
Abstract
Long noncoding RNAs (lncRNAs) have recently emerged as pivotal regulators that govern fundamental biological processes and disease pathogenesis. LncRNA MNX1-AS1 has been reported to promote cell proliferation and invasion in gallbladder cancer, but its biological role and regulatory mechanism in ovarian cancer are poorly defined. In this study, it was found that higher expression of lncRNA MNX1-AS1 is closely associated with International Federation of Gynecology and Obstetrics stage and lymphatic metastasis in ovarian cancer patients. RNA interference (RNAi) to downregulate the expression of lncRNA MNX1-AS1 was used in the ovarian cancer cell lines, OVCA433 and SKOV-3. CCK-8, EdU staining, and colony formation assays was used to test the viability and proliferation ability of these cells. Wound healing and transwell migration assays were performed to determine the migration ability of the cells. Cell cycle progression and apoptotic assays were carried out using flow cytometry. These in vitro loss-of-function experiments revealed that downregulation of lncRNA MNX1-AS1 suppressed cell proliferation, colony formation, cell migration ability, induced cell cycle arrest at the G0/G1 phase, and promoted apoptosis. Furthermore, MNX1-AS1 knockdown altered the protein expressions of CDK4, cyclin D, Bax, and Bcl-2. These findings demonstrated for the first time that lncRNA MNX1-AS1 functions as an oncogene in ovarian cancer and could be a potential target for this disease.
Introduction
Ovarian cancer is a highly lethal gynecological malignancy that affects women and is the leading cause of cancer-related death worldwide. 1 Despite improvements in surgery and chemotherapy, the overall survival of patients still remains poor. One reason for such a high mortality rate is that most ovarian cancer patients are usually diagnosed at a later stage 2 of the disease. Therefore, a thorough understanding of the molecular mechanisms involved in ovarian carcinogenesis is urgently needed for the diagnosis and therapy of ovarian cancer.
Long noncoding RNAs (lncRNAs >200 nucleotides in length) are increasingly recognized as pivotal cellular regulators that participate in cell fate determination and in a range of other biological processes. 3 Dysregulation of lncRNAs has been strongly linked to oncogenes, 4,5 or tumor suppressor genes 6,7 that are involved in carcinogenesis are therefore potential markers in diagnosing cancer. For example, MALAT-1 is overexpressed in ovarian cancer and promotes the growth and migration of ovarian cancer cells. 8 LncRNA BACE1-AS plays an antioncogenic role in ovarian cancer by suppressing ovarian cancer stem cell proliferation and invasion. 9 However, the functional involvement of lncRNAs in ovarian carcinogenesis has not yet been fully elucidated.
MNX1-AS1 (MNX1 antisense RNA1) is an lncRNA located on chromosome 7 that makes transcripts from the antisense strand near the 5′ end of the protein-coding gene. 10 Interestingly, microarray analysis by Wu identified many differentially expressed lncRNAs in gallbladder cancer (GBC), where MNX1-AS1 is highly expressed in 92.3% of GBC tissues. Furthermore, they showed that silencing MNX1-AS1 significantly inhibits cell proliferation, migration, and invasion in GBC in vitro. 11 Up until now, there have been no relevant reports describing the relationship between MNX1-AS1 and other cancers, including ovarian cancer.
Therefore, in this study, MNX1-AS1 expression levels were compared between ovarian cancer and normal ovarian tissue and investigated the relationship between MNX1-AS1 expression and clinicopathological features. Furthermore, functional experiments were carried out to evaluate the effects of MNX1-AS1 on ovarian cancer cell proliferation and migration. This study provides a new perspective on the diagnosis and therapeutic target of this deadly disease.
Materials and Methods
Sample collection
A set of 20 ovarian cancers and adjacent normal tissue (located >5 cm away from cancer tissue) were collected from patients who underwent surgery at the Department of Obstetrics and Gynecology, at the Qilu Hospital of Shandong University, between March 2014 and January 2016. None of the patients had received preoperative chemotherapy or had any concomitant gynecological or other primary cancer. Patients signed an informed consent and their clinical information is summarized in Table 1. All resected tissue samples were flash frozen in liquid nitrogen after resection and stored at −80°C before RNA was extracted. This study was approved by the institutional review board of Qilu Hospital of Shandong University.
p < 0.05.
