
Abstract
Abstract
Innovative catalysts of manganese oxides supported on Ce0.6Zr0.4O2 prepared by different methods (e.g., citric acid method, coprecipitation method, and combustion method) were studied for low-temperature selective catalytic reduction of NO x with NH3 at 80°C–260°C. Compared with MnO x /Ce0.6Zr0.4O2 (coprecipitation method) and MnO x /Ce0.6Zr0.4O2 (combustion method), MnO x /Ce0.6Zr0.4O2 (citric acid method) exhibited the best performance on selective catalytic reduction activity even in the presence of H2O and SO2. X-ray diffraction, N2 adsorption, X-ray photoelectron spectroscopy, and temperature-programmed experiments (H2-TPR and NH3-TPD) were used to characterize properties of the catalysts. Large surface area, small crystalline size, high surface chemisorbed oxygen, and high dispersion of manganese oxides might be the main reasons for the excellent performance of the catalysts.
Introduction
Manganese (MnO x )-based catalysts (Qi and Yang, 2003) have been reported to be highly active for low-temperature SCR of NO x with NH3 at a similar space velocity to that of V2O5-WO3/TiO2 catalysts. Therefore, it could be expected that high NO x conversion was achieved over the similar volume of low-temperature SCR catalysts compared with V2O5-WO3/TiO2 catalysts. Unsupported MnO x prepared by a precipitation method using sodium carbonate exhibited excellent activity for SCR of NO x with NH3 in the temperature range of 75°C–200°C (Kang et al., 2006). Supporting MnO x catalysts, such as MnO x /TiO2 (Ettireddy et al., 2007; Wu et al., 2008a; Jiang et al., 2009), MnO x /Al2O3 (Singoredjo et al., 1992; Kijlstra et al., 1997), and MnO x /ACF (Yoshikawa et al., 1998; Marbán and Fuertes, 2001), were also active for low-temperature SCR of NO x with NH3. The nature of the support was found to be a very important factor in SCR of NO x with NH3 (Smirniotis et al., 2006).
Because of high redox property, thermal stability, and catalytic activity, Ce x Zr1−xO2–mixed oxide has attracted much attention as a candidate for noble metal (Pd, Pt, or Rh) support in automotive three-way catalysts (Anderson et al., 2006; Guo et al., 2007, 2008). Ce x Zr1−xO2–supported other transition metals, such as CuO/Ce0.8Zr0.2O2 (Ma et al., 2003), CuO-CoO x /Ce0.67Zr0.33O2 (Liu et al., 2009b), and CuO/WO3/Ce0.5Zr0.5O2 (Li et al., 2005), were also investigated for abatement of NO and CO from automobile exhausts. Recently, Ce x Zr1−xO2–based catalysts were studied for SCR of NO with NH3; for example, WO3/CeO2-ZrO2 (Li et al., 2008) and V2O5/CeO2-ZrO2 (Putluru et al., 2009) have shown to be promising catalysts for SCR of NO with NH3 in the medium temperature range (200°C–450°C). However, there were few reports on the application of Ce x Zr1−xO2–based catalysts in low-temperature SCR of NO x with NH3 (80°C–260°C).
In this work, the innovative catalysts of MnO x supported on Ce0.6Zr0.4O2 prepared by different methods (citric acid method, precipitation method, and combustion method) were investigated for low-temperature SCR of NO x with NH3 under similar reaction conditions to that of MnO x -based catalysts (Qi and Yang, 2003). The H2O and SO2 tolerance of the catalysts under low-temperature SCR conditions was also considered.
Experimental
Preparation of Ce0.6Zr0.4O2
Three different preparation methods for Ce0.6Zr0.4O2 were introduced, namely citric acid method, coprecipitation method, and combustion method. The preparation methods are described below.
Citric acid method
A certain amount of Ce(NO3)3·6H2O and ZrO(NO3)2·2H2O were dissolved in water at 80°C, and the solution of citric acid was slowly added into the mixture solution under vigorous stirring. The molar ratio of Ce:Zr:citric acid was 0.6:0.4:1. The mixture was stirred at room temperature for 1 h. Then the solution was dried at 80°C for 12 h, and some white powders were obtained. These powders were calcined at 200°C for 6 h to produce the oxide and then calcined at 550°C for 6 h under static air in muffle furnace. The obtained sample was denoted as Ce0.6Zr0.4O2 (CA).
Coprecipitation method
After dissolving Ce(NO3)3·6H2O and ZrO(NO3)2 · 2H2O (Ce:Zr = 0.6:0.4) in water at 80°C, the ammonium hydroxide solution (25% ammonia) was slowly dropped into the mixture under vigorous stirring until pH 10. The blend was stirred at room temperature for 3 h and then aged for 1 h. The precipitation was washed with deionized water, filtered in vacuo, and dried at 80°C for 12 h. The obtained solid was calcined at 550°C for 6 h under static air in muffle furnace. The obtained sample was denoted as Ce0.6Zr0.4O2 (CP).
Combustion method
The solution (20 mL) of Ce(NO3)3·6H2O, ZrO(NO3)2 · 2H2O, and CO(NH2)2 (urea) with a molar ratio of Ce:Zr:CO(NH2)2 of 0.6:0.4:2.5 was directly calcined in a muffle furnace preheated to 550°C. The combustion mixture boiled and frothed, followed by the appearance of flame, yielding voluminous yellow solid materials within 5 min. The obtained sample was denoted as Ce0.6Zr0.4O2 (CB).
