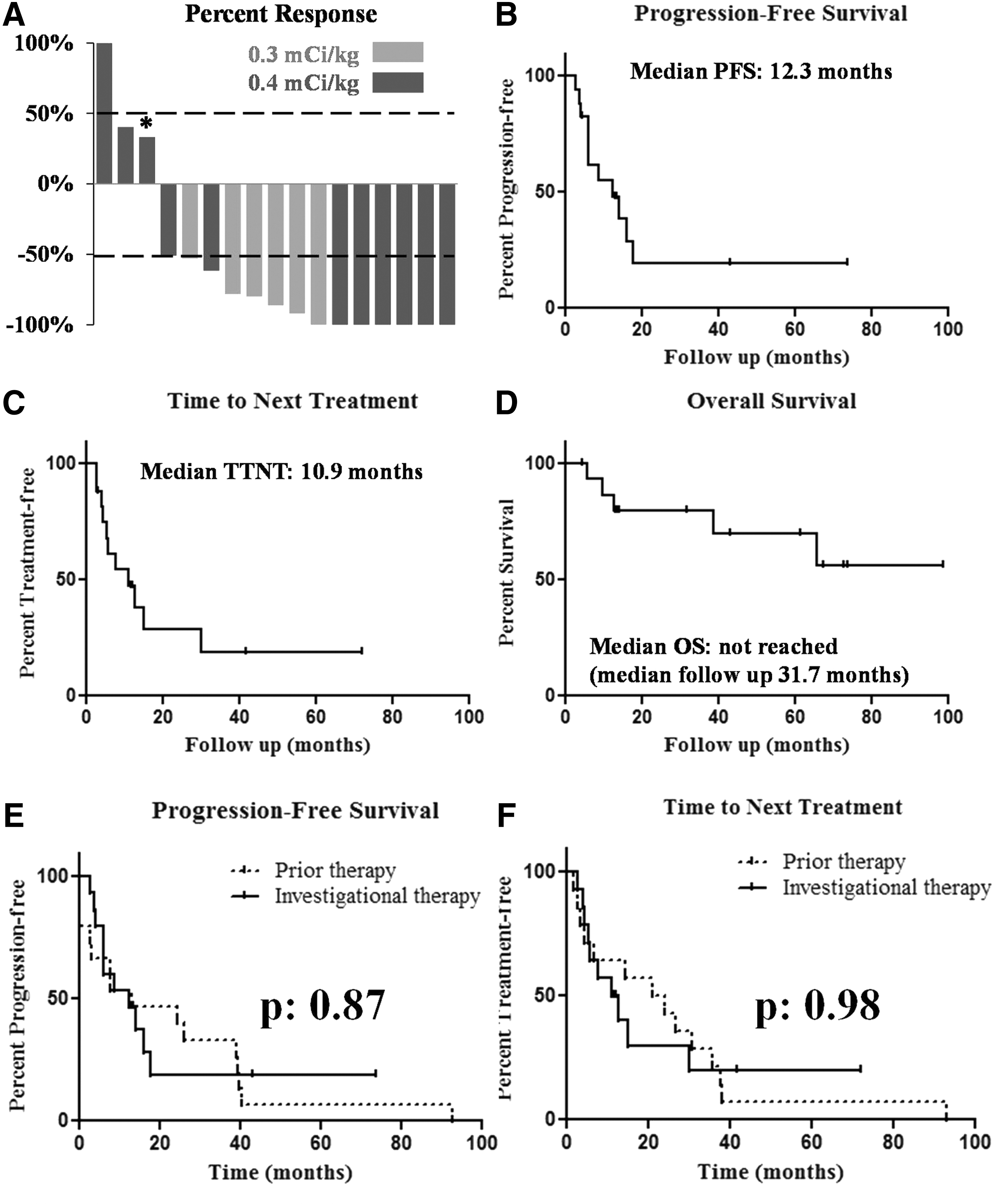
Clinical endpoints
With a median follow-up of 31.7 months (range, 4.3–98.7), the median progression-free survival and time to next treatment were 12.3 and 10.9 months, respectively (Fig. 4B, C). The median overall survival has not been reached (Fig. 4D). In 15 patients with available dates of prior treatment, the median progression-free survival achieved with the investigational therapy was not significantly different from the one achieved with the prior treatment (12.3 vs. 13.1 months for the investigational and prior treatment, respectively; p: 0.871) (Fig. 4E). Although the time to next treatment was shorter with the investigational therapy, it was not statistically different compared with the interval achieved with the prior therapy (10.9 vs. 20.9 months for the investigational and prior treatment, respectively; p: 0.98) (Fig. 4F).