Cell culture and siRNA transfection
Human ovarian cancer cell lines OVCA433 and SKOV-3 were purchased from the American Type Culture Collection. OVCA433 cells were cultured in Dulbecco's modified Eagle's medium, whereas SKOV-3 cells were grown in RPMI-1640 medium (Gibco-BRL). Culture media were supplemented with 10% fetal bovine serum (FBS) and 1% penicillin, which were replaced with fresh media every 2–3 days. All cells were maintained in a humidified atmosphere containing 5% carbon dioxide at 37°C.
Specific siRNAs targeting lncRNA MNX1-AS1 and a scrambled oligo as the negative control (NC) were designed and synthesized by GenePharma (Shanghai, China). Cells were seeded in six-well plates and transfected with 10 nM siRNA using Lipofectamine® RNAiMAX (Invitrogen) according to the manufacturer's instructions. Cells were transfected with an empty vector as controls. Forty-eight hours after transfection, cells were used for in vitro assays.
RNA extraction and quantitative real-time polymerase chain reaction
Total RNA was extracted from either tissue or transfected cells using TRIzol® reagent (Invitrogen), and cDNA was synthesized from RNA using a Reverse Transcription Reagent Kit (DBI), according to the manufacturer's instructions. The polymerase chain reaction (PCR) primer sequences used for analyses were as follows:
MNX1-AS1 (forward: 5′-CCCGCATTTTCAGATTCAC-3′) and (reverse: 5′-GCTCTCAGCCTCGCCATA-3′); GAPDH (5′-TGTTCGTCATGGGTGTGAAC-3′) and (reverse: 5′-ATGGCATGGACTGTGGTCAT-3′).
The quantitative real-time PCR (qRT-PCR) analyses were carried out using 2 × SYBR green PCR Master Mix (TOYOBO, Japan) and performed on an ABI PRISM 7500 Sequence Detection system. The relative levels of MNX1-AS1 mRNA were normalized to the expression of glyceraldehyde-3-phosphate dehydrogenase by the 2−ΔΔCt method. Each sample was repeated at least three times.
CCK-8 assay
Cell Count Kit-8 (Beyotime, China) assay was performed to determine the viability of OVCA433 and SKOV-3 cells after siRNA transfection. In brief, cells from different treatments were reseeded into 96-well plates and cultured for 12, 24, or 48 hours, respectively. The cells were then digested, resuspended, and incubated with 10% CCK-8 solution. After incubation for 1 hour, the absorbance at 450 nm was determined on an ELISA reader (Thermo MK3).
Colony formation assay
To evaluate monolayer colony formation, stably transfected cells (500 cells per well) were plated into six-well plates and incubated for 48 hours. Then cells were fixed with paraformaldehyde and stained with 1% crystal violet (Beyotime) for 20 minutes. The colonies formed (>50 cells per colony) were observed under the microscope.
EdU flow cytometry assay
After 48 hours transfection, cells were seeded onto 96-well plates and grown till well adherent. The cell proliferative rates were detected using the Cell Light™ EdU Apollo®488 In Vitro Imaging Kit (Ribobio, Guangzhou, China) according to the manufacturer's protocol. Finally, the percentage of EdU-positive cells was determined by flow cytometry with the Cytomics FC 500 MCL (Beckman Coulter, Brea, CA).
Hoechst 33258 staining
Cells were cultured in six-well plates for 48 hours after transfection and fixed with 4% formaldehyde for 10 minutes. The cells were then washed with PBS and stained with Hoechst 33258 (10 μg/mL; Sigma) for 20 minutes at room temperature in the dark. Cellular changes in nuclear morphology were examined under a fluorescence microscope (Zeiss Axioplan, Germany).
Flow cytometry analysis
Cells from different treatments were collected and seeded on 6-cm dishes at a density of 2 × 105 cells/dish. Cell cycle analysis was carried out using Propidine Iodide staining, as described previously. For cell apoptosis, cells were stained using the Annexin V/PI apoptosis detection kit (KeyGEN Biotech, Nanjing, China) following the previous report. Finally, stained cells were analyzed by an FACSCalibur flow cytometer (BD, New Jersey).
Wound healing assay
After transfection, cells were seeded in six-well plates and scratched with a pipette tip, when the culture reached 90% confluency. The cell layer was then washed twice with medium and cultured for up to 48 hours. At 0 and 48 hours, photographic images of the plates were obtained by microscopy at 0 and 24 hours after scratching. The wound healing effects were determined by measuring the percentage of the wound area at 48 hours compared with the wound area at 0 hour. Each sample was analyzed in triplicates. The experiment was also performed three times.