Preparation of MnOx/Ce0.6Zr0.4O2
MnO x /Ce0.6Zr0.4O2 was prepared by incipient wetness impregnation of Ce0.6Zr0.4O2 with an aqueous solution of Mn(NO3)2. The prepared samples were dried in a water bath at 80°C for 12 h and then calcined at 550°C for 6 h under static air in a muffle furnace. The obtained catalysts were denoted as MnO x /Ce0.6Zr0.4O2 (CA), MnO x /Ce0.6Zr0.4O2 (CP), and MnO x /Ce0.6Zr0.4O2 (CB), respectively. In all the catalysts, the molar ratio of Mn:Ce:Zr was 0.4:0.6:0.4. P25-TiO2 and γ-Al2O3, common SCR supports, were also used to prepare MnO x /P25-TiO2 and MnO x /γ-Al2O3 catalysts by the same procedure. The molar ratio of Mn:Ti (or Al) was designated as 0.4:1.
Catalytic activity test
The SCR activity of the catalysts for NO
x
removal with NH3 was carried out in a fixed-bed flow reactor. Six gas streams, 0.06 vol% NO, 0.06 vol% NH3, 3 vol% O2, 3 vol% H2O (when used), 0.01 vol% SO2 (when used), and pure N2 in balance, were used to simulate the flue gas. In all the runs, the total gas flow rate was maintained at 300 mL/min over 0.75 g of catalyst (40–60 mesh). The feed gases were mixed and preheated in a chamber before entering the reactor. The water vapor was generated by passing N2 through a heated gas-wash bottle containing deionized water (80°C). During the measurements, the concentrations of NO and NO2 at the inlet and outlet of the reactor were monitored by a Flue Gas Analyzer (KM940; Kane International Ltd.) equipped with NO, NO2, and SO2 sensors. The activities of the catalysts were measured by the conversion of NO
x
calculated as follows:
with [NO x ] = [NO] + [NO2].
Characterization of the catalysts
The surface areas (Brunauer-Emmett-Teller [BET] method) were determined by N2 isotherms at −196°C using a NOVA 2000 automated gas sorption system. The powder X-ray diffraction (XRD) patterns were recorded on a Rigaku D/Max 2500 system using Cu Kα radiation (40 kV and 100 mA). The X-ray photoelectron spectroscopy (XPS) was used to analyze the surface atomic state of the catalysts using a Kratos Axis Ultra DLD spectrometer equipped with a monochromatic Al Kα radiation (1,486.6 eV).
The temperature-programmed reduction (H2-TPR) and temperature-programmed desorption (NH3-TPD) experiments were performed on a tp-5080 automated chemisorption analyzer using 0.1 g catalysts. Prior to H2-TPR experiments, the samples were first pretreated in pure N2 at 500°C for 1 h and then cooled down to room temperature. TPR runs were carried out in a flow of H2 (5%) in N2 (30 mL/min) from room temperature to 900°C, with a heating rate of 10°C/min. For NH3-TPD experiments, after a pretreatment in pure N2 at 500°C for 1 h, the catalyst was cooled down to room temperature in pure N2 and then saturated for 30 min with a stream of pure NH3 (total flow rate = 1 mL/min [STP]). After saturation, the catalysts were flushed in a pure N2 flow for 30 min at 100°C. Finally, the TPD of NH3 was carried out in pure N2 at a heating rate of 10°C/min from 100°C to 500°C. The NH3 desorption (or H2 uptake) was measured quantitatively by a thermal conductivity detector.
Results
Structural and textural properties
The XRD patterns of Ce0.6Zr0.4O2 and MnO x /Ce0.6Zr0.4O2 for each method are shown in Fig. 1. For Ce0.6Zr0.4O2 in Fig. 1a, all profiles are attributed to the cubic, fluorite structure of CeO2 [(111), (200), (220), (311), (222), (400), and (311)]. The diffraction peaks for CeO2 phase became sharper in the order of Ce0.6Zr0.4O2 (CA) < Ce0.6Zr0.4O2 (CP) < Ce0.6Zr0.4O2 (CB). Figure 1b exhibits the XRD patterns of all three MnO x /Ce0.6Zr0.4O2 samples. The diffraction peaks for CeO2 phase could also be seen on all three MnO x /Ce0.6Zr0.4O2 XRD curves. Besides the CeO2 phase, several weak peaks assigned to Mn2O3 phase [(211), (400), (332), (440), and (622)] are observed as the secondary phase for MnO x /Ce0.6Zr0.4O2 (CB) and MnO x /Ce0.6Zr0.4O2 (CP). However, no Mn2O3 phase appeared for MnO x /Ce0.6Zr0.4O2 (CA).

X-ray diffraction patterns of fresh samples:
BET surface areas, crystallite sizes, and lattice parameters of all three MnO x /Ce0.6Zr0.4O2 samples are presented in Table 1 and compared with Ce0.6Zr0.4O2 samples. MnO x /Ce0.6Zr0.4O2 (CA) exhibited the largest surface area among all three MnO x /Ce0.6Zr0.4O2 samples. The crystallite sizes and lattice parameters of all samples were calculated according to Bragg's law and Debye–Scherrer equation. MnO x /Ce0.6Zr0.4O2 (CA) had small crystallite size compared with MnO x /Ce0.6Zr0.4O2 (CP) and MnO x /Ce0.6Zr0.4O2 (CB). The lattice parameter of MnO x /Ce0.6Zr0.4O2 (CA) was smaller than that of Ce0.6Zr0.4O2 (CA), whereas no variation was observed for Ce0.6Zr0.4O2 (CP) and Ce0.6Zr0.4O2 (CB) after the loading of manganese oxides.
Crystallite size and lattice parameters were both derived from (111) planes.