Discussion
This study was the first to evaluate the safety and efficacy of a 4-weekly course of rituximab preceding radioimmunotherapy with 90Y-ibritumomab tiuxetan in relapsed or refractory low-grade or follicular or transformed CD20+ B-cell NHL. The rationale of combining these two effective treatments was that the elimination of bone marrow infiltration with CD20+ cells would allow for higher doses of radioimmunotherapy to be tolerated, optimize biodistribution, and thereby mitigate the myelotoxicity and enhance the efficacy of 90Y-ibritumomab tiuxetan. Previous studies 25 have shown that a 4-weekly course of rituximab preceding radioimmunotherapy optimized the biodistribution of the radioimmunoconjugate and led to high response rates. 25 Excluding 1 patient in our study who developed human anti-murine antibodies and in whom paired comparisons could not be performed, the splenomegaly volume was reduced (median 21%; range 0%–82%), and 5 of 27 tumors were no longer detectable by gamma-camera imaging. 19
In this study, however, we observed that a 4-weekly course of rituximab did not particularly mitigate myelotoxicity. Although a 4-weekly course of rituximab may have reduced the localization of the radioimmunoconjugate to the bone marrow, grade-4 neutropenia and/or thrombocytopenia was seen in 44% of patients (16% in the 0.3 mCi/kg cohort and 60% in the 0.4 mCi/kg cohorts) as compared with the 30%–35% reported in previous studies. 26 Hematologic toxicity was observed even in patients with bone marrow clearance after a 4-weekly course of rituximab, probably secondary to the unchanged or increased radiation dose to the bone marrow from blood radioactivity. Although the increase in the marrow dose from blood before and after a 4-weekly course of rituximab was not statistically significant, 19 75% of patients with at least grade-3 cytopenia had an increase in the marrow dose from blood. On the other hand, among the few patients with no such cytopenias, the marrow dose from blood decreased before and after a 4-weekly dose of rituximab. Since treatment with 90Y-ibritumomab tiuxetan was held if the predicted radiation dose to the bone marrow exceeded 3 Gy, other reasons besides increased radiation to the marrow may account for the significant myelotoxicity. A strategy that may mitigate the myelotoxicity of this regimen without compromising its efficacy may involve administration of the 90Y-ibritumomab tiuxetan dose in two fractions, as shown with 131I-rituximab radioimmunotherapy. 25
It is noteworthy though that 50% of patients who received 0.3 mCi/kg of 90Y-ibritumomab tiuxetan did not experience any grade-3 or 4 hematologic toxicity, and myelotoxicity in the expansion cohort was less frequent compared to the initial cohort. Dosimetry data from this study did not show major differences in the radiation dose to the bone marrow from blood between the 0.3 and 0.4 mCi/kg cohorts 19 ; therefore, the lower myelotoxicity in the 0.3 and 0.4 mCi/kg expansion cohort may be due to careful patient selection with evidence of good performance status, adequate bone marrow reserve, and exclusion of patients with rituximab-resistant disease. Also, neutropenia associated with radioimmunotherapy does not have the same implications of that associated with cytotoxic chemotherapy, as in the former case, mucositis is infrequent, and hence neutropenic fever is less frequent as well.
