
Abstract
Abstract
Using data collected at both room temperature and higher temperatures, this study examined the mineralogy of, and iron coordination shell in, polluted sediments from drainage and irrigation channels. As a part of the study, mineralogical characteristics of iron phases of canal sediments were examined to determine if these sediments could be used in place of common clays for the production of bricks. While such use would reduce the impact of these wastes on the environment, an excess of iron in these sediments could affect some important features of bricks. Therefore, this study examined the oxidation state of iron as well as its structural location in the mineral matrix. X-ray powder diffraction, thermal analysis, and X-ray absorption spectroscopy were used to examine sediments and bricks manufactured from irrigation and drainage channels of the Po River region in northeast Italy. Results indicated that canal sediments can be used as row materials in clay brick production. In fact, the process temperature (950°C) that characterized brick production was similar to the temperature used for thermal treatment of polluted sediments in this study.
Introduction
Using brick production to recycle contaminated sediments addresses two important environmental issues: the depletion of natural resources and the disposal of industrial wastes. Furthermore, the traditional clay-based materials used in brick production are heterogeneous products that can accommodate different inorganic wastes without significantly affecting the production process or the technical properties of final products (Malferrari et al., 2009; Caligaris et al., 2000; Romero et al., 2009). In the search for waste materials that can be used in brick making, researchers have assessed everything from sewage sludge ash (Weng et al., 2003; Lin et al., 2006), waterworks sludge (Samara et al., 2009), and natural stone wastes (Acchar et al., 2006; Menezes et al., 2005; Blanco Garcìa et al., 2005), to fly and bottom ashes (Lin, 2006; Olgun et al., 2005; Balgaranova et al., 2003; Cheeseman et al., 2003), metallurgical wastes (Vieira et al., 2006; Shih et al., 2004), and boron waste (Uslu and Arol, 2004).
However, it is not just organic and inorganic pollutants that engender strong environmental concerns over the use of sediments in bricks. Sediments from drainage channels contain iron (Fe), which, depending on its oxidation, coordination, and bonding conditions, can affect a fired product's properties (e.g., color and hardness). Thus, a knowledge of the specific chemical state that iron can reach in sediments needs to be considered (Kuno et al., 1999; Zaw et al., 2002; O'Day et al., 2004). Although Mössbauer spectroscopy can be used to detect and discriminate between iron oxides and hydroxides and iron-bearing phyllosilicates from sediments and soils (Stucki, 2006), the quantitative determination of such phases is more difficult, particularly when the iron is in tetrahedral coordination (Manceau et al., 2000b). This study, then, will discuss the mineralogy and local Fe ordering, oxidation state, and local coordination shell topology in sediments from drainage and irrigation channels at both room temperature and brick production temperature (950°C).
Sampling
The sediments used in this study came from drainage and irrigation channels located in the east-central area of the Padana plain, a few kilometers south of the Po River in northeastern Italy (see Malferrari et al., 2009). Sediment core samples (60–70 mm in diameter and 200–250 mm in length) were collected along eight different canals at distances of approximately 10 m apart, for a span of 1 km per canal. The samples were taken primarily in winter. The top of each core (about 20 mm) was removed. After quartering, macroscopic organic matter was removed from samples, and they were suspended in water, shaken, dried at 30°C, and finally powdered. The samples were rich in organic matter, which may promote iron complexation (Violante et al., 2002). The mineralogical and chemical composition of the samples was very similar in the target area, so there were no significant differences between samples from sampling sites. For this reason, a general mineralogical description has been provided for all the collected samples without further note on the sampling point.
Sample homogeneity is a fundamental prerequisite for preventing mechanical defects (e.g., hardness, breaking, and tensile strength) in the final product. Sample S1 was been selected as the most representative for brick production. If not otherwise stated, all data reported here refer specifically to this sample. The label S1(25°C) refers to the sample at room temperature, whereas the label S1(950°C) refers to the sample thermally treated at the brick production temperature of 950°C.
Analytical Methods
Mineralogical composition at ambient and imposed thermal conditions was determined by X-ray powder diffraction (XRPD). XRPD patterns were collected in the temperature range of 25°C to 1200°C, using a Philips X'Pert PRO diffractometer equipped with X'Celerator area detector and an Anton Paar HTK 16 type high temperature camera (Cu-kα radiation, quartz as standard). The S1 sample was powdered, mixed with acetone, and then placed on an external platinum element (purity, 99.25%) that heated to a maximum of 1523K. In our study, the temperature range was defined as 25°C–950°C, and the diffraction patterns were collected from 3° to 75° 2θ.