XPS characterization
Table 2 shows the surface elemental concentrations and atomic ratio of MnO x /Ce0.6Zr0.4O2 determined by XPS. The surface Mn concentrations and Mn/(Ce+Zr) ratio for MnO x /Ce0.6Zr0.4O2 (CP) and MnO x /Ce0.6Zr0.4O2 (CB) increased significantly compared with MnO x /Ce0.6Zr0.4O2 (CA). Especially for MnO x /Ce0.6Zr0.4O2 (CB), the surface Mn concentrations and Mn/(Ce+Zr) ratio reached 70.18% and 2.35%, respectively. According to above XRD results, crystalline Mn2O3 phase was formed on the surface of MnO x /Ce0.6Zr0.4O2 (CB), which might result in a high Mn/(Ce+Zr) ratio for MnO x /Ce0.6Zr0.4O2 (CB).
The oxidation states of Ce, Zr, Mn, and O were investigated by analyzing the photoelectron spectra of Ce 3d, Zr 3d, Mn 2p, and O1s levels obtained from XPS measurements. As shown in Fig. 2a, the curves of Ce 3d spectra were fitted with eight peaks in accordance with the fitting and labeling method reported elsewhere (Wu et al., 2008b). The relative percentages of the cerium species obtained by the area ratios of the Ce4+ (Ce4+ = U′′′ + U′′ + U + V′′′ + V′′ + V)/Ce3+ (Ce3+ = U′ + V′) are given in Table 2. From the results, it could be seen that the cerium species were mainly in Ce4+ oxidation state in all three MnO x /Ce0.6Zr0.4O2 samples. The typical examples of observed Zr 3d spectra are presented in Fig. 2b. The binding energy of Zr 3d5/2 between 182.10 and 182.35 eV indicated that Zr was present only in Zr4+ oxidation state (Alifanti et al., 2003; Sun et al., 2003).
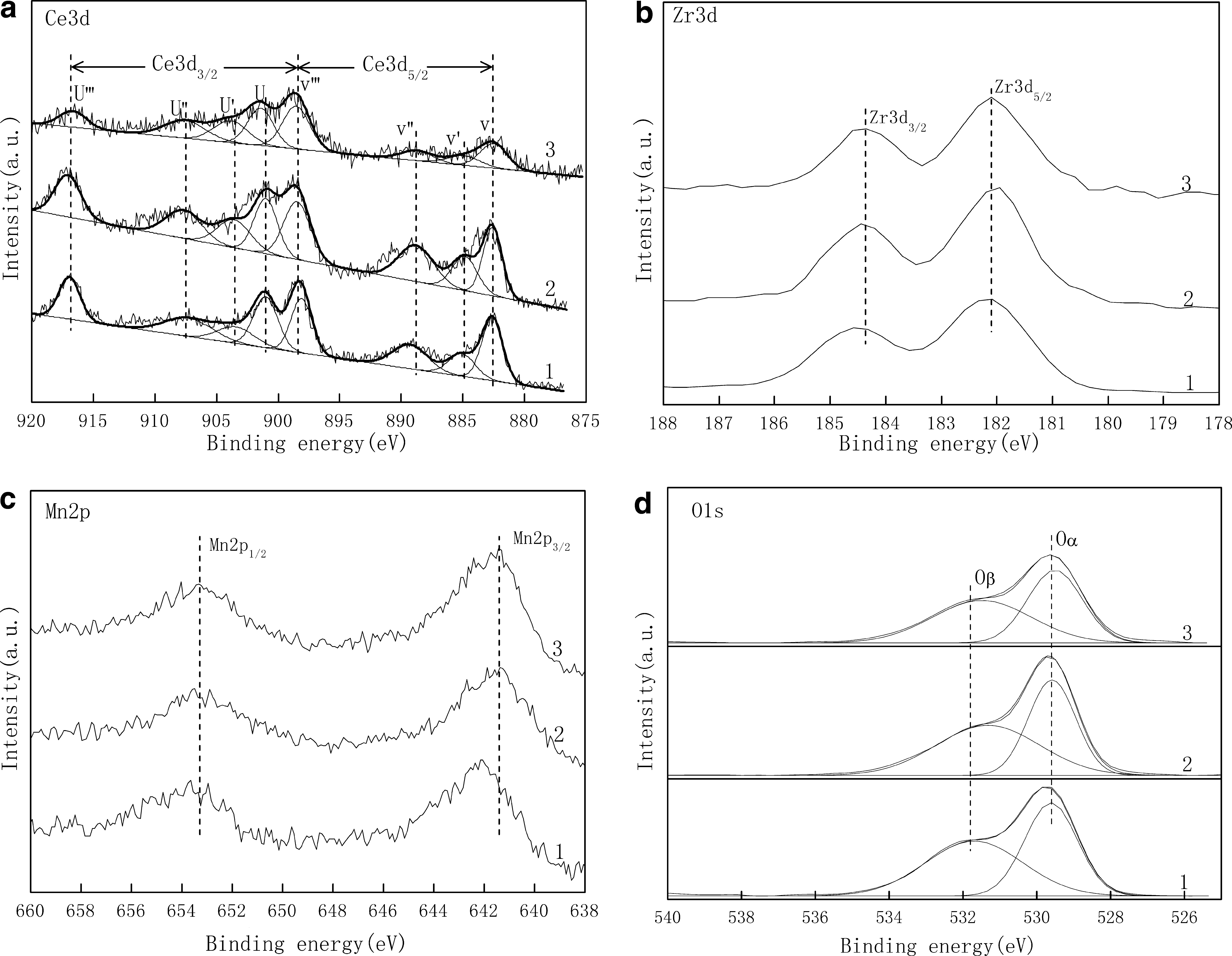
Figure 2c shows the Mn 2p photoelectron peaks of all three MnO x /Ce0.6Zr0.4O2 samples. For MnO x /Ce0.6Zr0.4O2 (CP) and MnO x /Ce0.6Zr0.4O2 (CB), the binding energies of Mn 2p3/2 fell in a very narrow range (641.3–641.5 eV). This value was in the range of Mn2O3 (641.3–641.7 eV) (Morales et al., 2006). For MnO x /Ce0.6Zr0.4O2 (CA), the binding energy of Mn 2p3/2 was 642.0 eV, which had been reported to be characteristic of a mixed-valence manganese system (Mn3+ and Mn4+) (Ettireddy et al., 2007).