Investigations on the pharmacokinetic properties of rituximab have indicated that its serum concentrations are lower in patients with high tumor burden and bulky disease. 27 –29 This was also shown with dosimetric calculations besides enzyme-linked immunosorbent assay measurements of serum concentrations in a similar study that combined a 4-weekly course of rituximab with 131I-rituximab radioimmunotherapy. 25 Unlike the latter study though that showed a consistent pattern of changes in T1/2 of radioimmunotherapy after a 4-weekly course of rituximab, 25 the pharmacokinetic parameters in our study changed variably. We, as well, observed an accelerated elimination (in other words, lower effective T1/2) of the radioimmunoconjugate in patients with high tumor burden, but the T1/2 did not consistently increase after a 4-weekly course of rituximab. This accelerated elimination of the radioimmunoconjugate in patients with high tumor burden may relate to increased CD20 availability and attendant cleavage and release of the radioactive compound. Pairwise comparisons of the AUC, T1/2, Cmax, and clearance before and after a 4-weekly course of rituximab did not indicate significant and consistent changes, despite the tumor responses achieved with rituximab alone, which may have led to a reduction of the CD20-binding sites. Considering that responses correlate with serum concentrations of rituximab 27 and that its serum concentrations are lower in patients with high tumor burden and bulky disease, 27 –29 our results indicate that successive weekly administrations of rituximab may not obviate the problem of accelerated elimination of the radioimmunoconjugate in cases of high tumor burden and bulky disease.
Although not the primary endpoint of this study, our results indicate that this investigational therapy was effective. The median progression-free survival exceeded 1 year, and the overall survival has not been reached. Considering that remission intervals in indolent lymphomas become shorter with successive lines of therapy, 2 progression-free survival and time to next treatment were not significantly different between the prior and the investigational therapy. The overall response rate was 82%, which is comparable to the 67%–83% reported in larger trials. 13 –16 As 6 patients (4 with partial and 2 with complete remission) achieved their best response in evaluations beyond the 19-week treatment period, it is possible that with longer median follow-up, the antitumor efficacy may be higher and more sustained as shown in other trials with 90Y-ibritumomab tiuxetan. 30 This observation indicates that radioimmunotherapy may have a delayed onset, but prolonged duration of activity. It may not reverse the clinical course in patients with a rapidly progressive disease: the patient in our study with progressive disease had transformed follicular lymphoma, with high tumor burden, and had progressed through her last cytotoxic chemotherapy administered 5 weeks before enrollment. Although refractoriness to rituximab by itself do not portend a lack of response to 90Y-ibritumomab tiuxetan, 15 her disease also progressed while receiving rituximab+CHOP. These observations regarding the efficacy of the investigational regimen should be interpreted with caution, as selection bias may influence the results, and the efficacy was not the primary endpoint of the study. Moreover, dosimetry analyses showed a decrease in the median tumor residence time of the radioimmunoconjugate after a 4-weekly course of rituximab. Besides tumor regression with rituximab, prior saturation of the tumor CD20-binding sites with rituximab compromising the binding of the radioimmunoconjugate may account for this observation. 19
In conclusion, the MTD of 90Y-ibritumomab tiuxetan after 4-weekly doses of rituximab in patients with relapsed or refractory low-grade or follicular or transformed CD20+ B-cell NHL and <25% bone marrow involvement is 0.4 mCi/kg. A 4-weekly course of rituximab decreased bone marrow involvement by NHL in all patients with complete clearance in 50%. This investigational therapy was associated with significant cytopenias secondary to unchanged or increased radiation to the bone marrow from 90Y-ibritumomab tiuxetan in blood. Cytopenias, however, were transient and reversed with supportive care. Despite the myelotoxicity, the response rates were high, and sustained complete and partial remissions were achieved. Responses were seen in cases of both bulky and nonbulky disease; however, pharmacokinetic analyses in our study showed that accelerated elimination of the radioimmunoconjugate in cases of bulky disease or high tumor burden has not been obviated with a 4-weekly course of rituximab.
Footnotes
Acknowledgment
We wish to acknowledge the contribution of Jeannie Connors, RN, study coordinator, in whose memory this manuscript is dedicated. This study has been funded by IDEC Pharmaceuticals (San Diego, California).
Authorship Contributions
C.V. and A.F. saw the patients, analyzed the data, and wrote the manuscript. R.F.M., S.S., S.J.K., I.N.M., and J.J.S. saw the patients and collected the data. A.F.L. contributed to the analysis.
Disclosure Statement
The authors have no conflicts of interest pertaining to this manuscript to disclose.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.