The sediments were analyzed using X-ray fluorescence (XRF) spectroscopy (Philips PW1480) and inductively coupled plasma optical emission spectroscopy (ICP-OES) (Perkin Elmer Optima 4200DV) after nitric–fluoridic total acid digestion. Instrument calibration was carried out using certified standards, and all chemical determinations were double-checked, always giving deviations <3.0%.
Thermal analyses and evolved gases mass spectrometry (EGMS) were carried out with ∼25 mg of each sample using a Seiko SSC 5200 thermal analyzer, equipped with an ESS GeneSys Quadstar 422 mass spectrometer (multiple ion detection mode). Signal integration was set to 1 s for water, nitric oxide, and carbon oxide compounds and 3 s for sulfur dioxide. Coupling with the mass spectrometer was performed by a ⅘-inch transfer line, heated to prevent gas condensing. Helium was used as purging gas, and its flow rate was set to 100 μL/min. Before each run, sample powders were loaded onto the platinum sample holder, and measurements were performed on the air-dried samples in the temperature range of 25°C–1100°C (heating rate, 10°C/min). Thermally treated kaolinite was used as reference material for differential thermal analyses (DTA) interpretation. The derivative thermo-gravimetric (DTG) curve profile was fitted by Origin 6.0 software. The quality of the approximation was estimated by the R factor.
Fe K-edge X-ray absorption spectroscopy (XAS) spectra were collected at the European Synchrotron Radiation Facility (ESRF, Grenoble, France) on the Spanish beamline SpLine, BM25, Branch A. The storage ring conditions were 6 GeV in the current range 180 mA to 200 mA, using a Si(III) double crystal monochromator. A metallic Fe reference foil was used to provide an energy calibration for the monochromator. Data were collected in fluorescence mode at room conditions, and each XAS spectrum represents the merged result from three scans. The spectra were recorded over a range of 500 eV across the Fe absorption K-edge with 1 eV energy step in the edge region (7100–7150 eV) and 3 eV in the extended X-ray absorption fine structure (EXAFS) region (7150–7500 eV). To compare structural information from XAS spectra on our samples, such Fe-rich minerals as Fe-rich montmorillonite (Brigatti et al., 2000), (Fe2+, Fe3+)-rich phlogopite (Tombolini et al., 2002), tetra-ferriphlogopite (Tombolini et al., 2002), (Fe2+)-rich phlogopite (Pini et al., 2008), and hematite (Petit et al., 2001) were selected as reference phases. These minerals were chosen as standards because they are well characterized: Fe-rich montmorillonite is characterized by its predominant octahedral Fe3+ content [Fe3+/(Fe3++Fe2+)=0.939]; (Fe2+, Fe3+)-rich phlogopite contains both Fe2+ and Fe3+ in the octahedral sites, with a slight predominance of Fe2+ [Fe3+/(Fe3++Fe2+)=0.474]; tetra-ferriphlogopite is mainly characterized by tetrahedrally coordinated Fe3+(Fe3+≈24% of tetrahedral occupancy); (Fe2+)-rich phlogopite contains Fe2+ in octahedral coordination only (Fe2+≈40% of total octahedral occupancy).
EXAFS spectra were quantitatively analyzed using the ATHENA software package (Ravel and Newville, 2005). From each averaged and energy-calibrated spectrum, a polynomial pre-edge function was subtracted, and the data was normalized. Normalized and background-subtracted data were weighted by k2. The radial structure function was obtained from the Fourier transforming χ2-weighted EXAFS spectra. Interatomic distances (R) and number of atomic neighbors (N) for the first shells (Fe–O, Fe–Fe) of the reference compounds were determined by models derived from known structures. For the S1(25°C) and S1(950°C) samples, the coordination numbers for Fe atoms were fixed to be integers.
Results and Discussion
According to the XRF analyses (see Table 1), the major components of the sediments were silicon dioxide (SiO2) and aluminium oxide (Al2O3), along with iron(III) oxide (Fe2O3) and calcium oxide (CaO). Table 2 lists heavy metals with concentrations above legal limits detected in the sediments. Zinc (Zn) is the most commonly encountered heavy metal in nature, whereas cadium (Cd) is the most potentially dangerous. XRPD patterns show the presence of minerals very common in Po River sediments, such as smectite (a peak at ∼15.2–15.5 Å on air-dried samples or at 17.5–18.0 Å on samples treated with ethylene glycol), illite (peaks at 10.1–10.2 Å), kaolinite (peak at 7.4 Å), and sometimes negligible amounts of chlorite (14.0–14.5 Å), quartz and feldspar. Variable amounts of calcite and dolomite phases were observed, possibly as a consequence of a localized variation of environmental parameters, such as a lowering of pH resulting in a selective dissolution of carbonates. S1 sample (Fig. 1) well represents common mineralogical composition of Po River sediments, at least for what concerns main mineral phases.