The XPS spectra for O1s showed that the O1s spectra gave two distinct peaks (Fig. 2d). The peak at 529.6–530.0 eV was assigned to lattice oxygen (denoted as O α ) from metal oxides, whereas the one at 531.3–531.7 eV was assigned to chemisorbed oxygen (denoted as O β ), which had been reported to have a positive effect on SCR reaction (Wu et al., 2008b). As shown in Table 2 and Fig. 2d, the chemisorbed oxygen on the surface of all three MnO x /Ce0.6Zr0.4O2 samples increased in the order of MnO x /Ce0.6Zr0.4O2 (CB) < MnO x /Ce0.6Zr0.4O2 (CP) < MnO x /Ce0.6Zr0.4O2 (CA).
H2-TPR and NH3-TPD
The H2-TPR for Ce0.6Zr0.4O2 and MnO x /Ce0.6Zr0.4O2 samples are shown in Fig. 3a. Figure 3a shows Ce0.6Zr0.4O2 (CA), Ce0.6Zr0.4O2 (CP), and Ce0.6Zr0.4O2 (CB) have reduction peaks centered at 570°C, 534°C, and 591°C, respectively, which might be attributed to the reduction of Ce4+ in Ce0.6Zr0.4O2 samples (Wei et al., 2008). However, a shoulder peak at 827°C was observed in the TPR profiles of Ce0.6Zr0.4O2 (CP). As pointed out by recent studies, the shoulder peak at high temperature was assigned to the bulk reduction of cerium phase (Guo et al., 2007).
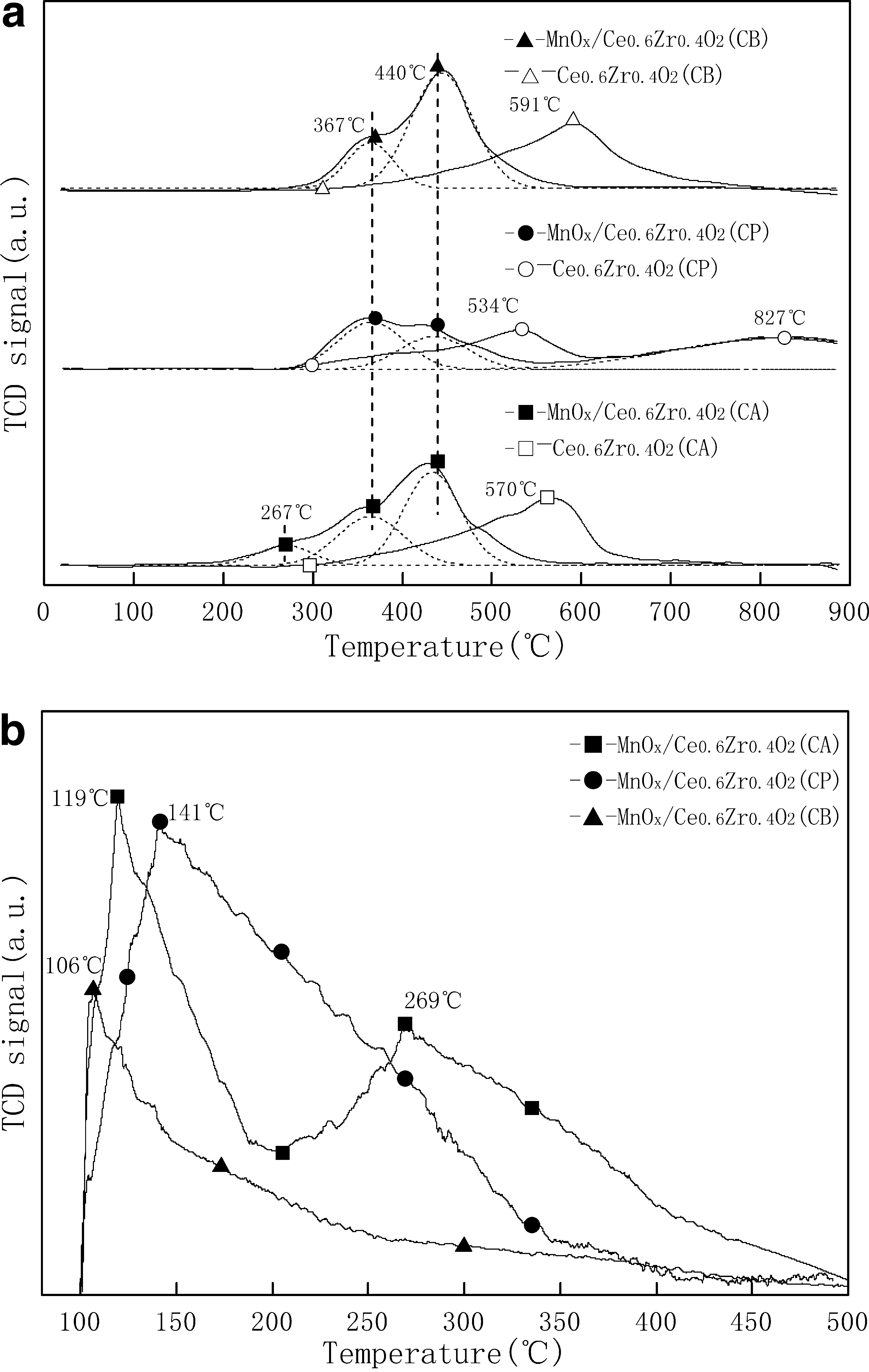
Compared with Ce0.6Zr0.4O2 for each method, the reduction peaks of corresponding MnO x /Ce0.6Zr0.4O2 shifted to low temperature. A similar behavior was also observed for Mn- and Cu-doped CeO2-ZrO2 solid solutions (Terribile et al., 1999). For MnO x /Ce0.6Zr0.4O2 (CB) and MnO x /Ce0.6Zr0.4O2 (CP), the reduction peaks emerged at 367°C and 440°C. A three-step reduction profile was observed for MnO x /Ce0.6Zr0.4O2 (CA). An additional shoulder peak appeared at 267°C except the other two peaks at 367°C and 440°C. The H2 consumption values of all three MnO x /Ce0.6Zr0.4O2 samples are given in Table 3. MnO x /Ce0.6Zr0.4O2 (CA) gave almost same H2 consumption for MnO x /Ce0.6Zr0.4O2 (CB) and MnO x /Ce0.6Zr0.4O2 (CP) samples.