Cell migration assay
Cell migration was detected using transwell chambers (8.0 μm pore size; Costar) following manufacturer's instructions. In brief, cells were seeded into the upper chamber in serum-free medium. Ten percent FBS medium was added to the lower chamber of the transwell. After incubation for 24 hours, cells migrated to the bottom chamber and were fixed with 4% paraformaldehyde and stained with 0.5% crystal violet. The number of migratory cells was observed and counted under a microscope in five random fields.
Western blotting
Total protein was extracted from cells using RIPA lysis buffer and protein concentration was determined using the bicinchoninic acid protein assay reagent kit (Pierce) according to the manufacturer's instructions. Equal amounts of protein were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to a polyvinylidenedifluoride (PVDF) membrane (Bio-Rad). The membrane was blocked with 10% nonfatty milk for 1 hour and then probed with primary antibodies that included anti-CDK4, anticyclin D1, anti-Bax, and anti-Bcl-2 (all from Cell Signaling Technology) overnight at 4°C. GAPDH was used as a loading control. The PVDF membrane was washed with Tris Buffered Saline with Tween three times and incubated with an appropriate horseradish peroxidase-conjugated secondary antibody (Invitrogen) at room temperature for 1 hour. The membrane was washed three times with TBST and observed by an enhanced chemiluminescence system (Bio-Rad Laboratories).
Statistical analysis
All experiments were independently performed three times and quantitative data are presented as mean ± SD. Student's t test and χ2 test were used to evaluate the statistical significance in GraphPad Prism 5 software (GraphPad, CA). All statistical tests were two sided and a p value <0.05 was considered statistically significant.
Results
Expression of MNX1-AS1 in ovarian cancer tissues and its clinical significance