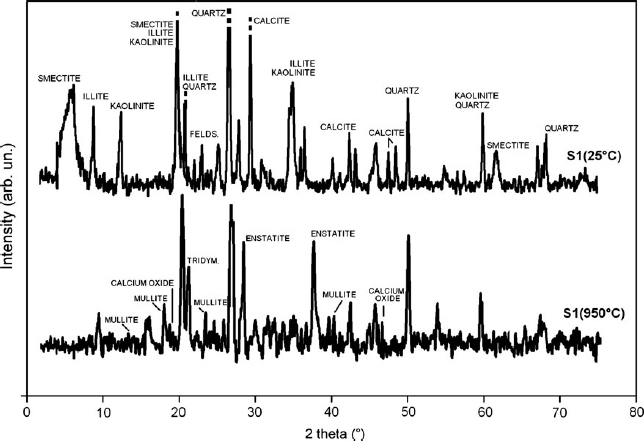
X-ray powder diffraction patterns of S1(25°C) and S1(950°C) obtained through an in-situ heating apparatus. See Malferrari et al. (2009) for further details.
Source: Malferrari et al. (2009).
As specified in D.Lgs. (2006).
Thermal analysis plots are shown in Fig. 2. The dehydration reactions, related to the interlayer sites of smectite, can be observed in the 25°C–100°C temperature range and the 100°C–200°C temperature range. These observations are consistent with the observations in Malferrari et al. (2009). The reactions that occurred in the 200°C–400°C temperature range are associated with the thermal decomposition of organic matter, and those that occurred in the 400°C–800°C range with dehydroxilation and decarbonizing reactions (Fig. 2a). Comparing these data with those collected by mass spectrometry, the broad reaction between 400°C and 600°C can be ascribed unequivocally to water (H2O), whereas the reaction at 716°C may be attributed to both H2O and carbon dioxide (CO2) loss.

Derivative thermo-gravimetric (DTG) curve of sample S1(25°C) in , DTG curve; 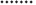 , calculation of the deconvoluted curves;
, calculation of the deconvoluted curves;  , fitting between DTG and deconvoluted curves.
, fitting between DTG and deconvoluted curves.
The best fit obtained from three Gauss-Voigt deconvolution curves in the temperatures ≥400°C is shown in Fig. 2b. The best fit in the 400°C–600°C temperature range shows the contribution of three main outcomes (420°C, 500°C, and 518°C) and suggests dehydroxylation reactions related to amorphous hydroxide, disordered kaolinite, and dioctahedral trans-vacant 2:1 layer silicates, respectively (MacKenzie, 1970). The best fit at 716°C was obtained with three Gaussian fitting curves with maxima at 665°C, 695°C and 710°C. The effects at 665°C and 695°C may be attributed to the dehydoxylation of cis-vacant sites pertaining to layers with different ordering (Drits et al., 1998). The effect at 710°C is clearly related to CO2 loss. SO2 release occurs in the 200°C–400°C temperature range and may be attributed to both organic matter decomposition (thiol groups) and thermal decomposition of sulfates or sulfurs not detected by X-ray diffraction because of their microcrystalline or amorphous nature. In the DTA curve, which is not shown in Fig. 2, the high temperature phase patterns occur at temperatures >950°C.
Phase transitions were also confirmed by XRD data. In the XRD pattern collected at 950°C (Fig. 1), the peaks observed at room temperature could no longer be observed, while newly generated ones associated with tridymite, mullite, calcium oxide (CaO), and enstatite-like phases could be clearly identified.
These chemical and mineralogical compositions, present at room temperature and high temperatures, suggest that sediments can be used as secondary raw material for a traditional ceramic paste (Worrall, 1986). Indeed, quartz, feldspar, and clay are the main components in ceramic pastes. The only issue that could limit the technological exploitation of the sediments may be the presence of organic matter and high Fe content. The high Fe content could be ascribed to smectite, chlorite, and the amorphous phases. Iron oxidation, coordination, and bonding can affect the fired product's properties.