The NH3-TPD was carried out to investigate the catalyst acidity for all three MnO x /Ce0.6Zr0.4O2 samples. The results of NH3-TPD runs performed over all three samples are shown in Fig. 3b. With increasing desorption temperature at 100°C–500°C, MnO x /Ce0.6Zr0.4O2 (CB) and MnO x /Ce0.6Zr0.4O2 (CP) exhibited similar NH3-TPD curves with one strong peak centered at 106°C and 141°C, respectively. The NH3-TPD curve for MnO x /Ce0.6Zr0.4O2 (CA) showed different features, with two desorption peaks centered at 119°C and 269°C, respectively. The peaks centered at 106°C, 119°C, and 141°C were assigned to the NH3 molecules adsorbed by weak acid sites, whereas the one centered at 269°C was assigned to the NH3 molecules adsorbed by moderate acid sites. This indicated that the chemisorbed NH3 molecules adsorbed by moderate acid sites might play a more important role in the adsorption of NH3 on MnO x /Ce0.6Zr0.4O2 (CA) than on the other two catalysts. According to the total acidity of all three MnO x /Ce0.6Zr0.4O2 samples as shown in Table 3, MnO x /Ce0.6Zr0.4O2 (CA) and MnO x /Ce0.6Zr0.4O2 (CP) had similar chemisorbed NH3 amount, which was comparatively higher than that of MnO x /Ce0.6Zr0.4O2 (CB).
Activity test
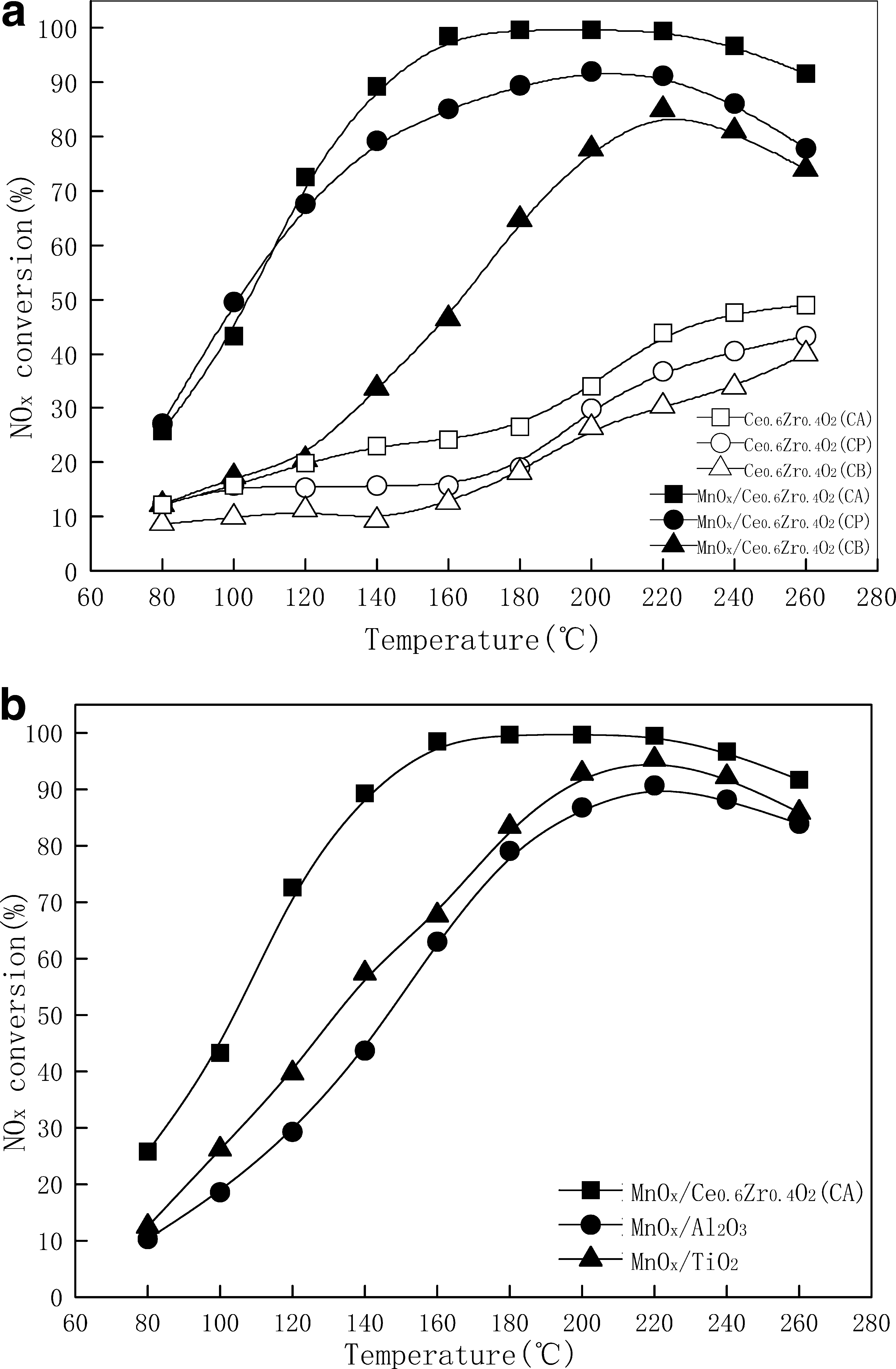
The catalytic activities for NO x removal with NH3 over all three MnO x /Ce0.6Zr0.4O2 samples were investigated at a temperature of 80°C–260°C. As shown in Fig. 4a, MnO x /Ce0.6Zr0.4O2 (CA) performed most efficiently, and nearly 100% NO x conversion was obtained at 160°C–220°C. MnO x /Ce0.6Zr0.4O2 (CP) showed nearly 90% NO x conversion at 160°C–220°C. For MnO x /Ce0.6Zr0.4O2 (CB), only 85% NO x conversion was obtained when the temperature reached 220°C. It could also been seen from Fig. 4a that low NO x -SCR activities were observed over all three Ce0.6Zr0.4O2 samples at a temperature of 80°C–180°C. However, the NO x -SCR activities were improved significantly when the temperature was higher than 180°C. Figure 4b shows the comparison of NO x conversion as a function of temperature over MnO x /Ce0.6Zr0.4O2 (CA), MnO x /P25-TiO2, and MnO x /γ-Al2O3. The results indicated that MnO x /Ce0.6Zr0.4O2 (CA) could provide higher NO x conversion than MnO x /P25-TiO2 and MnO x /γ-Al2O3, especially at a temperature of 80°C–180°C.