Ovarian cancer tissue and corresponding normal tissue (n = 20) were used to examine the expression of MNX1-AS1 using qRT-PCR analysis. MNX1-AS1 levels in ovarian cancer tissue were significantly upregulated nearly fourfold the normal tissue (Fig. 1, p < 0.001). To further understand the significance of MNX1-AS1 expression in ovarian cancer, MNX1-AS1 expression was correlated with various clinicopathological features. As shown in Table 1, higher MNX1-AS1 expression in ovarian cancer was significantly correlated with the FIGO stage and lymphatic metastasis, which suggests that MNX1-AS1 might be a potential prognostic biomarker in ovarian cancer patients.
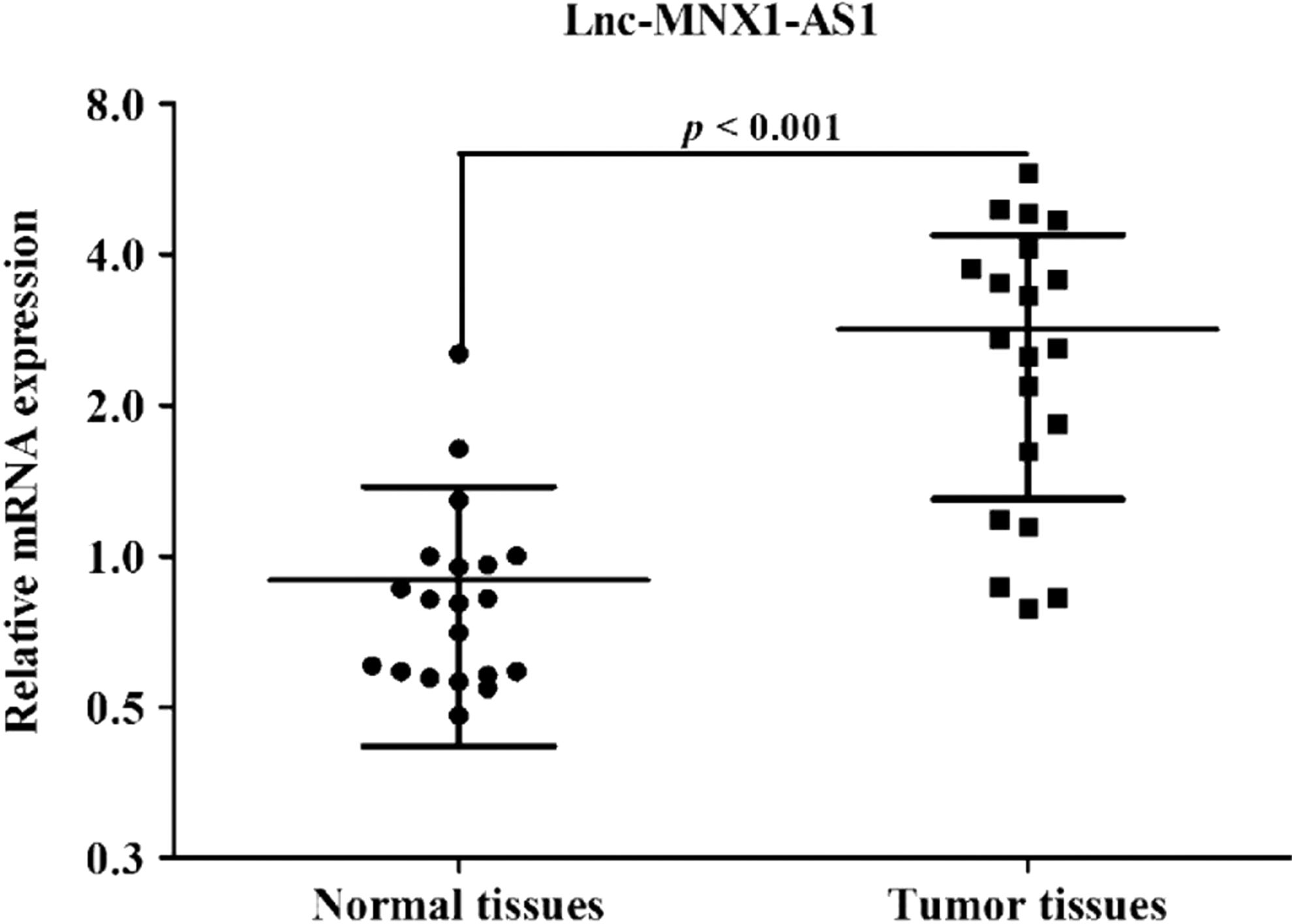
Expression of MNX1-AS1 in ovarian cancer tissues and adjacent tissues. MNX1-AS1 expression was significantly upregulated in ovarian cancer tissues relative to their corresponding ovarian normal tissues in 20 ovarian cancer patients. p < 0.001 versus controls.
The expression of MNX1-AS1 was effectively decreased in ovarian cancer cells
As MNX1-AS1 was overexpressed in ovarian cancer, it was speculated that it might have an important biological function in ovarian cancer cells. To examine this possibility, the expression of MNX1-AS1 was knocked down by siRNA transfection in ovarian cancer cell lines, OVCA433 and SKOV-3. Knockdown efficiency was evaluated by qRT-PCR analysis. The results (Fig. 2A, B) indicated that endogenous MNX1-AS1 mRNA was significantly reduced in the siRNA group compared with that in the NC group in OVCA433 and SKOV-3 cells (p < 0.001).
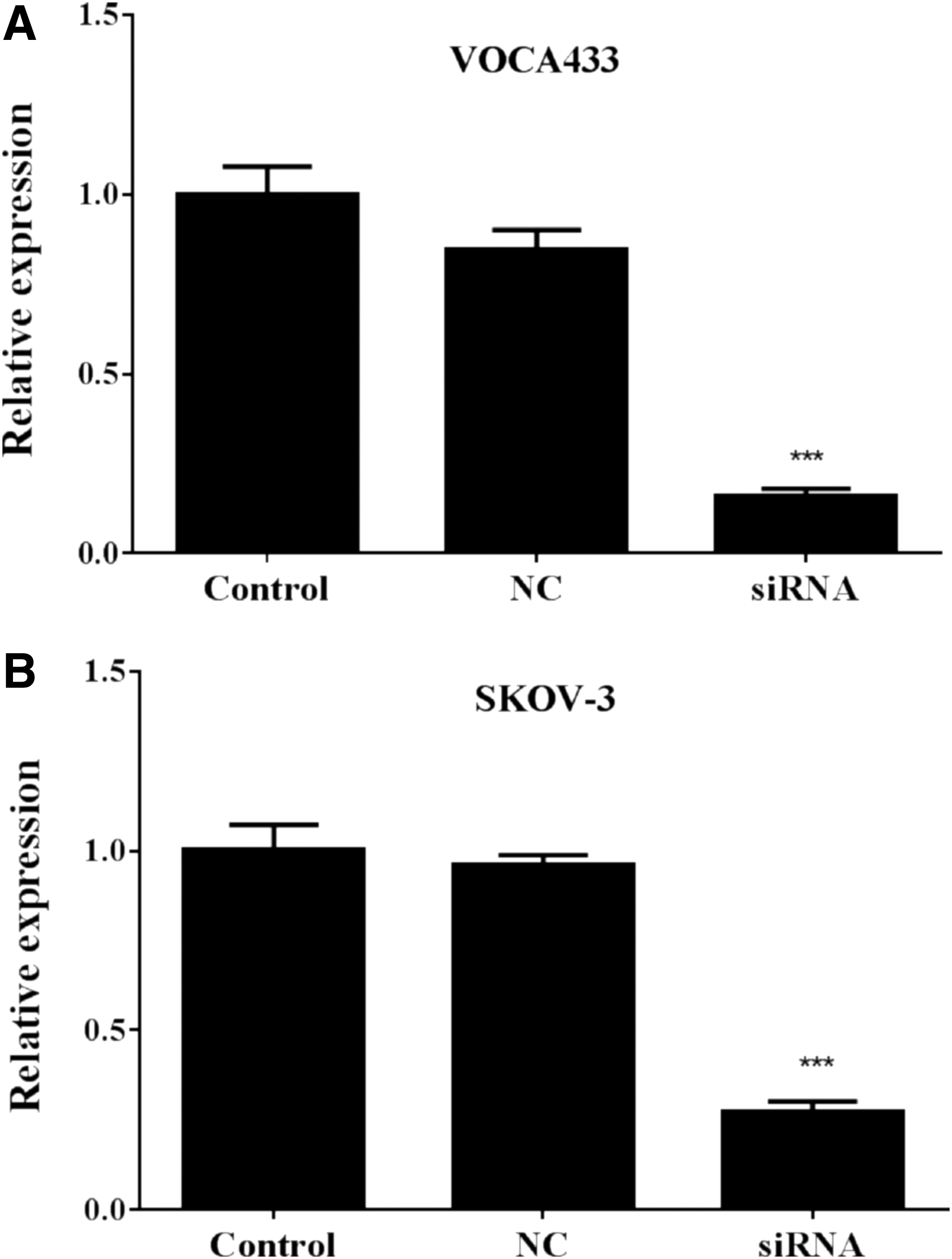
Knockdown of MNX1-AS1 through siRNA transfection. MNX1-AS1 mRNA levels were measured by quantitative real-time polymerase chain reaction in three groups (Control, NC, and siRNA) in OVCA433
siRNA-mediated knockdown of MNX1-AS1 inhibits ovarian cancer cell proliferation