Fig. 3 introduces XAS spectra of S1 samples before and after thermal treatment, which reached a temperature typical of the temperature reached during the industrial firing process. Remarkable changes can be observed after thermal treatment, especially in the pre-edge and edge regions (Figs. 3b and 3c). The differences between the two spectra are clearly ascribable to thermal treatment.
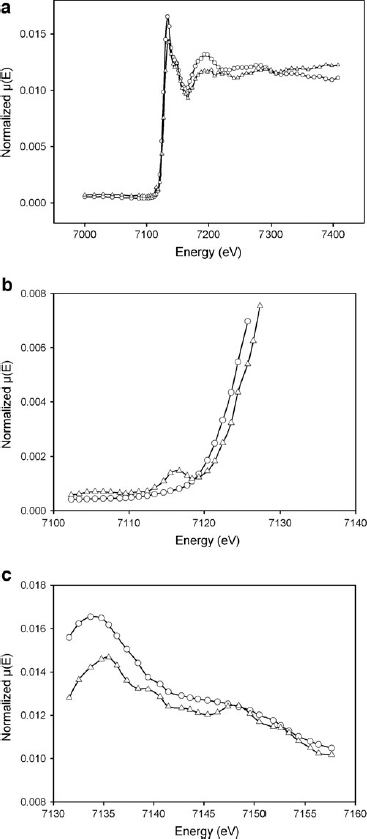
The best fit of the X-ray absorption near edge structures (XANES) region for S1(25°C) was obtained with the reference spectra associated with a mixture of 60% Fe-rich montmorillonite and 40% (Fe2+, Fe3+)-rich phlogopite (Fig. 4). Fe-rich montmorillonite is characterized by predominant octahedral Fe3+ content [Fe3+/(Fe3++Fe2+)=0.939], whereas (Fe2+, Fe3+)-rich phlogopite contains both Fe2+ and Fe3+ in the octahedral sites, with a slight predominance of Fe2+ [Fe3+/(Fe3++Fe2+)=0.474]. XANES spectra of S1(950°C) (heating at 950°C and fast cooling) are shown in Fig. 5a and can be reproduced with a mixture of Fe-rich montmorillonite (68%), tetra-ferriphlogopite (20%), and (Fe2+, Fe3+)-rich phlogopite (12%). Tetra-ferriphlogopite shows a significant Fe3+ for Si tetrahedral substitution, which is consistent with an effect in the pre-edge region as a consequence of the 1s→3d orbital transition (Fig. 5b). The modification of the pre-edge feature is also present in the S1(950°C) sample, thus confirming that Fe is present in a tetrahedral coordination, except in the S1(25°C) sample. The energy value, which characterizes the maximum of the pre-edge effect, is at ∼7116 eV in both the tetra-ferriphlogopite and S1(950°C) sample. This is consistent with a trivalent oxidation state (Berry et al., 2003), which better matches tetrahedral coordination requirements. These data are also in agreement with research that has found that tetrahedral Fe coordination is favored by fast cooling after heating at high temperature (Farges et al., 2005; Wilke et al., 2007).
Normalized, background-subtracted XAS spectra at the Fe K-edge for the S1(25°C) sample compared to

, tetra-ferriphlogopite; ----, fitting between S1(950°C) and the considered reference compounds.
A shoulder at 7124 eV in the edge region, evident in the S1(950°C) sample but not present in the S1(25°C), is also present in tetra-ferriphlogopite and may be associated with the forbidden transition 1s→4s (Fig. 5b).
The maximum associated with the 1s→4p transition can be observed at 7133 eV in the sample S1(25°C) and in Fe-rich montmorillonite. Slightly higher values (7135 eV) (Fig. 5c) characterize sample S1(950°C) and the tetra-ferriphlogopite.
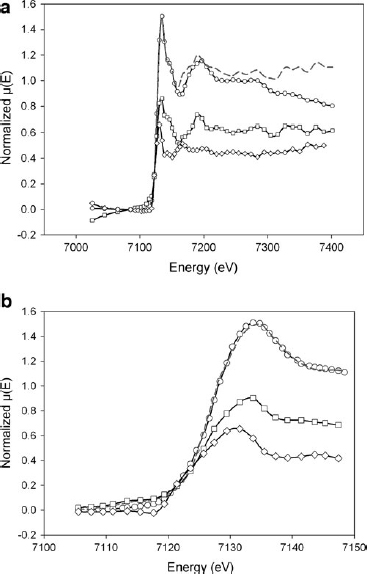
Interatomic distances in the S1(25°C) and S1(950°C) have been obtained by calculating the radial distribution function and comparing these data with several reference compounds that have extensive structural information available from X-ray single crystal refinements. Phase shift corrected radial structure functions of normalized and background-subtracted Fe K-edge EXAFS spectra, weighted by χ2, are reported in Fig. 6.