The NO oxidation activities of all three MnO x /Ce0.6Zr0.4O2 samples with the variation of temperature were also investigated (Fig. 5). For all three MnO x /Ce0.6Zr0.4O2 samples, the NO oxidation activity increased with the increasing temperature, especially at 180°C–260°C. It could also be seen from Fig. 5 that the NO oxidation activities of all three MnO x /Ce0.6Zr0.4O2 samples in the whole temperature range (80°C–260°C) increased in the following sequence: MnO x /Ce0.6Zr0.4O2 (CB) < MnO x /Ce0.6Zr0.4O2 (CP) < MnO x /Ce0.6Zr0.4O2 (CA).
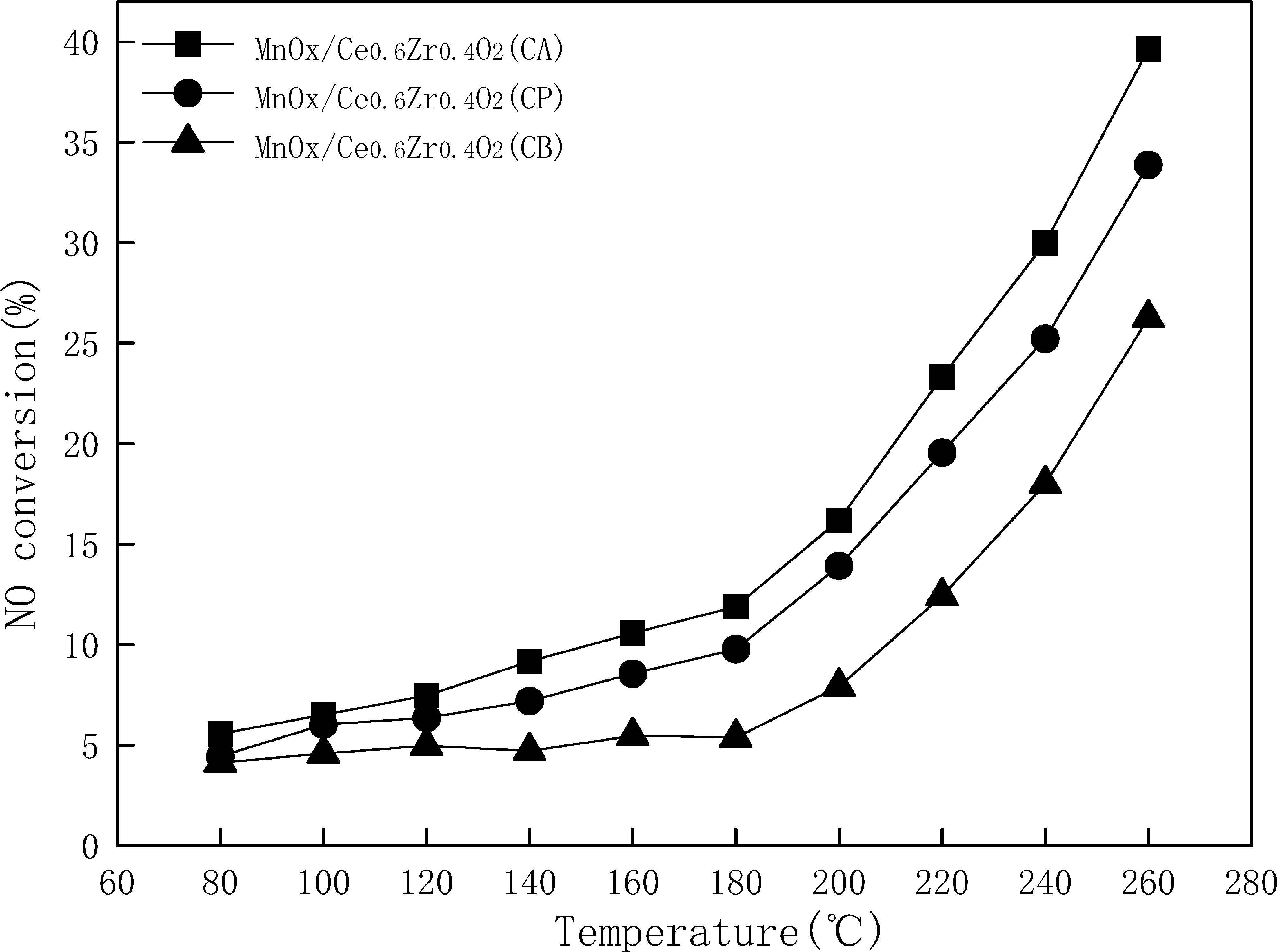
Oxidation activity of NO to NO2 by O2 over MnO x /Ce0.6Zr0.4O2 samples. Reaction conditions: 0.06 vol% NO, 3 vol% O2, balance N2, W/F = 2.5 × 10−3 g/(mL min).
Figure 6 shows the effect of H2O and SO2 on activities of all three samples for NO x removal with NH3 at 180°C. MnO x /Ce0.6Zr0.4O2 (CA) exhibited the best resistance to H2O and SO2 in the presence of 3 vol% H2O and 100 ppm SO2. A barely detectable decrease in NO x conversion over MnO x /Ce0.6Zr0.4O2 (CA) was observed in 6 h in the presence of 3 vol% H2O and 100 ppm SO2. The NO x conversion over MnO x /Ce0.6Zr0.4O2 (CP) decreased slowly to 79.1% and then stabilized under low-temperature SCR conditions including 3 vol% H2O and 100 ppm SO2. The effects of SO2 concentration on NO x conversion over MnO x /Ce0.6Zr0.4O2 (CA) and MnO x /Ce0.6Zr0.4O2 (CP) had also been studied and shown in the inset graph of Fig. 6. It could be seen that the effect of 3 vol% H2O and 200 ppm SO2 on NO x conversion over the catalysts was a little stronger than that of 3 vol% H2O and 100 ppm SO2. However, MnO x /Ce0.6Zr0.4O2 (CA) and MnO x /Ce0.6Zr0.4O2 (CP) exhibited excellent resistance to low concentrations of H2O and SO2 at 180°C anyway. For MnO x /Ce0.6Zr0.4O2 (CB), the catalytic activity decreased sharply upon 3 vol% H2O and 100 ppm SO2 addition into the flue gases, and the NO x conversion decreased from initial 65.6% to 13% in the first 6 h. The original SCR activity was not restored after cutting off of H2O and SO2 supply.