To explore the biological function of MNX1-AS1 on ovarian cancer, the CCK-8 assay was first used to evaluate the effect of MNX1-AS1 on cell viability. siRNA transfection-mediated MNX1-AS1 knockdown significantly decreased cell viability during 48 hours after transfection in both OVCA433 and SKOV-3 cells (Fig. 3A, p < 0.05). In addition, the proliferation rate of ovarian cancer cells was evaluated after MNX1-AS1 knockdown using EdU flow cytometry. As depicted in Figure 3B, the percentage of EdU-positive cells significantly decreased from 24.31% in the NC group to 15.27% in the siRNA group in OVCA433 cells (p < 0.01). Similarly, siRNA-mediated knockdown of MNX1-AS1 reduced the percentage of EdU-positive cells from 28.84% to 12.57% in SKOV-3 cells (p < 0.01). Furthermore, the effect of MNX1-AS1 knockdown on colony formation in OVCA433 and SKOV-3 cells was determined by crystal violet staining. As shown in Figure 3C, both the size and number of colonies formed in siRNA-infected cells exhibited an obvious decrease in both OVCA433 and SKOV-3 cells (p < 0.001). These data provide evidence that MNX1-AS1 promotes cell growth and proliferation in ovarian cancer cells.

Effect of MNX1-AS1 on cell viability, proliferation rate, and colony formation ability.
siRNA-mediated knockdown of MNX1-AS1 induced cell cycle arrest in ovarian cancer cells
To assess the potential mechanism of MNX1-AS1 in ovarian cancer cell proliferation, the cell cycle in the OVCA433 and SKOV-3 cells was tested, by flow cytometry using PI staining. siRNA-mediated knockdown of MNX1-AS1 resulted in an increase in percentage of cells at the G0/G1 phase, but a decrease in the percentage of cells in the S phase, in both OVCA433 (Fig. 4A, p < 0.01) and SKOV-3 (Fig. 4B, p < 0.01) cells. Moreover, Western blotting further confirmed that knockdown of MNX1-AS1 induced cell cycle arrest at G0/G1 phase by significantly decreasing the expression of CDK4 and cyclin D1 in OVCA433 (Fig. 4C, p < 0.01) and SKOV-3 (Fig. 4D, p < 0.01) cells.