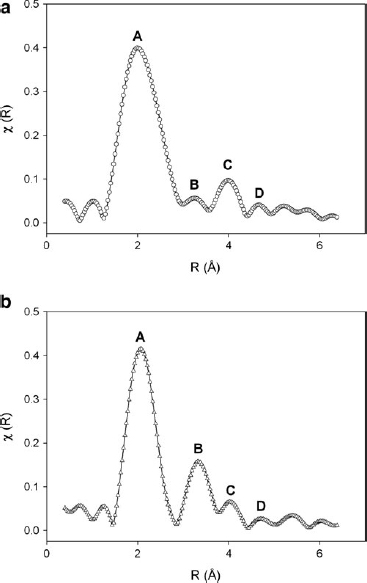
Phase-shift–corrected radial structure functions for normalized and background-subtracted Fe K-edge extended X-ray absorption fine structure spectra weighted by χ2 for
As previously noted, the studied sediments are very complex systems in which Fe local ordering can vary significantly. Thus, reported distances are mean values, derived from varying Fe coordination shells. Table 3 reports Fe–O and Fe–Fe distances refined for the first coordination shells. Fe–O mean distance values do not vary significantly before and after the thermal treatment. In both samples, three other shells follow the first one (shell A). In the S1(25°C) sample, these are shell B at 3.22 Å, shell C at 3.96 Å, and shell D at 4.63 Å; in the S1(950°C) sample, these are shell B at 3.28 Å, shell C at 4.02 Å, and shell D at 4.66 Å (Fig. 6).
Aa-Sa, relationships between central absorber and scattering atom; N, coordination number; R, refined interatomic distance; σ2, Debye–Waller factor; ΔE0, energy shift.
According to Manceau et al. (2000a, 2000b), shell B results from the combined effects of the nearest tetrahedral and octahedral cations. Shell C may result from the contribution of tetrahedral cations in the adjacent layers. Shell D originates from the contribution of two Fe atoms in cis-sites separated by a vacant site. The greater intensity of shell B in S1(950°C) may thus suggest an increased octahedral occupancy. With X-ray diffraction, these results consistently evidence the formation of enstatite-like phases.
Conclusion
The presence of iron in mineral structures is common. In soils and sediments, iron may form oxides and hydroxides that can precipitate as separate phases and/or be adsorbed on clay mineral surfaces. The distribution of iron among these various phases and its structural position may lead to the definition of the chemical and physical parameters relevant for remediation and recycling procedures. It is well known that the presence of trivalent iron in raw material as iron (hydr)oxide and/or in the octahedral sheets of 1:1 and 2:1 clay minerals considerably limits the overall quality of the final product, primarily because of the dark-red color imparted to the fired material. Even at low temperatures, a slowdown of the firing process is required to attain complete thermal decomposition of iron hydroxides. On the other hand, trivalent iron in tetrahedral coordination and divalent iron in octahedral coordination do not necessarily represent a severe limitation in the employment of sediments as raw material to obtain medium-high quality products.
The results of this study demonstrate that the presence of iron in smectite- and chlorite-like minerals does not affect the physical properties and use of sediments as secondary material in brick production. In the sediments used in this study, Fe was bonded primarily to such phyllosilicates such as smectite and illite, suggesting an octahedral, rather than tetrahedral, occupancy, and the presence of Fe3+ rather than Fe2+. This evidence was taken into account during firing. The fired products, which were structurally characterized, were the typical red color without macroscopic aesthetic defects.
Footnotes
Acknowledgments
The authors thank Italy's Ministero dell'Istruzione Università e Ricerca (MIUR) for its financial support in the project “Effect of petrologic variables on mica crystal chemistry” (PRIN 2006), the European Synchrotron Radiation Facility (ESRF) for the beam-time availability, and Dr. German Castro and the SpLine (BM25, Spanish Beamline) staff for their valuable help during the acquisition of XAS spectra. The support of Centro Interdipartimentale Grandi Strumenti (CIGS) of Modena and Reggio Emilia University and its staff are also gratefully acknowledged.
Author Disclosure Statement
No competing financial interests exist.