Effect of H2O and SO2 on NO x conversion over MnO x /Ce0.6Zr0.4O2 samples. Reaction conditions: 0.06 vol% NO, 0.06 vol% NH3, 3 vol% O2, balance N2, reaction temperature = 180°C, W/F = 2.5 × 10−3 g/(mL min).
Discussion
The XRD patterns of Ce0.6Zr0.4O2 samples displayed only diffraction peaks corresponding to fluorite structure of CeO2, and no diffraction peaks for ZrO2 phase were detected. As shown in Table 1, the lattice parameters of all three Ce0.6Zr0.4O2 samples were smaller than that of CeO2 (5.410 Å, JCPDS 78-0694). Because of the smaller ionic radius for Zr4+ (0.084 nm) compared with that of Ce4+ ions (0.097 nm), the incorporation of Zr4+ into CeO2 phase might decrease the lattice parameters of all Ce0.6Zr0.4O2 samples.
In the preparation of Ce0.6Zr0.4O2 (CA), citric acid was used to chelate various cations (Ce4+ and Zr4+) to form homogeneous metal chelates of citric acid, which might favor the small crystallite size and high surface area. Ce0.6Zr0.4O2 (CA) with large surface area and small crystallite size was favorable to be a catalyst support in the SCR of NO x with NH3. For Ce0.6Zr0.4O2 (CB), the small surface area (5.90m2/g) and large crystallite size (22.52 nm) indicated the agglomeration by combustion method.
According to XRD results, no intense peaks attributable to manganese oxides appeared for MnO x /Ce0.6Zr0.4O2 (CA), which indicated the high dispersion and/or low crystalline nature of manganese oxides in this sample. Crystalline Mn2O3 was formed using MnO x /Ce0.6Zr0.4O2 (CB) and MnO x /Ce0.6Zr0.4O2 (CP) catalysts. Therefore, manganese oxides dispersed well in MnO x /Ce0.6Zr0.4O2 (CA) compared with MnO x /Ce0.6Zr0.4O2 (CP) and MnO x /Ce0.6Zr0.4O2 (CB). It has been reported that amorphous manganese oxide performed well in SCR reaction, whereas the crystalline one contributed little to the activity (Wu et al., 2008a; Jiang et al., 2009).