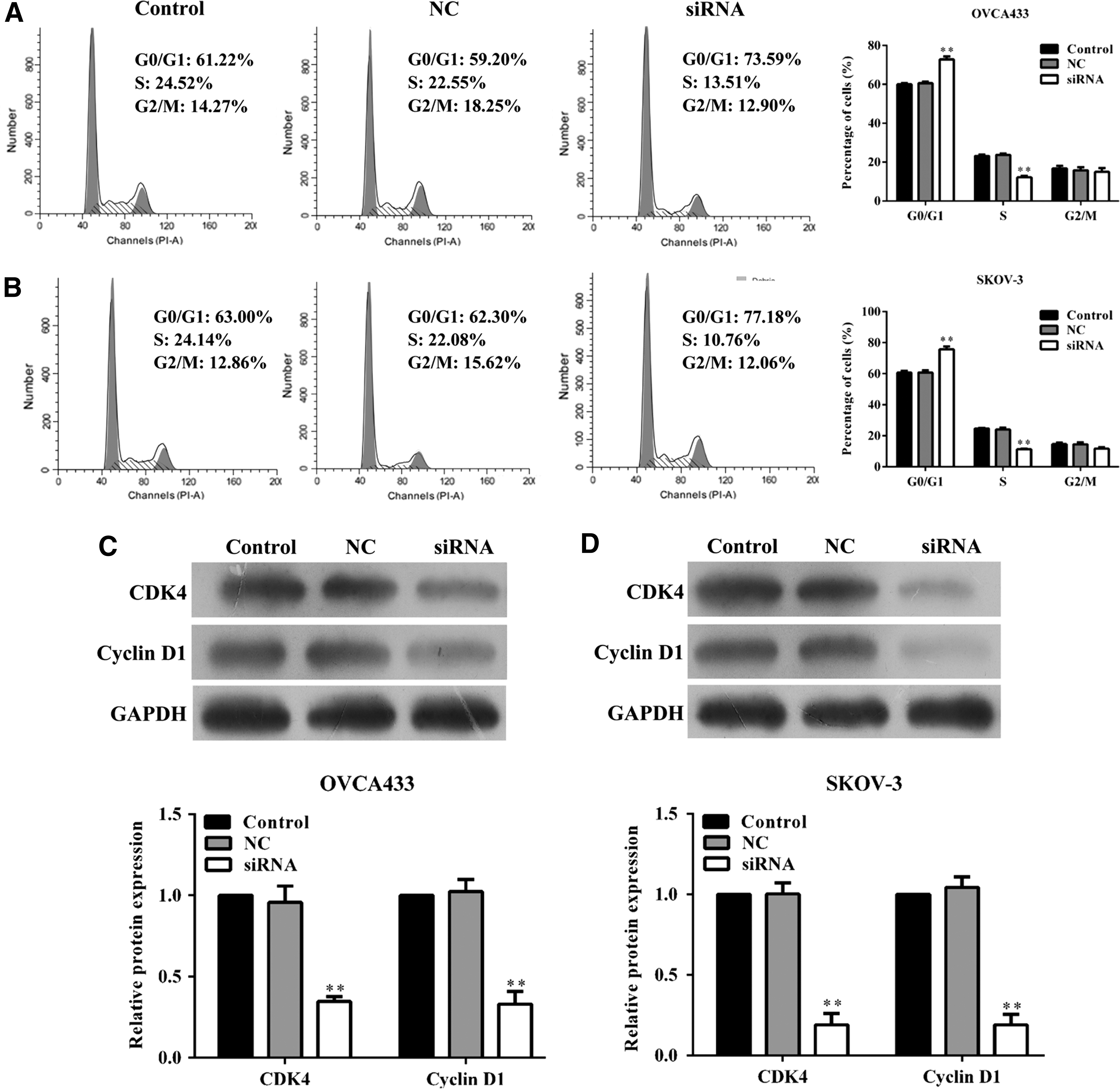
Effect of MNX1-AS1 on ovarian cancer cell cycle progression.
siRNA-mediated knockdown of MNX1-AS1 promoted ovarian cancer cell apoptosis
Next, Annexin V/PI double staining was applied to investigate the effects of MNX1-AS1 knockdown on apoptosis. As shown in Figure 5A, the percentage of apoptotic cells was significantly increased in the siRNA group in both OVCA433 and SKOV-3 cells compared with that in the corresponding NC group (p < 0.01). In addition, Hoechst staining was used to intuitively assess cell apoptosis induced by MNX1-AS1 knockdown. As shown in Figure 5B, more cells had condensed and fragmented nuclei (indicating early apoptotic cell fraction) in siRNA-transfected OVCA433 and SKOV-3 cells. Furthermore, proapoptotic Bax was upregulated, whereas antiapoptotic Bcl-2 was downregulated in OVCA433 and SKOV-3 cells after MNX1-AS1 knockdown using Western blotting (Fig. 5C, p < 0.01). These results suggested that siRNA-mediated knockdown of MNX1-AS1 causes a strong apoptotic effect in human ovarian cancer cells.

Effect of MNX1-AS1 on ovarian cancer cell apoptosis.
siRNA-mediated knockdown of MNX1-AS1 suppressed ovarian cancer cell migration
Furthermore, a wound healing assay was utilized to determine whether downregulation of MNX1-AS1 affected the migratory ability of ovarian cancer cells. As expected, MNX1-AS1 knockdown in OVCA433 (Fig. 6A) and SKOV-3 (Fig. 6B) resulted in a slower closing of the scratch wound compared with the NC or controls (p < 0.01). Moreover, the transwell migration assay showed that suppression of MNX1-AS1 significantly inhibited the migration ability of OVCA433 (Fig. 6C, p < 0.05) and SKOV-3 (Fig. 6D, p < 0.01) cells compared with the control and NC cells, as indicated by a marked decrease in the number of cells that migrated to the bottom well. There were no significant differences in the number of migrated cells between control and NC groups.
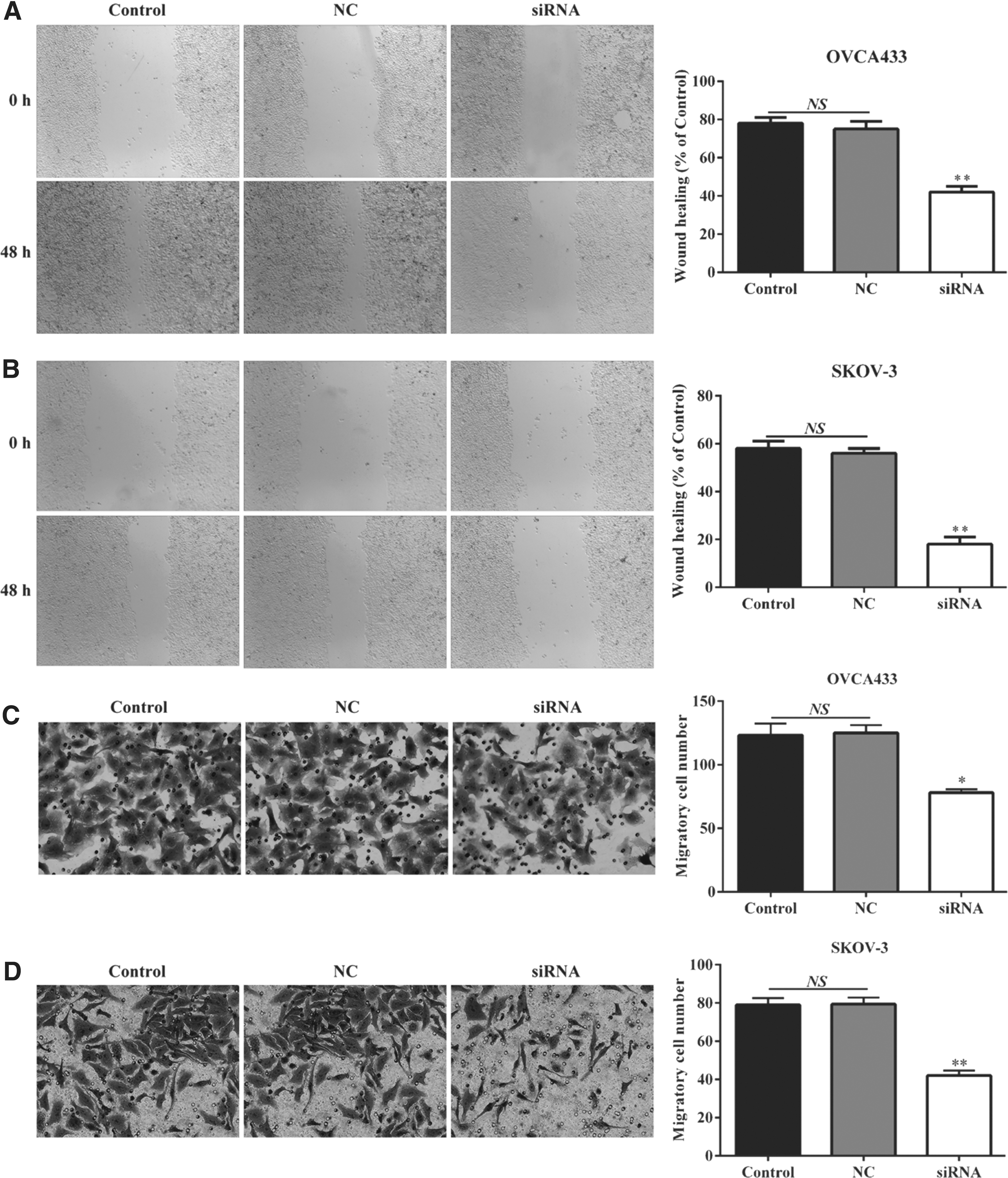
Effect of MNX1-AS1 on ovarian cancer cell migration. Downregulation of MNX1-AS1 resulted in a slower closing of the scratch wound compared with NC or control in
Discussion
It is now widely accepted that lncRNAs are key players in gene regulatory processes and are involved in the pathogenesis of cancer. 3,12 –14 These will potentially help us in constructing a novel platform for the diagnosis and treatment of cancer. Previous work demonstrated that lncRNA MNX1-AS1 plays an important role in cell proliferation and migration in GBC. 11 However, the functions of MNX1-AS1 in ovarian cancer were unknown. In this study, the first evidence that MNX1-AS1 is significantly upregulated in ovarian cancer tissues compared with adjacent normal tissues is provided, and that high expression of MNX1-AS1 is associated with the FIGO stage of lymphatic metastasis in ovarian cancer patients. These findings suggest that MNX1-AS1 might be used as a diagnostic and prognostic ovarian cancer marker.
To investigate the functions of MNX1-AS1 in ovarian cancer, antihuman siRNA was used for MNX1-AS1 knockdown in two ovarian cancer cell lines, OVCA433 and SKOV-3. Loss-of-function assays indicated that knockdown of MNX1-AS1 resulted in diminished cell proliferation and migration. These data demonstrated that MNX1-AS1 plays an important role in ovarian cancer cell growth and metastasis. To test whether MNX1-AS1 expression influenced cell cycle progression and apoptosis, flow cytometry and Hoechst 33258 staining were used to detect cell cycle progression and apoptosis in OVCA433 and SKOV-3 cells after treatment with either an siRNA or an empty vector. The results showed that knockdown of MNX1-AS1 induced significant arrest in the G0/G1 phase and an obvious increase in apoptosis.