From the H2-TPR of all three MnO x /Ce0.6Zr0.4O2 samples, the low-temperature reduction features attributable to the reduction of manganese oxides could be observed. According to XPS results, manganese oxides in MnO x /Ce0.6Zr0.4O2 (CP) and MnO x /Ce0.6Zr0.4O2 (CB) are exclusively in Mn3+ state, whereas Mn3+ and Mn4+ coexist in MnO x /Ce0.6Zr0.4O2 (CA). This might be the reason why MnO x /Ce0.6Zr0.4O2 (CA) presented a three-step reduction profile different from that of MnO x /Ce0.6Zr0.4O2 (CP) and MnO x /Ce0.6Zr0.4O2 (CB). The first reduction peak at 267°C might be attributed to the reduction of Mn4+ to Mn3+, which has also been observed for MnO x /TiO2 catalysts at 261°C because of the interaction between titania and manganese oxide (Ettireddy et al., 2007). The other two peaks (at 367°C and 440°C) could be speculated to be assigned to the reduction of Mn2O3 to Mn3O4, and Mn3O4 to MnO. Because of the exclusive presence of Mn2O3 in Ce0.6Zr0.4O2 (CB) and Ce0.6Zr0.4O2 (CP), the reduction peaks emerged only at 367.0°C and 440°C, which were due to the reduction of Mn2O3 to Mn3O4, and Mn3O4 to MnO, respectively. However, the reduction of Ce0.6Zr0.4O2 phase could not be distinguished clearly in H2-TPR curves, which indicated the complicated interaction between manganese oxide and Ce0.6Zr0.4O2 phase in all three catalysts.
XPS results indicated that there were different amounts of chemisorbed oxygen on the surface of all three samples. The chemisorbed oxygen has been reported to be helpful to the oxidation of NO to NO2, which could lead to enhanced SCR activities because of the occurrence of the “fast SCR”: NO + NO2 + 2NH3 → 2N2 + 3H2O (Liu et al., 2009a). It could be seen from Fig. 5 that the oxidation activities of all three samples are in good agreement with the variation in surface chemisorbed oxygen shown in Table 2. Therefore, it could be expected that MnO x /Ce0.6Zr0.4O2 (CA) with the highest chemisorbed oxygen in all three samples performed well in the low-temperature SCR reaction.
Because the SCR catalyst is usually deactivated mainly by water vapor (H2O) and residue SO2 in the flue gases, the resistance of the catalysts to H2O and SO2 has also been investigated at 180°C, as shown in Fig. 6. MnO x /Ce0.6Zr0.4O2 (CA) and MnO x /Ce0.6Zr0.4O2 (CP) performed well in the presence of 3 vol% H2O and 100 ppm SO2 in the flue gas. However, the deactivation of MnO x /Ce0.6Zr0.4O2 (CB) by 3 vol% H2O and 100 ppm SO2 was observed as some other SCR catalysts in previous literatures (Wu et al., 2009; Shen et al., 2010). At a low temperature, SO2 could be adsorbed by metal oxides in the catalysts, producing metal sulfates. On the other hand, some ammonium sulfates [such as NH4HSO4 and (NH4)2SO4] would be formed by the reaction between SO2 and the reactants (NH3 and O2). Because of small surface area, the active sites of MnO x /Ce0.6Zr0.4O2 (CB) would be occupied and deactivated more easily by the formed metal sulfates and ammonium sulfates than that of MnO x /Ce0.6Zr0.4O2 (CA) and MnO x /Ce0.6Zr0.4O2 (CP).
The lattice parameters of Ce0.6Zr0.4O2 (CP) and Ce0.6Zr0.4O2 (CB) did not change with the loadings of manganese oxides. However, the lattice parameter of MnO x /Ce0.6Zr0.4O2 (CA) was found to be smaller than that of Ce0.6Zr0.4O2 (CA). Some manganese ions with small ion radius (e.g., 0.067 nm for Mn3+ and 0.05 nm for Mn4+) might incorporate into the CeO2 phase, which led to the shrinkage of the lattice parameters for MnO x /Ce0.6Zr0.4O2 (CA). However, there were no more evidence to support the presumption for the behaviors of manganese oxides in MnO x /Ce0.6Zr0.4O2 (CA). Large surface area, small crystallite size, high dispersion of manganese oxide, and high surface chemisorbed oxygen observed for MnO x /Ce0.6Zr0.4O2 (CA) might be related to its high activity and good resistance to SO2 and H2O.
Conclusions
Among all three MnO x /Ce0.6Zr0.4O2 samples, MnO x /Ce0.6Zr0.4O2 (CA) exhibited the highest activity for SCR of NO x with NH3, and nearly 100% NO x conversion was obtained at 160°C–220°C. Compared with MnO x /Ce0.6Zr0.4O2 (CP) and MnO x /Ce0.6Zr0.4O2 (CB), MnO x /Ce0.6Zr0.4O2 (CA) showed excellent resistance to H2O and SO2 under low-temperature SCR conditions. According to the results of BET, XRD, and XPS, large surface area, small crystalline size, high dispersion of manganese oxides, and high surface chemisorbed oxygen were observed for MnO x /Ce0.6Zr0.4O2 (CA), which might be the main reasons for the excellent performance of MnO x /Ce0.6Zr0.4O2 (CA) catalyst.
Footnotes
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (NSFC 90610018 and 50976050) and the Key Special Technologies R&D Program of Tianjin (09ZCKFSH01900).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.