Cell cycle regulation is a precise physiological process and primarily participates in cellular proliferation, which consists of four distinct phases (G0/G1, S, and G2/M). 15,16 Different CDK/cyclin complexes play a central role in controlling the cell cycle phases and their deregulation is observed in a variety of human tumors. 17,18 CDK4 and cyclin D1 are cell cycle regulators that control the transition from G1 to S phase of the cell cycle. 19 To uncover how MNX1-AS1 regulates the cell cycle, the expression of CDK4 and cyclin D1 was determined in ovarian cancer cells after MNX1-AS1 knockdown. Knockdown of MNX1-AS1 downregulated the expression of CDK4 and cyclin D1. These data suggest that CDK4/cyclin D1 might be downstream targets of MNX1-AS1 in ovarian cancer. In addition, cell apoptosis plays a crucial role in maintaining cellular homeostasis, which is regulated by various apoptotic-related proteins, including the Bcl-2 family of proteins. 20 Bcl-2 and Bax are an antiapoptotic protein and a proapoptotic protein, respectively, and their levels are directly related to the occurrence of apoptosis. 21 Bcl-2 binds to Bax to inhibit apoptosis. Consistent with this, increased Bax expression promotes apoptosis by forming homodimers. 22 In this study, a notable decrease in Bcl-2 protein expression and an increase in Bax protein expression were observed in ovarian cancer cells after MNX1-AS1 knockdown, suggesting that downregulation of MNX1-AS1 promotes apoptosis in ovarian cancer cells by affecting Bcl-2 and Bax expression.
In summary, this is the first report showing that MNX1-AS1 is highly expressed in ovarian cancer tissue and is closely associated with clinicopathological parameters. Furthermore, it is demonstrated that MNX1-AS1 functions as an ovarian cancer oncogene by presenting evidence that knockdown of MNX1-AS1 inhibits cell proliferation, migration, and induces cell cycle arrest and apoptosis. These findings set the foundation to test whether MNX1-AS1 might be a potential new target and an early diagnostic tool in ovarian cancer.
Footnotes
Disclosure Statement
No competing financial interests exist.